

Abstract
Evidence from biological and human studies strongly supports a role for MMP and TIMP genes as candidate genes for non-syndromic cleft lip with or without cleft palate (NSCL/P). We previously showed the association of promoter polymorphisms in MMP3 (rs3025058 and rs522616) and TIMP2 (rs8179096) with NSCL/P. In this study, we examined the functional significance of these polymorphisms. A specific DNA-protein complex for MMP3 rs522616 A was detected, and this allele by itself showed greater promoter activity than the G allele. However, the effect of rs522616 was ultimately regulated by the rs3025058 allele on the background. For TIMP2 rs8179096, the T allele showed a 2.5-fold increase in promoter activity when compared with allele C, whereas both C and T alleles were found to bind to nuclear factor kappa B. Our results provide new evidence that promoter polymorphisms in MMP3 and TIMP2 are functional and may affect gene transcription with possible effects on craniofacial development leading to NSCL/P.
Keywords: MMP, TIMP, luciferase, EMSA, oral cleft, single nucleotide polymorphism
Introduction
Non-syndromic cleft lip with or without cleft palate (NSCL/P) accounts for 65% of all birth defects (Gorlin et al., 2001). In addition to the impact on quality of life for affected individuals, the presence of a cleft imposes a substantial burden in terms of health care costs on the families and the society. Lifelong treatment including surgical, dental, nutritional, and behavioral interventions is often required (Strauss, 1999).
The etiology of NSCL/P is complex, with genetic and environmental risk factors. Studies have estimated that from 3 to 14 genes, acting individually and/or in concert, may be involved (Schliekelman and Slatkin, 2002). Traditionally, numerous research approaches – including studies of candidate genes suggested by family-based linkage or linkage disequilibrium studies, association studies using case-parent trios or case-control samples, identification of chromosomal abnormalities in cases, direct sequencing of affected individuals, or by animal models displaying a cleft phenotype – have been used in NSCL/P studies (Dixon et al., 2011). A combination of genome-wide linkage studies and genome-wide association studies led – after years of studies - to major breakthroughs in the identification of NSCL/P risk loci.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteases collectively responsible for extracellular matrix (ECM) and tissue remodeling. They are comprised of 23 enzymes that share sequence homologies and substrate specificities, and are classified as collagenases, gelatinases, stromelysins, matrilysins, or membrane-type MMPs (Nagase and Woessner, 1999). Together with their endogenous tissue inhibitors (TIMPs), MMPs have key roles during the embryogenesis and homeostasis of adult tissues (Nagase et al., 2006). MMPs and TIMPs have been suggested as potential candidate genes for NSCL/P based on observations of their spatial and temporal expression patterns during palatogenesis in mice and because absence of MMP activity resulted in cleft palate (Morris-Wiman et al., 2000; Blavier et al., 2001; Brown et al., 2002; Shi et al., 2008; de Oliveira Demarchi et al., 2010).
We have previously shown the association of 2 MMP3 promoter polymorphisms (rs3025058 and rs522616) with NSCL/P phenotypes in Brazilian and US populations; haplotypes containing these variants were also associated with NSCL/P (Letra et al., 2007, 2012a). A promoter variant in the TIMP2 (rs8179096) gene was also associated with NSCL/P (Letra et al., 2012a), corroborating previous association findings in Europeans (Nikopensius et al., 2011).
MMP3 transcription is tightly regulated and influenced by the rs3025058 5A/6A polymorphism, characterized by the presence of 5 or 6 adenine residues, with the 5A allele having greater promoter activity than the 6A allele (Ye et al., 1996; Zhu et al., 2006; Souslova et al., 2010). Differential transcriptional activity was also recently reported for the rs522616 A/G variant, where allele G showed lower promoter activity than allele A (Huai et al., 2013). Nevertheless, the relationship between rs522616 and rs3025058 and their combined effects on MMP3 transcription remain unknown. Similarly, the regulatory effects of TIMP2 rs8179096 are still unclear.
