

Abstract
Hereditary diseases affecting the skeleton are heterogeneous in etiology and severity. Though many of these conditions are individually rare, the total number of people affected is great. These disorders often include dental-oral-craniofacial (DOC) manifestations, but the combination of the rarity and lack of in-depth reporting often limit our understanding and ability to diagnose and treat affected individuals. In this review, we focus on dental, oral, and craniofacial manifestations of rare bone diseases. Discussed are defects in 4 key physiologic processes in bone/tooth formation that serve as models for the understanding of other diseases in the skeleton and DOC complex: progenitor cell differentiation (fibrous dysplasia), extracellular matrix production (osteogenesis imperfecta), mineralization (familial tumoral calcinosis/hyperostosis hyperphosphatemia syndrome, hypophosphatemic rickets, and hypophosphatasia), and bone resorption (Gorham-Stout disease). For each condition, we highlight causative mutations (when known), etiopathology in the skeleton and DOC complex, and treatments. By understanding how these 4 foci are subverted to cause disease, we aim to improve the identification of genetic, molecular, and/or biologic causes, diagnoses, and treatment of these and other rare bone conditions that may share underlying mechanisms of disease.
Keywords: fibrous dysplasia of bone, osteogenesis imperfecta, familial hypophosphatemic rickets, hypophosphatasia, hyperphosphatemic familial tumoral calcinosis, Gorham-Stout disease
Introduction
Hereditary diseases affecting the skeleton, which we define to include the craniofacial and dental structure, are heterogeneous in etiology, onset, and severity. Although many of these conditions are individually rare (affecting less than 1 in 200,000), the total number of people affected is great (see the National Organization for Rare Disorders at www.rarediseases.org and the Rare Bone Disease Patient Network at http://www.usbji.org/projects/RBDPN_op.cfm). Rare disorders are difficult to diagnose and are easily misdiagnosed; those of the skeleton are no exception. While progress has been made in our understanding of the underlying causes, pathologies, and mechanisms of bone diseases, those of the dental-oral-craniofacial (DOC) complex are frequently overlooked or superficially reported. This has implications in terms of understanding, diagnosing, and treating affected individuals, in turn affecting health and quality of life.
Proper formation of the skeleton and dentition requires integration of numerous processes beginning in early embryonic development. These include patterning of the head, limbs, and skeletal/dental elements, cell migration and proliferation, differentiation to specialized cells, matrix secretion, biomineralization of bones and teeth, and remodeling of bone. For purposes of this review, we discuss 4 processes directly affecting the formation and function of craniofacial bones and teeth (Fig. 1) that can serve as models for an understanding of basic pathophysiologic processes in the broader sense: progenitor cell differentiation, extracellular matrix production, mineralization, and resorption, a key step in remodeling of bone, but not teeth. For each of these processes, we highlight a disease or small number of diseases that occur(s) when that stage goes awry. By discussing mechanisms of how bone and tooth functions are negatively affected in these specific cases, we aim to identify patterns that can ultimately be applied to improve the identification of genetic, molecular, and/or biologic causes of additional diseases by their shared skeletal and dental etiopathologies. Appendix Table 1 summarizes additional conditions under each disease process.
Figure 1.

Diseases affecting specific stages of bone formation and remodeling. This model outlines key stages of bone development (differentiation of stem/progenitor cells, matrix production, mineralization, and resorption) affected by the genetic disorders highlighted in this review. For each developmental process (indicated by text boxes), associated diseases caused by defects in that process are identified. Tooth development depends on parallel processes outlined for bone formation, though notably does not undergo physiological resorption as part of a remodeling process.
Defective Stem/Progenitor Cell Differentiation
Fibrous Dysplasia
Fibrous dysplasia of bone (FD; MIM 174800) is caused by somatic activating mutations in the GNAS gene encoding the G-protein-coupled receptor-associated 3′, 5′-cyclic adenosine monophosphate (cAMP)-regulating protein, Gsα. Mutations result in ligand-independent activation of the cAMP signaling pathway (Weinstein et al., 1991). In skeletal tissues, overproduction of cAMP impairs the ability of bone-forming mesenchyme to transition from stem-like immature cells to mature osteogenic cells. There is increased secretion of interleukin-6 and expression of receptor activator for NFκB ligand (RANKL), both of which contain cAMP response elements in their promoters (Riminucci et al., 2003). The outcome is abnormal bone formation and remodeling characterized by fibrous tissue proliferation and expansion of the marrow or diploë by fibrous tissue that contains immature, structurally abnormal woven bone (Riminucci et al., 1997). The phenotype is broad, ranging from a single skeletal site (monostotic) to massive disease affecting multiple bones (polyostotic) (Fig. 2A). The phenotype is determined by when, and in which clonal cell in the inner cell mass, the sporadic mutation arises during development. The degree of clinical sequelae likewise varies, from asymptomatic to disfigurement with loss of function, including blindness and deafness. When extra-skeletal sites are involved, such as skin (café-au-lait macules) (Fig. 2B) and/or gonads (precocious puberty) (Fig. 2C), the disease is known as McCune-Albright syndrome (MAS). Hyperthyroidism, hypophosphatemia, and growth hormone excess can also occur in FD/MAS (Figs. 2D, 2E). Craniofacial bones are the most commonly involved skeletal sites in FD, especially the skull base, maxilla, and midface (Lee et al., 2002; Kelly et al., 2008) (Figs. 2A, 2F, 2G). There can be extensive expansion of the skull, maxilla, and mandible, tooth displacement, severe malocclusion, and facial disfigurement (Akintoye et al., 2003, 2013) (Figs. 2G-2I). When associated with pituitary hypersecretion of growth hormone, overgrowth of craniofacial FD is associated with greater morbidity. Panoramic radiographs of jaw FD display ground glass trabeculation that may progress to mixed radiolucent/radiopaque lesions (Figs. 2J, 2K). Dental disorders associated with FD include enamel hypoplasia and hypomineralization, dentin dysplasia, odontoma, taurodontic pulp, and high caries index (Figs. 2I, 2L, 2M). It is unclear whether FD-related cAMP excess affects tooth development, but taurodontism (Fig. 2K) seen in FD may be the combined effect of FD and endocrine dysfunction.
Figure 2.