In this study, we examined the functional effects of MMP3 rs522616 and rs3025058, and TIMP2 rs8179096 polymorphisms previously associated with human NSCL/P.
Materials & Methods
Reporter Constructs
Details on the reporter constructs and insert sequences are available in the online Appendix.
Transient Transfection and Luciferase Reporter Assays
Human fibrosarcoma cells (HT-1080) were purchased from the American Type Culture Collection (Gaithersburg, MD, USA), cultured in Eagle’s Minimum Essential Medium (Gibco, Grand Island, NY, USA), supplemented with 10% fetal calf serum and penicillin-streptomycin, and maintained at 37°C in 5% CO2. For transfection, cells were seeded at a density of 15,000 cells/well in a 96-well plate in culture medium for 24 hr to reach 80% confluence. Cells were transfected with 50 ng of each construct in Opti-MEM medium (Life Technologies, Grand Island, NY, USA) and 0.15 μL of FuGENE HD (Invitrogen, Carlsbad, CA, USA), and incubated for 24 hr. Cells underwent lysis at -80°C overnight, were thawed and then incubated with 100 μL LightSwitch Assay solution for 30 min. Luciferase activity was measured with Tecan Infinite 200 PRO reader (Tecan Systems, San Jose, CA, USA). Empty vector, RO1 negative control, and RPL10 positive control constructs were assayed in parallel for evaluation of transfection efficiency. The mean luciferase activity of each construct was compared by t test with Welch’s correction in GraphPad Prism (GraphPad Inc., La Jolla, CA, USA). Data are presented as the mean ± standard error from three independent experiments. Differences were significant if p ≤ .05.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed according to the LI-COR IRDye® protocol (LI-COR, Lincoln, NE, USA). In brief, double-stranded oligonucleotides harboring MMP3 rs522616 and TIMP2 rs8179096 were synthesized (details in the online Appendix), labeled with infrared at the 5′ end, and used as probes. Hela S3 cell nuclear extracts (Active Motif, Carlsbad, CA, USA) were incubated with 50 fmol of each probe along with binding buffer (100 mM Tris, 500 mM KCl, 10 mM DTT; pH 7.5) and 25 mM DTT/2.5% Tween in 20 μL reaction volume for 20 min at room temperature. Unlabeled probes at various concentrations (1- to 100,000-fold of labeled probe) were used as specific competitors. Reaction mixtures were loaded on a 4% native polyacrylamide gel and run in 1X TBE buffer. Gels were scanned by means of a LI-COR Odyssey CLx scanner.
Supershift Assays
Our previously published in silico analyses revealed caudal-type homeobox 1 (CDX1) and nuclear factor-κ beta (NFκB) as potential binding partners for MMP3 rs522616 A and TIMP2 rs8179096 C/T alleles, respectively (Letra et al., 2012a). Antibodies for CDX1 (Abgent, San Diego, CA, USA) and NFκB (Millipore, Billerica, MA, USA) were used in supershift assays to verify their ability to bind respective alleles. In brief, 1 μL of antibody was added to 20 μL of DNA-protein-binding reaction mixture, then loaded on a 4% native polyacrylamide gel for electrophoresis. Reagents and procedures were as described in the EMSA section. Rabbit IgG antibody was used as isotope control. Unlabeled wild-type and mutant NFκB consensus oligonucleotides were used to confirm rs8179096 binding to NFκB protein (details in the online Appendix).
Mass Spectrometry
Hela S3 nuclear extracts (Active Motif) were incubated with 200 fmol of MMP3 A or G probes in a DNA-protein-binding reaction, and run on a 4% to 12% TBE gel electrophoresis. Gels were scanned and silver-stained. Gel bands containing DNA-protein complexes were excised and sent for mass spectrometry analysis. Results were viewed with Scaffold 3 software (Proteome Software Inc., Portland, OR, USA).
Results
Binding of Nuclear Proteins to MMP3 rs522616 A/G
Both A and G probes produced a specific DNA-protein complex (Y) (Fig. 1A, lanes 3 and 4), which was abolished by the lowest competitor DNA levels (16X) (Figs. 1B, 1C, lanes 3 and 12), suggesting that the protein(s) involved bind equally to the A- or G-containing motifs. MMP3 A also produced an additional complex (X) (Fig. 1A, lane 3), which was abolished by the unlabeled A-competitor (Fig. 1B, lane 5), whereas the G-competitor failed to do so at even higher concentrations (Fig. 1B, lanes 10-18), suggesting that complex X is specific to the A probe. The binding in complex Z was not blocked by either the A or G competitor, and thus was considered non-specific (Figs. 1B, 1C). Supershift assays for CDX1 did not confirm the published in silico results (data not shown); nonetheless, mass spectrometry analysis identified 34 proteins as unique for the A probe binding complex (online Appendix).
Figure 1.

Electrophoretic mobility shift assays of MMP3 rs522616. (A) Double-stranded, infrared-labeled A and G probes incubated in the absence (lanes 1, 2) or presence of cell nuclear extract (lanes 3, 4). (B) Competition assay with double-stranded, infrared labeled A probe incubated with cell nuclear extract and 1-, 4-, 16-, 80-, 400-, 2,000-, 10,000-, 50,000-, and 100,000-fold molar excess of unlabeled A (lanes 1-9) and G probes (lanes 10-18). (C) Competition assay with double-stranded, infrared-labeled G probe incubated with cell nuclear extract and 1-, 4-, 16-, 80-, 400-, 2,000-, 10,000-, 50,000-, and 100,000-fold molar excess of unlabeled G (lanes 1-9) and A probes (lanes 10-18). Note that complex Y was produced by both A and G probes and abolished by the lowest competitor DNA levels (16X) (Fig. 1C, lanes 3, 12), suggesting that the protein(s) involved bind equally to the A- or G-containing motifs. MMP3_A also produced an additional complex (X) (Fig. 1A, lane 3), which was abolished by unlabeled A-competitor (Fig. 1B, lane 5), but not G-competitor at even higher concentrations (Fig. 1B, lanes 10-18), suggesting that complex X is specific to the A probe.
Differential MMP3 Gene Promoter Activity Based on rs522616 A/G and rs3025058 5A/6A Variant Haplotypes
MMP3 rs522616 A resulted in a 3.7-fold (p = .004) increase in activity when compared with G. When investigating the background effect of rs3025058 on the activity of rs522616, we observed highest promoter activity with the 6A_G haplotype, followed by 5A_G, 5A_A, and 6A_A. Haplotype 5A_G resulted in a 1.5-fold increased promoter activity compared with 5A_A (p = .01), whereas 6A_G activity was four-fold higher than 6A_A (p = .004) (Fig. 2).
Figure 2.
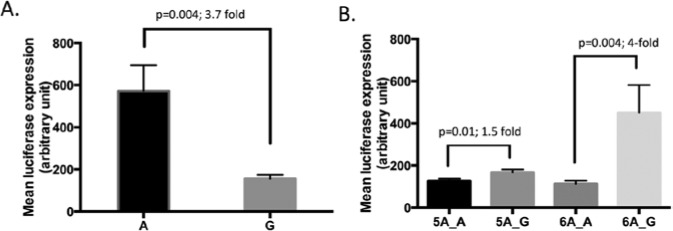
Luciferase reporter assay results for MMP3 promoter variants in HT-1080 cells. (A) Results for MMP3 rs522616 showing increased promoter activity with the A allele. (B) Results for rs522616 and rs3025058 haplotype combinations showing differential effects of rs522616 depending on rs3025058 background allele.