Fibrous dysplasia (FD). (A) Technetium-99 bone scintigraphy of a female patient with FD/McCune-Albright syndrome (MAS) shows patchy areas of increased radiotracer uptake by FD in the craniofacial bones (black arrowheads), pelvis, and long bones (black arrows). (B) Café-au-lait macules on the face of a boy with MAS macules display the typical jagged “coast of Maine” borders. (C) Testicular ultrasound of a patient with MAS displays heterogeneous mixed cystic and solid lesions characteristic of Leydig cell hyperplasia (white arrows). (D) Thyroid ultrasound of a patient with MAS and hyperthyroidism demonstrates characteristic heterogeneity with a cystic, “Swiss cheese”-like appearance (white arrow). (E) Magnetic resonance imaging of the brain of a patient with MAS-associated growth hormone excess displays a large, dumbbell-shaped growth-hormone- and prolactin-secreting pituitary macroadenoma (white arrows); the bright spot (black arrowhead) indicates the posterior pituitary. (F) Computed tomography (CT) of the head in this MAS patient shows right mandibular expansion by FD (white arrow). (G) Three-dimensional volume rendering of CT images of the same patient seen in panel F shows expansion of FD in the right mandible (white arrow), as well as smaller potential areas of FD in the right fronto-orbital region and midface (black arrowheads). (H) Severe expansion of the maxilla by FD (black stars) is associated with malocclusion and right-sided overbite (white arrow). (I) In another FD patient, malocclusion shows a right lingual crossbite (black arrows) and a left posterior open bite (white arrow), possibly from mandibular enlargement due to growth hormone excess. (J) Panoramic radiograph of mandibular FD shows radiolucent ground glass trabeculation (white arrow). (K) Another MAS patient with growth hormone excess displays generalized FD radiopacity, particularly in the maxilla, and taurodontic pulp chambers (black arrows). (L) Enamel hypomineralization (black arrowheads) and (M) dentin dysplasia (black arrowheads) are also dental features often seen in patients with FD/MAS. (N) Histologic features of FD shown in Goldner’s trichrome-stained section include immature irregular woven bone (WB) with Sharpey’s fibers (white arrows) in a fibrous tissue (FT) matrix of variable cellularity. Note the unmineralized osteoid (white star) indicating osteomalacia, and an osteoclast resorbing bone in Howship’s lacuna (black arrow). Images in C, D, F, E, and J reproduced with permission from Akintoye et al., 2013. Image in K reproduced with permission from Akintoye et al., 2003.
Diagnosis of FD and/or MAS involves careful clinical, radiographic, and occasionally histopathologic evaluation. Technetium-99 scintigraphy is useful for determining sites of skeletal involvement and disease burden. When this is combined with computed tomography (CT), the overall extent of the craniofacial disease can usually be determined in individuals by age 5 yr (Hart et al., 2007). Evaluation should include screening for hyperthyroidism, hypophosphatemia, and growth hormone excess. Histopathology of FD shows immature, irregular, woven bone, often with ectopic Sharpey’s fibers, abundant osteoid, in a fibrous stroma of variable cellularity, and often with active, osteoclast-mediated bone resorption (Fig. 2N).
The mainstay of FD treatment is surgery (Stanton et al., 2012). Intramedullary fixation devices are preferred for orthopedic procedures. However, craniofacial surgery should be undertaken with caution because of potential for the rapid regrowth of FD lesions. Optic nerve decompression is contraindicated in cases where nerves are constricted but vision is unaffected (Lee et al., 2002). Bisphosphonates are helpful to treat bone pain; however, no clear radiographic, functional, or histologic benefits have been demonstrated. There is evidence suggesting that, with age, mutant progenitor cells fail to self-renew, while surviving normal cells are able to form normal bone, thereby limiting the progression of FD lesions (Kuznetsov et al., 2008).
Defective Matrix Function
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a group of hereditary disorders characterized by variable bone fragility. Clinical diversity led to classification of OI into 4 subtypes based on clinical phenotype and mode of inheritance (Sillence et al., 1979). There are currently 15 OI subtypes caused by mutations involving 11 genes (Appendix Table 2). More than 90% of individuals with OI have heterozygous mutations in 1 of the 2 genes encoding type I collagen (CO1A1 and COL1A2), the most abundant matrix protein in bone, dentin, ligaments, sclera, and skin. OI types V-XV are caused by mutations in 1 of at least 9 different genes (Byers and Pyott, 2012). These OI types are rare (about 10% of total OI cases) compared with those caused by mutations in CO1A1 and COL1A2. There are hundreds of allelic mutations in these genes, and genotype-phenotype relationships remain unclear in many instances, although mutations affecting different collagen-coding genes and types of mutations (e.g., splice mutations vs. glycine substitutions) are associated with differing degrees of clinical severity (Marini et al., 2007). Other causative genes for OI affect crosslinking and stabilization of the collagen fibril (e.g. PLOD2, FKBP10) or mineralization of fibrils (e.g., BMP1, which cleaves the C-terminus of collagen propeptide) (Byers and Pyott, 2012; Martinez-Glez et al., 2012).
Craniofacial and dental features for OI types III and IV are summarized in Fig. 3. Affected individuals suffer from decreased bone mineral density, increased fracture risk, deformations, short stature, kyphoscoliosis, craniofacial deformities, and a tooth phenotype resembling dentinogenesis imperfecta (DI). Wormian bones (intrasutural bones with unusually situated centers of ossification) are associated with OI and have been correlated with disease severity (Semler et al., 2010). Additional clinical manifestations include blue sclera, easy bruising, hearing loss, joint hypermobility, and hernias (van Dijk et al., 2011). DI-affected teeth tend to be smaller than normal, and feature constriction at the cementum-enamel junction, diminished roots, blue-gray to yellow-brown discoloration, and pulp chambers that are progressively diminished and appear obliterated on radiographs (Majorana et al., 2010). It is unclear why some OI mutations are associated with a DI phenotype, while others are not.
Figure 3.