Binding of Nuclear Proteins to TIMP2 rs8179096 C/T
Two similar DNA-binding complexes (L and M) were observed for C and T probes (Fig. 3A, lanes 3, 4). Both probes presented similar affinities for these complexes, being blocked by 4- or 16-fold molar excess of either C or T competitor (Figs. 3B, 3C, lanes 2, 8). Supershift assay confirmed the presence of a NFκB-binding site (complex K) for both C and T probes (Fig. 3D), as predicted in our previous work (Letra et al., 2012a). No bands were observed in isotype control lanes, indicating that the supershift bands reflected NFκB-probe interaction (Appendix Fig.). Furthermore, we confirmed the specificity of rs8179096 to bind to NFκB by comparing wild-type and mutant NFκB oligonucleotides introduced in EMSA reaction. We observed decreased binding of C/T probes to nuclear extract when NFκB wild-type oligo was increased (Fig. 3E, lanes 6, 10). In contrast, no reduction in binding was observed upon increase in mutant NFκB (Fig. 3E, lanes 8, 12). This suggests that the mutant NFκB oligonucleotide was unable to bind to NFκB protein and therefore freed all of the specific protein to bind to other oligos (in this case, C/T probes). This is further indication that rs8179096 binds to NFκB protein.
Figure 3.

Electrophoretic mobility shift assays with TIMP2 rs8179096 variant. (A) Double-stranded, infrared-labeled C and T probes incubated in the absence (lanes 1, 2) or presence of cell nuclear extract (lanes 3, 4). (B) Competition assay with double-stranded, infrared labeled C probe incubated in the presence of cell nuclear extract and 1-, 4-, 16-, 80-, 400-, 2,000-, 10,000-, 50,000-, and 100,000-fold molar excess of unlabeled C (lanes 1-9) and T probes (lanes 10-18). (C) Competition assay with double-stranded, infrared-labeled T probe incubated in the presence of cell nuclear extract and 1-, 4-, 16-, 80-, 400-, 2,000-, 10,000-, 50,000-, and 100,000-fold molar excess of unlabeled T (lanes 1-9) and C probes (lanes 10-18). (D) Supershift assays were performed incubating double-stranded, infrared-labeled C and T probes with cell nuclear extract plus NFκB antibody (Complex K). (E) EMSA for verification of NFκB binding to rs8179096. Double-stranded, infrared-labeled C and T probes were incubated in the absence (lanes 1, 2) or presence of cell nuclear extract (lanes 3-12). One- or two-fold molar excess (in comparison with probe concentration) of NFκB consensus unlabeled oligonucleotide was added in lanes 5, 6, 9, and 10 (solid frames), whereas mutant NFκB consensus unlabeled oligonucleotide was added in lanes 7, 8, 11, and 12 (dotted frames). No bands were observed in isotype control lanes, indicating that the supershift bands reflected NFκB-probe interaction.
TIMP2 rs8179096 T Shows Higher Promoter Activity
The presence of the T allele resulted in a 2.5-fold increase in promoter activity when compared with the C allele (p < .0001) (Fig. 4).
Figure 4.
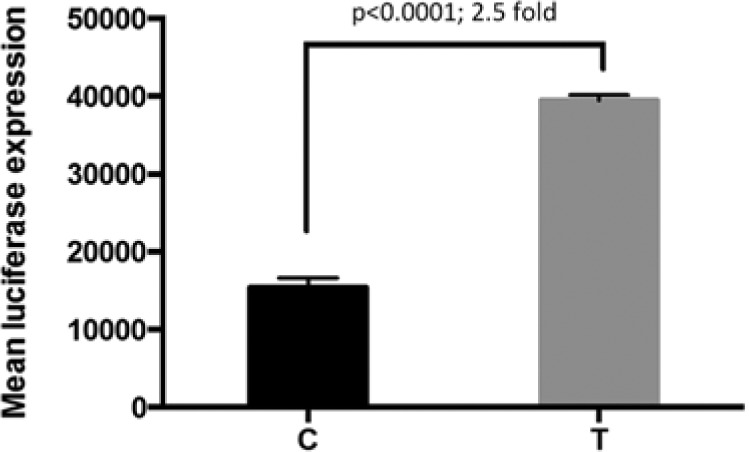
Luciferase reporter assay results for TIMP2 rs8179096 in HT-1080 cells. Results showed increased promoter activity with the T allele.