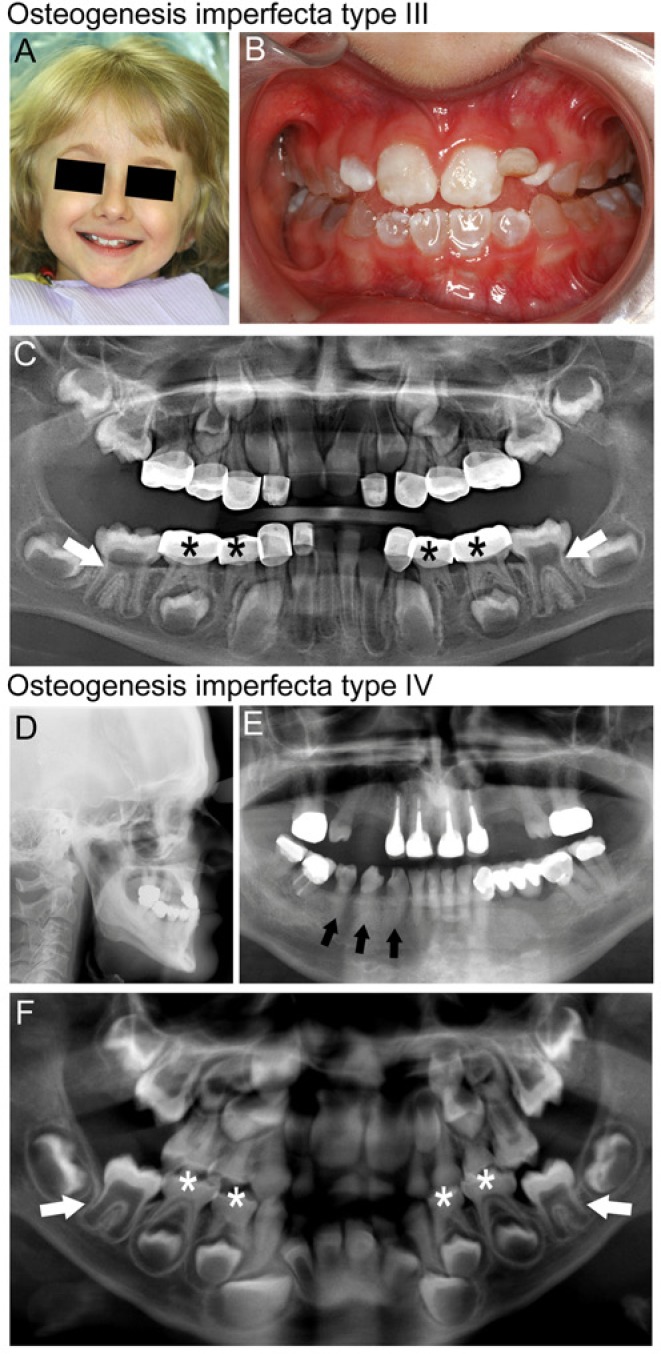
Osteogenesis imperfecta (OI). Clinical and radiographic findings in individuals with OI types III (A-C) and IV (D-F). (A) Facial appearance of child with OI type III shows a triangular face with prominent forehead and blue sclera. (B) Dentition of this child shows blue-gray discoloration characteristic of dentinogenesis imperfecta (DI). Discoloration is less pronounced in the maxillary permanent incisors than in the primary teeth. (C) A panoramic radiograph of the dentition of this child with OI type III shows cervical constriction and large pulp chambers in the six-year molars (white arrows), and diminished pulp space in the primary dentition (black stars) that has been restored with stainless steel crowns. (D) A cephalometric radiograph of an adult individual with OI type IV shows midface deficiency and a steep mandibular plane angle. (E) A panoramic radiograph of the dentition of the same individual displays obliteration of the pulp space and poor contrast of the roots to the bone due to the hypomineralized dentin (for example, mandibular teeth indicated by black arrows). This individual has required multiple root canals as a result of pulpal necrosis associated with DI. (F) A panoramic radiograph of an individual with OI type IV showing delayed dentin formation and large pulp chambers in the early-developing permanent dentition (see white arrows for examples), while the primary dentition is undergoing pulpal obliteration due to increased dentin formation (see white stars for examples).
Clinical manifestations of OI are managed by a combination of intra-osseous rods to reduce long bone fractures (Mulpuri and Joseph, 2000) and bisphosphonate infusions to improve bone density and manage bone pain (Ruck et al., 2011; Nicolaou et al., 2012). Oral bisphosphonates have been used with some success in milder OI cases. Despite concerns about bisphosphonate-induced osteonecrosis of the jaw, to date there are no reports of this condition in patients with OI treated with bisphosphonates (Milano et al., 2011; Christou et al., 2013).
Management of the dentition when DI is present depends on the severity, and whether enamel fracturing occurs and results in excessive tooth breakdown. Crowning of the teeth is the treatment of choice in these cases, since intra-coronal restorations are not retained in DI teeth that are affected by enamel loss and excessive dentin loss.
Mineralization Disorders
Familial Tumoral Calcinosis/Hyperostosis-Hyperphosphatemia Syndrome
Familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome (FTC/HHS; MIM 211900) is an autosomal-recessive disorder arising from functional deficiency of intact fibroblast growth factor 23 (iFGF23), a phosphaturic hormone produced primarily by osteocytes and osteoblasts. Prior to secretion into the circulation, iFGF23 undergoes O-linked glycosylation by the galactosyl transferase, ppGalNacT3, encoded by UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3). O-linked glycosylation protects iFGF23 from cleavage into inactive fragments by furin-like pro-protein convertases (Kato et al., 2006). iFGF23 signaling requires Klotho, a co-receptor that converts a generic fibroblast growth factor (FGF) receptor into a specific FGF23 receptor (Urakawa et al., 2006). The underlying genetic causes of decreased iFGF23 action include loss-of-function mutations in FGF23 (Benet-Pages et al., 2005), GALNT3 (Topaz et al., 2004), or KL (Ichikawa et al., 2007), which encodes Klotho (Farrow et al., 2011).
Decreased iFGF23 leads to stimulation of sodium phosphate co-transporters (NaPi-2a and-2c) and 25-OH-vitamin D3-1-α–hydroxylase in the proximal renal tubule. Hyperphosphatemia ensues from increased renal tubular reabsorption and increased gastrointestinal absorption of phosphate (Pi). In addition, inappropriately normal or frankly elevated 1,25-dihydroxyvitamin D3 [1,25(OH)2D] may enhance gastrointestinal absorption of Pi and calcium (Ca2+) (Farrow et al., 2011). The increased Ca2+ x Pi product contributes to ectopic calcification (Yamaguchi et al., 1995).
Clinical manifestations include periarticular, subcutaneous, and soft-tissue calcifications that tend to occur in areas of repeat trauma (Figs. 4A-4D). Calcifications may be associated with inflammation – ulceration of the overlying skin with milky drainage in severe cases. In addition to visible and palpable ectopic calcifications, radiographs often reveal cortical hyperostosis (Fig. 4E), which may cause diaphyseal pain of the long bones. Age-inappropriate calcification of the thyroid cartilage (Fig. 4F) has been described in one patient (Dumitrescu et al., 2009).