Discussion
In this study, we examined the function of MMP3 (rs522616 and rs3025058) and TIMP2 (rs8179096) promoter variants, previously associated with NSCL/P (Letra et al., 2007, 2012a). Our findings indicate that each of these variants has a different regulatory effect on gene transcription depending on the allele present.
MMP-3, also known as stromelysin-1, is a key member of the MMP family, which participates in regulating the accumulation of extracellular matrix and activating other MMPs. MMP-3 has proteolytic activity on types III, IV, and V collagen, proteoglycans, laminin, fibronectin, and elastin, all of which are abundantly present in the palatal matrix (Nagase et al., 2006). During mouse palatogenesis, MMP-3 expression was detected subjacent to the medial-edge epithelia following palatal shelf contact (Morris-Wiman et al., 2000; Blavier et al., 2001), and significantly increased in fused palates than in palates that failed to fuse (Brown et al., 2002). Further, induction of MMP-3 expression has been shown to mediate the process of epithelial-mesenchymal transformation, an important event during palatal fusion (Lochter et al., 1997). These findings imply that the MMP3 gene is critical for proper palate development, and that perturbed expression may result in cleft palate.
Transcription of MMP3 is tightly regulated and influenced by a polymorphism in the promoter region (rs3025058). This polymorphism, located at -1171 of the transcription start site (Lei et al., 2002), is characterized by the presence of 5 or 6 adenine residues (5A/6A), and higher transcription has been described for the 5A allele (Ye et al., 1996; Zhu et al., 2006; Souslova et al., 2010). Recently, differential transcriptional activity was also reported for the rs522616 A/G variant. MMP3 activity with the A-allele was up-regulated in HEK293 and HUVEC cells, with ~2.7- and 5.9-fold changes, respectively (Huai et al., 2013). Further, the sequence near rs522616, located upstream of the MMP3 gene, may include a binding site for one or more transcriptional factors, and the presence of A or G may change the binding affinity of those factors (Huai et al., 2013). Corroborating these findings, our results also indicate that both rs522616 and rs3025058 are functional, with effects on MMP3 transcription. Our luciferase assays suggest a positive regulatory element at rs522616, since significantly higher promoter activity was found for the A allele in comparison with the G allele. This suggests that the A allele can enhance promoter activity, possibly by augmenting transcription factor binding. Indeed, electrophoretic mobility shift assays showed that both A and G alleles produced 2 specific DNA-protein complexes, whereas allele A produced an additional specific complex. Mass spectrometry analyses revealed 34 proteins that were exclusively bound to the A allele; nevertheless, further investigation is needed to verify if these proteins are authentic binding partners.
We also investigated the effect of rs522616 in the background of the rs3025058 variant. While we expected to see higher activity with the 5A_A promoter haplotype, based on the higher transcription activities of the A and 5A alleles individually (Ye et al., 1996; Souslova et al., 2010), the activity of this haplotype was decreased by 1.5-fold when compared with that of 5A_G. Although speculative, this finding may represent a negative feedback loop effect, in an attempt to limit transcription in the presence of the 2 ‘high transcription’ alleles, 5A and A. Conversely, we observed a four-fold increased activity with the 6A_G promoter. This suggests that while rs522616 can affect the activity of the MMP3 promoter by itself, it is ultimately regulated by the rs3025058 alleles in the background. A previous study assessing the association of MMP3 variant haplotypes in coronary stenosis found that haplotypes containing the 5A allele were associated with myocardial infarction susceptibility, and elicited higher promoter activity in macrophages (Beyzade et al., 2003). Taken together, these results indicate that the rs3025058 variant influences MMP3 transcription due to differential effects on additional promoter variants. This highlights the complexity of the MMP3 gene promoter, and correlates with the many functions of the MMP3 enzyme. Other cis-regulatory elements may also be responsible for the increased transcription promoted by the A allele.