Figure 4.

Familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome (FTC/HHS). Clinical, radiographic, and histologic findings of patients with FTC/HHS. (A) Erythematous, inflamed subcutaneous calcifications of the neck and (B) periarticular ectopic calcification of the elbow in a 9-year-old girl. (C) Radiograph with extensive periarticular ectopic calcification of the right hip (white arrows). (D) Histology of ectopic calcification with amorphous calcium deposits (arrowheads) surrounded by a thick infiltration of mono- and poly-nucleated macrophages (“foreign body macrophages”), associated with a chronic inflammatory reaction. (E) Radiograph showing periarticular ectopic calcification of the right elbow (white arrow) and diaphyseal hyperostosis of the radius and humerus (white arrowheads). (F) Lateral neck radiograph showing age-inappropriate calcification of the thyroid cartilage in a 36-year-old woman (white arrow). (G) Panoramic dental radiograph of a 9-year-old girl with blunted, bulbous roots and obliteration of the pulp chamber in the maxillary and mandibular permanent central and lateral incisors. The erupting permanent premolars exhibit calcification at the root apices. Sclerotic bony changes are also present around the erupting teeth (white arrows). (H) Periapical radiograph of mandibular right sextant showing thistle-shaped roots (white arrows), midroot bulges (white arrowheads), and mineralized molar pulps and root canals (white star) in a 36-year-old woman. Images in F and H reproduced with permission from Dumitrescu et al., 2009.
Dental findings include short bulbous teeth, shortened roots, root dilacerations, thistle-shaped dental pulps, pulp chamber and root canal obliteration, and pulp stones (Figs. 4G, 4H) (Burkes et al., 1991; Dumitrescu et al., 2009). Radiographs of younger individuals suggest that abnormal calcification of the pulp occurs prior to root formation. Histologically, the root phenotype resembles dentin dysplasia II (MIM 125420), but the timing of calcification differs (Burkes et al., 1991). Alterations in gingiva and “velvety-red” lesions on the palate, mucosa, and tongue were noted in one family (Gal et al., 1994). However, other reports found no soft-tissue changes in the oral mucosa of patients with FTC/HHS. Large bilateral mandibular tori and bony sclerosis of the mandible, maxilla, and skull were reported in one patient (Krstevska et al., 2012).
Treatments for FTC/HHS have not been well-studied, and there is no standard regimen. Most reported therapies focus on lowering serum Pi. A low-Pi diet is generally encouraged, although difficult for many to achieve. Pi binders such as sevelamer and aluminum hydroxide may decrease Pi absorption in the gastrointestinal tract. Medications that promote renal excretion of Pi, such as acetazolamide and probenecid, are also used (Yamaguchi et al., 1995; Lammoglia and Mericq, 2009). Clinical responses vary, ranging from complete resolution of calcific tumors to development of additional ectopic calcifications. When inflammation is present, anti-inflammatory medications may be beneficial. Surgical debulking is sometimes necessary in patients with functional impairment or severe pain, but is not routinely done, since tumors often recur.
Hypophosphatemic Rickets
Rickets arises from defects in factors that regulate mineral ion homeostasis, including 1,25(OH)2D and FGF23, described above. 1,25(OH)2D functions in biomineralization by stimulating gastrointestinal absorption of Ca2+ and Pi, and initiating cell signaling via the intracellular vitamin D receptor (VDR). While the causes of rickets can be classified as primarily calciopenic or phosphopenic, evidence suggests that growth plate defects are primarily due to hypophosphatemia. In calciopenic forms of rickets, both nutritional vitamin D deficiency and rare inherited conditions affecting vitamin D action [loss-of-function of 1α(OH)ase from CYP27B1 mutations in vitamin D-dependent rickets type 1, VDDR-I (MIM 264700), or loss of VDR function in VDDR-IIa (MIM 277440)], lead to hypocalcemia and hypophosphatemia (the latter induced by secondary hyperparathyroidism).
Many hereditary forms of primary hypophosphatemic rickets arise from mutations affecting renal Pi handling. The most common type of hypophosphatemic rickets is X-linked hypophosphatemia (XLH; MIM 307800), caused by inactivating mutations in PHEX (phosphate-regulating endopeptidase homolog, X-linked). More recently, mutations in the gene for dentin matrix protein 1 (DMP1) were linked with type 1 autosomal-recessive hypophosphatemic rickets (ARHR1; MIM 241520). This was a surprise, since DMP1 was known as an extracellular matrix protein in dentin and bone. ARHR also results from mutations in the ENPP1 gene encoding ectonucleotide pyrophosphatase phosphodiesterase 1 (ARHR2; MIM 613312), a membrane-bound ectoenzyme. Loss-of-function of ENPP1 was previously associated with generalized arterial calcification of infancy. It remains unclear why the disparate mutations responsible for XLH, ARHR1, and ARHR2 all result in elevated iFGF23. Increased iFGF23 is thought to be the driving force for hypophosphatemia and reduced 1,25(OH)2D, which contribute to disturbances in skeletal and dental mineralization. Autosomal-dominant hypophosphatemic rickets (ADHR; MIM 193100), with a similar clinical and biochemical phenotype, is caused by missense mutations in the FGF23 gene, resulting in a proteolytically resistant FGF23, thus increasing circulating iFGF23.
The skeletal manifestations of these disorders can be separated into rickets, growth plate abnormalities that occur in children, and osteomalacia, defective mineralization that occurs throughout the skeleton in children and adults. Rickets manifests clinically as widening of the distal long bones, often evident at the wrists, knees, ankles, and ribs, radiographically as widened growth plates with frayed and cupped metaphyses (Fig. 5A). Osteomalacia, causing bowing deformities, is a tissue diagnosis where staining demonstrates unmineralized (osteoid) matrix (as in Fig. 2N or 5I). Craniofacial manifestations include enlarged cranial sutures, delayed fontanelle closure, craniotabes (skull bone thinning and softening), and malformations of the cranium, including parietal and occipital flattening and frontal bossing. Craniosynostosis has been documented in VDDR and XLH, often in the second or third year of life, leading to scaphocephaly (anterior-posterior elongation of the cranium), elevated intracranial pressure, and papilledema (optic disc swelling) (Glass et al., 2011) (Figs. 5B, 5C). Cephalometric analysis in patients with osteomalacia shows alterations in bones of both endochondral and intramembranous origin (Gjørup et al., 2011). While the mineralization disorders and malformations are explained by hyperosteoidosis, the etiology for craniosynostosis remains unclear. One hypothesis involves FGF23-FGFR2 signaling in the areas of the sutures.