Our hypothesis for the involvement of MMP3 in NSCL/P is that down-regulation of MMP3 would contribute to the phenotype because of decreased tissue remodeling and/or epithelial-mesenchymal transformation during palatal fusion. In a recent study, we found a strong association for rs522616 A with NSCL/P, while strong associations were observed for haplotypes including the A and 6A alleles (Letra et al., 2012b). Interestingly, the results of the present study showed that the 6A_A haplotype was the least active promoter and may reflect gene down-regulation.
Our findings for TIMP2 rs8179096 also suggest distinct allele-dependent effects. The T allele resulted in a 2.5-fold increased activity, and this allele may harbor binding sites for additional transcriptional enhancers. We also confirmed that both C and T alleles bind to NFκB, a key transcription factor involved in the innate immune system. While C and T alleles were losing binding capability when NFκB consensus binding oligo diverges from protein in the same reaction, introduction of a mutant NFκB immunized C and T alleles from binding abolition. However, it is unlikely that NFκB is the single binding partner at this locus. The TIMP2 promoter has several consensus binding sequences for transcription factors such as Sp1, AP-1, AP-2, and NFκB (Hegab et al., 2005), and our results support these observations. In our previous study, we found evidence of gene-gene interaction between MMP3 and TIMP2, where individuals carrying at least 1 copy of MMP3 rs522616 A and 1 or 2 copies of TIMP2 rs8179096 C presented an increased risk for NSCL/P (Letra et al., 2012a). Intriguingly, NFκB also interacts with rs3025058 to modulate MMP3 transcription (Souslova et al., 2010); hence, possible interaction between MMP3 and TIMP2 variants could be explained by the fact that NFκB has regulatory effects on both variants.
In summary, the findings of this study provide insights into the functional effects of MMP3 and TIMP2 promoter polymorphisms associated with NSCL/P. Of note, the results herein presented reflect the use of cell lines instead of actual palate tissues; therefore, different results could be observed if real facial mesenchyme cells were used. There is a constellation of molecules engaged in complex interactions to drive palate development in a concerted mode of action. Some of these molecules act as repressors, others as activators, whereas some display dual effects depending on the temporal and spatial context. However, we still know relatively little about the regulation and targets of many molecules identified as playing pivotal roles during lip and palate development. Knowing targets of key regulators of palate development ensures the identification of novel risk factors for NSCL/P. Careful in vitro functional studies provide a wealth of information, although they have limitations and cannot be directly extrapolated to in vivo conditions.
Supplementary Material
Acknowledgments
Thanks to R. Rylands and D. Knight for technical assistance and to K. Posey, W. Fakhouri, and S. Wendell for many helpful discussions.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
A.L. and R.M.S. were supported by National Institute of Dental and Craniofacial Research (NIDCR) grants K99/R00-DE018954 and K99/R00-DE018913, respectively.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Beyzade S, Zhang S, Wong YK, Day IN, Eriksson P, Ye S. (2003). Influences of matrix metalloproteinase-3 gene variation on extent of coronary atherosclerosis and risk of myocardial infarction. J Am Coll Cardiol 41:2130-2137. [DOI] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. (2001). TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell 12:1457-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NL, Yarram SJ, Mansell JP, Sandy JR. (2002). Matrix metalloproteinases have a role in palatogenesis. J Dent Res 81:826-830. [DOI] [PubMed] [Google Scholar]
- de Oliveira Demarchi AC, Zambuzzi WF, Paiva KB, da Silva-Valenzuela Md, Nunes FD, de Cássia Sávio Figueira R, et al. (2010). Development of secondary palate requires strict regulation of ECM remodeling: sequential distribution of RECK, MMP-2, MMP-3, and MMP-9. Cell Tissue Res 340:61-69. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nature Rev Genetics 12(3):167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Cohen M, Jr, Hennakam RC. (2001). Syndromes of the head and neck. 4th ed. Oxford, UK: Oxford University Press. [Google Scholar]
- Hegab AE, Sakamoto T, Uchida Y, Nomura A, Ishii Y, Morishima Y, et al. (2005). Promoter activity of human tissue inhibitor of metalloproteinase 2 gene with novel single nucleotide polymorphisms. Respirology 10:27-30. [DOI] [PubMed] [Google Scholar]
- Huai C, Song J, Ma Z, Qin X, Li P, Chen H, et al. (2013). Allelic variation of the MMP3 promoter affects transcription activity through the transcription factor C-MYB in human brain arteriovenous malformations. PloS One 8:e57958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Zaloudik J, Vorechovsky I. (2002). Lack of association of the -1171 (5A) allele of the MMP3 promoter with breast cancer. Clin Chem 48:798-799. [PubMed] [Google Scholar]
- Letra A, Silva RA, Menezes R, Astolfi CM, Shinohara A, de Souza AP, et al. (2007). MMP gene polymorphisms as contributors for cleft lip/palate: association with MMP3 but not MMP1. Arch Oral Biol 52:954-960. [DOI] [PubMed] [Google Scholar]
- Letra A, Silva RM, Motta LG, Blanton SH, Hecht JT, Granjeiro JM, et al. (2012a). Association of MMP3 and TIMP2 promoter polymorphisms with nonsyndromic oral clefts. Birth Defects Res A Clin Mol Teratol 94:540-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letra A, Silva RM, Rylands RJ, Silveira EM, de Souza AP, Wendell SK, et al. (2012b). MMP3 and TIMP1 variants contribute to chronic periodontitis and may be implicated in disease progression. J Clin Periodontol 39:707-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. (1997). Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139:1861-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Wiman J, Burch H, Basco E. (2000). Temporospatial distribution of matrix metalloproteinase and tissue inhibitors of matrix metalloproteinases during murine secondary palate morphogenesis. Anat Embryol (Berl) 202:129-141. [DOI] [PubMed] [Google Scholar]
- Nagase H, Woessner JF., Jr (1999). Matrix metalloproteinases. J Biol Chem 274:21491-21494. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. (2006). Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69:562-573. [DOI] [PubMed] [Google Scholar]
- Nikopensius T, Kempa I, Ambrozaityte L, Jagomagi T, Saag M, Matuleviciene A, et al. (2011). Variation in FGF1, FOXE1, and TIMP2 genes is associated with nonsyndromic cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol 91:218-225. [DOI] [PubMed] [Google Scholar]
- Schliekelman P, Slatkin M. (2002). Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet 71:1369-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Son MY, Yamada S, Szabova L, Kahan S, Chrysovergis K, et al. (2008). Membrane-type MMPs enable extracellular matrix permissiveness and mesenchymal cell proliferation during embryogenesis. Dev Biol 313:196-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souslova V, Townsend PA, Mann J, van der Loos CM, Motterle A, D’Acquisto F, et al. (2010). Allele-specific regulation of matrix metalloproteinase-3 gene by transcription factor NFkappaB. PloS One 5:e9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss RP. (1999). The organization and delivery of craniofacial health services: the state of the art. Cleft Palate Craniofac J 36:189-195. [DOI] [PubMed] [Google Scholar]
- Ye S, Eriksson P, Hamsten A, Kurkinen M, Humphries SE, Henney AM. (1996). Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J Biol Chem 271:13055-13060. [DOI] [PubMed] [Google Scholar]
- Zhu C, Odeberg J, Hamsten A, Eriksson P. (2006). Allele-specific MMP-3 transcription under in vivo conditions. Biochem Biophys Res Commun 348:1150-1156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.