Figure 5.

X-linked hypophosphatemic rickets (XLH) and hypophosphatasia (HPP). Representative images of individuals with hypophosphatemic rickets (A-G) and hypophosphatasia (H-O). (A) Skeletal pathology resulting from osteomalacia includes bowing of the femur (arrow) due to softening of the bone. (B) The computed tomography (CT) scan of a 3-year-old female with XLH shows scaphocephaly (elongation of the skull), suggestive of premature fusion of the sagittal suture. (C) Brain CT (left image) from the same patient shows enlargement of optic nerve sheaths (red arrows) due to papilledema (optic disc swelling), while orbital CT shows ventricular narrowing and decreased size of ventricles (white arrows) due to synostoses, contributing to elevated intracranial pressure. (D) Enamel discoloration indicating hypoplasia in a juvenile individual diagnosed with XLH, and (E) dental radiograph from the same individual features thin dentin and wide pulp chambers (white arrows). (F) Histologic staining reveals interglobular dentin hypomineralization, reflecting inhibition of mineralization foci to fuse into a unified mineralization front, a pattern replicated in (G) scanning electron microscopy from an adult individual with XLH. (H) Radiograph of pseudofracture of the proximal femur (white arrow) in adult diagnosed with HPP. (I) Goldner’s trichrome stain of iliac crest biopsy from adult diagnosed with HPP, exhibiting excess osteoid (red layer indicated by black arrow). (J) Magnetic resonance imaging (MRI) of the skull of a six-year-old individual diagnosed with childhood HPP exhibiting craniosynostosis and resulting bregmatic eminence (white arrow). (K) Radiograph of a four-year-old HPP individual with hypomineralization of cranial vault exhibiting severe “copper beaten” skull appearance, as well as deformations of cranial vault shape. Alterations in cranial shape in J and K are indicative of increased cranial pressure. (L) Clinical photograph of a fourteen-year-old individual with HPP who experienced spontaneous exfoliation of the lower mandibular incisor during toothbrushing. (M) Panoramic radiograph of the same individual revealing delayed eruption of several permanent teeth (e.g., lower premolars), enlarged pulp chambers, and thin dentin. (N) Periapical radiograph of the same patient at 20 yr old, showing loss of lower incisor, endodontic treatment, and splinting of remaining incisors, with generalized loss of alveolar bone. (O) Scanning electron microscopy of incisor root from HPP patient indicates lack of cementum and exposure of dentin (*), as well as extensive resorption over the root surface. Images in B and C adapted from Glass et al. (2011); reproduced under open access policy. Images in D-F are adapted from Pereira et al. (2004) and are reproduced with permission. Image in G was adapted from Foster et al. (2013) and is reproduced by permission. Images in H and I are adapted from Berkseth et al. (2013) and are reproduced with permission. Images in J and K are adapted from Collmann et al. (2009) and are reproduced with permission. Images in L, M, and O are adapted from Rodrigues et al. (2012) and are reproduced with permission. Image in N is adapted from Rodrigues et al. (2012), and is reproduced by permission.
Delayed tooth eruption is frequently cited in cases of XLH (Foster et al., 2014). There have been several case reports on dental manifestations of VDDR and XLH, and limited numbers of publications on ARHR and ADHR are consistent with these pathologies. The rachitic tooth features defects in all the dental hard tissues. These include hypoplastic enamel that is easily abraded and fractured and subject to caries, thin and poorly mineralized dentin, enlarged pulp chambers, predisposition to dental abscesses, short and malformed roots, periodontal disease, and sometimes malocclusion (Figs. 5D-5G). Animal studies show that enamel defects arise from a combination of altered vitamin D metabolism along with hypocalcemia and hypophosphatemia (Berdal et al., 2011). Dentin mineralization is consistently affected across all forms of hypophosphatemic rickets, with the histologic presence of interglobular dentin indicating inhibition of growth of mineralization foci, likely arising from a combination of hypophosphatemia, elevated levels of circulating mineral inhibitors, and reduced active 1,25(OH)2D. The hypomineralized dentin is prone to fractures and predisposes teeth to infection and abscess. Periodontal problems likely arise from a combination of osteomalacia in the alveolar bone and root surface defects in the cementum (Foster et al., 2014).
Vitamin D disorders are treated with vitamin D analogues and Ca2+; VDDR-II is refractory to calcitriol due to impaired VDR function. Children with hypophosphatemic rickets are typically administered calcitriol and Pi, which can improve skeletal (and dental) manifestations (Chaussain-Miller et al., 2007). In cases of craniosynostosis, surgical interventions such as cranial vault expansion are necessary to allow for cranial growth and to alleviate intracranial pressure (Murthy, 2009).
Hypophosphatasia
Hypophosphatasia (HPP) is characterized by poor bone mineralization resulting from loss-of-function mutations in ALPL, encoding tissue-nonspecific alkaline phosphatase (TNAP) (Whyte, 1994). More than 267 HPP-associated mutations in ALPL have been identified, passed in dominant or recessive mode (http://www.sesep.uvsq.fr/03_hypo_mutations.php). TNAP is an ectoenzyme that hydrolyzes (thus reducing) inorganic pyrophosphate (PPi), a potent mineralization inhibitor. In addition to expression by chondrocytes and osteoblasts, TNAP is found in the dentoalveolar complex, with wide expression in the periodontium, dentin-pulp complex, and enamel organ (Foster et al., 2012).
Six clinical forms of HPP have been identified, varying in severity and age of onset (Reibel et al., 2009). Infantile and childhood HPP (MIM 241500, 241510) causes a widely hypomineralized skeleton (including rickets), short stature, and waddling gate. Adult HPP (MIM 146300) presents during middle age and features osteomalacia, bone pain, and stress fractures (Figs. 5H, 5I) (Berkseth et al., 2013). An intriguing aspect of HPP is the tooth-specific clinical isotype, odontohypophosphatasia, in which serum ALP is decreased and urinary phosphoethanolamine increased, but skeletal development is not (evidently) impaired, and tooth loss is the primary clinical sign (Mornet, 2007).
Severe forms of HPP feature hypomineralized regions of calvarial bone that radiographically appear as widely open fontanelles, though functional craniosynostosis (closure) can occur, leading to increased intracranial pressure and a bulging anterior fontanel (Whyte, 1994; Collmann et al., 2009) (Figs. 5J, 5K). Wormian bones are sometimes present in infantile and childhood HPP.
Observations of premature tooth loss led to the finding that root acellular cementum is defective in HPP, resulting in compromised periodontal attachment (Figs. 5L-5O). Reduction in alveolar bone height reflects bone loss due to poor periodontal attachment. Defects in dentin and enamel mineralization have been reported, though not consistently. The dentin phenotype includes widened pulp chambers, “shell teeth” with thin root dentin, and short, abnormally shaped roots (Figs. 5L-5N), related to developmental defects in the initial mantle dentin layer (Foster et al., 2012), likely parallel to effects causing osteomalacic bone and similar to effects in hypophosphatemic rickets, described above.
Results from translational studies and clinical trials with a recombinant bone-targeting TNAP suggest that this is a potentially life-saving treatment for severe HPP, with potential to improve skeletal and dental function in less-impaired individuals with HPP (Millán et al., 2008; Whyte et al., 2012).
Bone Resorption
Gorham-Stout Disease
Gorham-Stout disease (GSD), also known as massive osteolysis, vanishing or disappearing bone disease, is an extremely rare disorder (MIM 123880). The first patient was described in 1938, and in 1955, Gorham and Stout described the disease as “angiomatosis,” or proliferation of small, thin-walled blood vessels (Gorham and Stout, 1955). With about 200 cases reported in the literature, much about GSD remains unknown.
Histopathologically, the disease is a benign vascular proliferation of endothelial channels within bone, leading to extreme thinning of bony trabeculae, osteoclast-mediated resorption, and replacement of bone with fibrous tissue. Gorham and Stout postulated that hemangiomatosis led to hyperemia, causing excessive bone destruction. Hypotheses have included local hypoxia/acidosis and endothelial dysplasia (Heyden et al., 1977; Young et al., 1983), enhanced osteoclastic activity or increased sensitivity of osteoclast precursors to humoral factors (e.g., IL-1, IL-6, and TNF-α) (Devlin et al., 1996; Moller et al., 1999; Hirayama et al., 2001), or trauma (Al-Jamali et al., 2012). Another hypothesis proposes GSD as a form of lymphangiomatosis, a disease of soft tissue secondary to aberrant proliferation of lymphatic vasculature, placing these diseases on the same pathophysiologic spectrum.
GSD occurs sporadically, without apparent predilection for age, race, or gender. Most cases occur in individuals under the age of 40 yr, affecting mostly children and young adults. The disease can be progressive (Fig. 6) and lethal when associated with extensive involvement of the ribs, vertebrae, and pleural effusions. However, in some cases, it may be self-limiting. GSD can be monostotic or polyostotic; affected areas include the shoulder, pelvis, scapula, clavicle, ribs/sternum, femur, and skull. About 30% of reported patients with GSD have maxillofacial involvement, most frequently in the mandible (Kiran and Anupama, 2011). Pain is not a common finding; however, during the acute resorptive phase, pain, swelling, and progressive deformity may be present. In the dentoalveolar region, findings may include mobile teeth, malocclusion, mandibular deviation, bony deformity, and pathologic fractures (Fig. 6).
Figure 6.

Gorham-Stout disease (GSD). Progressive destruction of the mandible in an individual with GSD, initially presenting at age 4 yr with mobility of left mandibular primary teeth, and premature exfoliation of the left mandibular primary first molar. (A) Panoramic radiograph, age 6 yr. Note normal complement of permanent teeth (left mandibular primary first molar #L missing) and abnormal left mandibular condyle. (B) Computed tomography (CT), age 6 yr. Destruction of normal cortical architecture of mandibular condyle, ramus, and body, bilaterally (white arrows). (C) Panoramic radiograph, age 7 yr (post-treatment with Interferon-α2b and zoledronic acid). Further bony changes with complete loss of left mandibular angle (white arrow). (D) CT, age 7 yr. Remodeling changes of the left mandibular condyle and ramus (white arrows). (E) Panoramic radiograph, age 10 yr. Loss of bony continuity in the left mandible (white arrow); teeth appear to be “floating.” Permanent tooth formation has progressed (left mandibular canine, white star). (F) CT, age 10 yr. Tooth buds appear to hold remaining bone together in left mandible. (G) CT, age 12 yr. Interval disease progression and bone loss of the bilateral mandible. Radiographs courtesy of Dr. Leonard B. Kaban, Massachusetts General Hospital, Department of Oral and Maxillofacial Surgery, Boston, MA, USA.
Radiographically, bone undergoes a series of changes in GSD. The first stage is marked by patchy bone with intramedullary and subcortical lucencies. In the second stage, new areas of lucency appear, with confluence of the lesions. The third stage involves adjacent soft tissues and cortical breakdown. The last stage results in complete resorption of bone and replacement with fibrous tissue. Magnetic resonance imaging (MRI) shows low signal intensity on T1-weighted imaging and high signal intensity on T2, with enhancement on contrast imaging (Ruggieri et al., 2011). Technetium bone scan is not a reliable modality, since intensity is stage-dependent. In the maxillofacial region, panoramic radiographs and CT scans are preferred for monitoring disease progression. While tooth resorption has not been reported, tooth loss may result from reduced alveolar bone.
Knowledge gaps in pathophysiology have prevented standardized treatment guidelines for GSD and lymphangiomatosis. Surgery is necessary for pathologic fractures and reconstruction after massive resorption. Prostheses may offer advantages over bone grafts because of the potential for recurrent osteolysis. Radiotherapy may limit disease spread by inhibiting the proliferation of disease-causing cells (Lowing et al., 2007). Medical therapies include bisphosphonates and interferon gamma; however, neither has demonstrated definitive benefit (Kuriyama et al., 2010). Additional studies have focused on the proliferation of endothelial cells, and the role of the mTOR pathway in lymphangiogenesis, leading to new potential therapeutic targets (Reinglas et al., 2011). Prognosis depends on the extent and location of affected areas. Mild disease may stabilize and ultimately reverse over time, while severe cases involving the craniofacial and/or thoracic areas may be fatal.
Conclusion
In this review, we outlined the effects of rare skeletal diseases on the DOC complex, focusing on 4 physiologic processes exemplified by specific genetic defects: FD as a defect in progenitor cell differentiation, OI caused by defective matrix production, FTC/HHS and HPP as mineralization disorders, and GSD as an example of excessive bone resorption (Fig. 1). From these examples, it is apparent that the formation and function of the skeleton and dentition are compromised when defects arise in any one of these processes. Other rare bone diseases with DOC effects are summarized in Appendix Table 1.
In addition to the conditions covered here, numerous undiagnosed skeletal diseases exist. Overlap between the underlying pathophysiology of these etiologically elusive conditions with that of known diseases may point to commonalities in their mechanisms. By understanding how defects in the development and maintenance of bones and teeth are subverted to cause disease, we aim to broaden the understanding, diagnosis, and treatment of rare skeletal diseases sharing underlying mechanisms. This is a paradigm currently used by the NIH Undiagnosed Disease Program (Office of Rare Disease Research; http://rarediseases.info.nih.gov/) and a goal of skeletal and craniofacial researchers elsewhere. This approach has led to the diagnosis of newly recognized diseases such as arterial calcification due to deficiency of CD73 (ACDC; MIM 211800), and identification of causative mutations in rare diseases including Raine syndrome (MIM 259775) and Winchester syndrome (MIM 277950).
When new rare skeletal diseases are recognized, it is important for these patients to be referred to a craniofacial/dental team for clinical evaluation, the collection of clinical materials for translational studies, the monitoring of development over time, and treatment when possible. Only through this careful approach can we accomplish 3 issues of paramount importance: (1) to provide answers and appropriate care to individuals suffering from conditions that have eluded diagnosis, (2) to advance medical knowledge about rare diseases of the skeleton and dentition, and (3) to develop and improve treatments for these diseases.
Supplementary Material
Acknowledgments
We thank Alan Hoofring (NIH) for Fig. 1 illustration. We recognize the International Association for Dental Research (IADR) and its Executive Director, Dr. Christopher Fox, for calling for this timely review, acknowledge the important support of patient advocacy groups, and thank affected individuals who have participated in studies and provided consent to be included in publications.
Footnotes
This research was supported by the National Institute of Dental and Craniofacial Research (NIDCR) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) Intramural Research Programs, and by NIDCR/ National Institutes of Health (NIH) grant 1R56DE022932 (SOA).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Akintoye SO, Lee JS, Feimster T, Booher S, Brahim J, Kingman A, et al. (2003). Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 96:275-282. [DOI] [PubMed] [Google Scholar]
- Akintoye SO, Boyce AM, Collins MT. (2013). Dental perspectives in fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol 116:e149-e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jamali J, Glaum R, Kassem A, Voss PJ, Schmelzeisen R, Schon R. (2012). Gorham-Stout syndrome of the facial bones: a review of pathogenesis and treatment modalities and report of a case with a rare cutaneous manifestations [sic]. Oral Surg Oral Med Oral Pathol Oral Radiol 114:e23-e29. [DOI] [PubMed] [Google Scholar]
- Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. (2005). An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 14:385-390. [DOI] [PubMed] [Google Scholar]
- Berdal A, Molla M, Descroix V. (2011). Vitamin D and oral health. In: Vitamin D. Feldman D, Pike JW, Adams JS, editors. San Diego, CA: Academic Press, pp. 521-532. [Google Scholar]
- Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. (2013). Clinical spectrum of hypophosphatasia diagnosed in adults. Bone 54:21-27. [DOI] [PubMed] [Google Scholar]
- Burkes EJ, Jr, Lyles KW, Dolan EA, Giammara B, Hanker J. (1991). Dental lesions in tumoral calcinosis. J Oral Pathol Med 20:222-227. [DOI] [PubMed] [Google Scholar]
- Byers PH, Pyott SM. (2012). Recessively inherited forms of osteogenesis imperfecta. Annu Rev Genet 46:475-497. [DOI] [PubMed] [Google Scholar]
- Chaussain-Miller C, Sinding C, Septier D, Wolikow M, Goldberg M, Garabedian M. (2007). Dentin structure in familial hypophosphatemic rickets: benefits of vitamin D and phosphate treatment. Oral Dis 13:482-489. [DOI] [PubMed] [Google Scholar]
- Christou J, Johnson AR, Hodgson TA. (2013). Bisphosphonate-related osteonecrosis of the jaws and its relevance to children—a review. Int J Paediatr Dent 23:330-337. [DOI] [PubMed] [Google Scholar]
- Collmann H, Mornet E, Gattenlöhner S, Beck C, Girschick H. (2009). Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst 25:217-223. [DOI] [PubMed] [Google Scholar]
- Devlin RD, Bone HG, 3rd, Roodman GD. (1996). Interleukin-6: a potential mediator of the massive osteolysis in patients with Gorham-Stout disease. J Clin Endocrinol Metab 81:1893-1897. [DOI] [PubMed] [Google Scholar]
- Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J, White KE, et al. (2009). A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int 20:1273-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow EG, Imel EA, White KE. (2011). Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and alphaKlotho). Best Pract Res Clin Rheumatol 25:735-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Nagatomo KJ, Tso HW, Tran AB, Nociti FH, Jr, Narisawa S, et al. (2012). Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J Bone Miner Res 28:271-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Nociti FH, Jr, Somerman MJ. (2014). The rachitic tooth. Endocr Rev 35:1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal G, Metzker A, Garlick J, Gold Y, Calderon S. (1994). Head and neck manifestations of tumoral calcinosis. Oral Surg Oral Med Oral Pathol 77:158-166. [DOI] [PubMed] [Google Scholar]
- Gjørup H, Kjaer I, Sonnesen L, Haubek D, Beck-Nielsen SS, Hintze H, et al. (2011). Craniofacial morphology in patients with hypophosphatemic rickets: a cephalometric study focusing on differences between bone of cartilaginous and intramembranous origin. Am J Med Genet A 155A:2654-2660. [DOI] [PubMed] [Google Scholar]
- Glass LR, Dagi TF, Dagi LR. (2011). Papilledema in the setting of x-linked hypophosphatemic rickets with craniosynostosis. Case Rep Ophthalmol 2:376-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham LW, Stout AP. (1955). Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); its relation to hemangiomatosis. J Bone Joint Surg Am 37A:985-1004. [PubMed] [Google Scholar]
- Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, et al. (2007). Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res 22:1468-1474. [DOI] [PubMed] [Google Scholar]
- Heyden G, Kindblom LG, Nielsen JM. (1977). Disappearing bone disease. A clinical and histological study. J Bone Joint Surg Am 59:57-61. [PubMed] [Google Scholar]
- Hirayama T, Sabokbar A, Itonaga I, Watt-Smith S, Athanasou NA. (2001). Cellular and humoral mechanisms of osteoclast formation and bone resorption in Gorham-Stout disease. J Pathol 195:624-630. [DOI] [PubMed] [Google Scholar]
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, et al. (2007). A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Musculoskelet Neuronal Interact 7:318-319. [PubMed] [Google Scholar]
- Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, et al. (2006). Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 281:18370-18377. [DOI] [PubMed] [Google Scholar]
- Kelly MH, Brillante B, Collins MT. (2008). Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int 19:57-63. [DOI] [PubMed] [Google Scholar]
- Kiran DN, Anupama A. (2011). Vanishing bone disease: a review. J Oral Maxillofac Surg 69:199-203. [DOI] [PubMed] [Google Scholar]
- Krstevska A, Gale S, Blair F. (2012). Tumoral calcinosis: a dental literature review and case report. Dent Update 39:416-418, 421. [DOI] [PubMed] [Google Scholar]
- Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. (2010). Treatment of Gorham-Stout disease with zoledronic acid and interferon-alpha: a case report and literature review. J Pediatr Hematol Oncol 32:579-584. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. (2008). Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res 23:1731-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammoglia JJ, Mericq V. (2009). Familial tumoral calcinosis caused by a novel FGF23 mutation: response to induction of tubular renal acidosis with acetazolamide and the non-calcium phosphate binder sevelamer. Horm Res 71:178-184. [DOI] [PubMed] [Google Scholar]
- Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Weintroub S, et al. (2002). Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 347:1670-1676. [DOI] [PubMed] [Google Scholar]
- Lowing K, Aström E, Oscarsson KA, Söderhall S, Eliasson AC. (2007). Effect of intravenous pamidronate therapy on everyday activities in children with osteogenesis imperfecta. Acta Paediatr 96:1180-1183. [DOI] [PubMed] [Google Scholar]
- Majorana A, Bardellini E, Brunelli PC, Lacaita M, Cazzolla AP, Favia G. (2010). Dentinogenesis imperfecta in children with osteogenesis imperfecta: a clinical and ultrastructural study. Int J Paediatr Dent 20:112-118. [DOI] [PubMed] [Google Scholar]
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28:209-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, Tenorio J, et al. (2012). Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat 33:343-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano M, Wright T, Loechner KJ. (2011). Dental implications of osteogenesis imperfecta: treatment with IV bisphosphonate: report of a case. Pediatr Dent 33:349-352. [PubMed] [Google Scholar]
- Millán J, Narisawa S, Lemire I, Loisel T, Boileau G, Leonard P, et al. (2008). Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res 23:777-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. (1999). The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br 81:501-506. [DOI] [PubMed] [Google Scholar]
- Mornet E. (2007). Hypophosphatasia. Orphanet J Rare Dis 2:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulpuri K, Joseph B. (2000). Intramedullary rodding in osteogenesis imperfecta. J Pediatr Orthop 20:267-273. [PubMed] [Google Scholar]
- Murthy AS. (2009). X-linked hypophosphatemic rickets and craniosynostosis. J Craniofac Surg 20:439-442. [DOI] [PubMed] [Google Scholar]
- Nicolaou N, Agrawal Y, Padman M, Fernandes JA, Bell MJ. (2012). Changing pattern of femoral fractures in osteogenesis imperfecta with prolonged use of bisphosphonates. J Child Orthop 6:21-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CM, de Andrade CR, Vargas PA, Coletta RD, de Almeida OP, Lopes MA. (2004). Dental alterations associated with X-linked hypophosphatemic rickets. J Endod 30:241-245. [DOI] [PubMed] [Google Scholar]
- Reibel A, Manière M, Clauss F, Droz D, Alembik Y, Mornet E, et al. (2009). Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinglas J, Ramphal R, Bromwich M. (2011). The successful management of diffuse lymphangiomatosis using sirolimus: a case report. The Laryngoscope 121:1851-1854. [DOI] [PubMed] [Google Scholar]
- Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P. (1997). Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol 151:1587-1600. [PMC free article] [PubMed] [Google Scholar]
- Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Gehron Robey P. (2003). Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL-6 expression. Bone 33:434-442. [DOI] [PubMed] [Google Scholar]
- Rodrigues TR, Georgetti AP, Martins L, Pereira Neto JS, Foster BL, Nociti FH., Jr (2012). Clinical correlate: case study of identical twins with cementum and periodontal defects resulting from odontohypophosphatasia. In: Mineralized tissues in oral and craniofacial science: biological principles and clinical correlates. McCauley LK, Somerman MJ, editors. Ames, IA: Wiley-Blackwell, pp 183-189. [Google Scholar]
- Ruck J, Dahan-Oliel N, Montpetit K, Rauch F, Fassier F. (2011). Fassier-Duval femoral rodding in children with osteogenesis imperfecta receiving bisphosphonates: functional outcomes at one year. J Child Orthop 5:217-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. (2011). Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol 40:1391-1397. [DOI] [PubMed] [Google Scholar]
- Semler O, Cheung MS, Glorieux FH, Rauch F. (2010). Wormian bones in osteogenesis imperfecta: correlation to clinical findings and genotype. Am J Med Genet A 152A:1681-1687. [DOI] [PubMed] [Google Scholar]
- Sillence DO, Senn A, Danks DM. (1979). Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16:101-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton RP, Ippolito E, Springfield D, Lindaman L, Weintroub S, Leet A. (2012). The surgical management of fibrous dysplasia of bone. Orphanet J Rare Dis 7(Suppl 1):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. (2004). Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet 36:579-581. [DOI] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. (2006). Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770-774. [DOI] [PubMed] [Google Scholar]
- van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PG, van Rijn RR, et al. (2011). Osteogenesis imperfecta: a review with clinical examples. Mol Syndromol 2:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. (1991). Activating mutations of the stimulatory G protein in the McCune-Albright syndrome [see comments]. N Engl J Med 325:1688-1695. [DOI] [PubMed] [Google Scholar]
- Whyte M. (1994). Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 15:439-461. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. (2012). Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 366:904-913. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sugimoto T, Imai Y, Fukase M, Fujita T, Chihara K. (1995). Successful treatment of hyperphosphatemic tumoral calcinosis with long-term acetazolamide. Bone 16(4 Suppl):247S-250S. [DOI] [PubMed] [Google Scholar]
- Young JW, Galbraith M, Cunningham J, Roof BS, Vujic I, Gobien RP, et al. (1983). Progressive vertebral collapse in diffuse angiomatosis. Metab Bone Dis Relat Res 5:53-60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.