

Abstract
Interleukin-1α and interleukin-1β aggravate neuronal injury by mediating the inflammatory reaction following ischemic/hypoxic brain injury. It remains unclear whether interleukin-1α and interleukin-1β are released by microglia or astrocytes. This study prepared hippocampal slices that were subsequently subjected to oxygen and glucose deprivation. Hematoxylin-eosin staining verified that neurons exhibited hypoxic changes. Results of enzyme-linked immunosorbent assay found that interleukin-1α and interleukin-1β participated in this hypoxic process. Moreover, when hypoxic injury occurred in the hippocampus, the release of interleukin-1α and interleukin-1β was mediated by the P2X4 receptor and P2X7 receptor. Immunofluorescence staining revealed that during ischemia/hypoxia, the P2X4 receptor, P2X7 receptor, interleukin-1α and interleukin-1β expression was detectable in rat hippocampal microglia, but only P2X4 receptor and P2X7 receptor expression was detected in astrocytes. Results suggested that the P2X4 receptor and P2X7 receptor, respectively, mediated interleukin-1α and interleukin-1β released by microglia, resulting in hippocampal ischemic/hypoxic injury. Astrocytes were activated, but did not synthesize or release interleukin-1α and interleukin-1β.
Keywords: neural regeneration, brain injury, inflammatory, P2X4 receptor, P2X7 receptor, interleukin-1β, interleukin-1β, microglia, astrocytes, oxygen-glucose deprivation, hippocampal slices, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
Purinergic receptors are associated with vascular responses, cell apoptosis and cytokine secretion.
-
(2)
P2X4 and P2X7 receptors participated in ischemic/hypoxic injury, but the precise mechanisms of this kind of injury remain poorly understood.
-
(3)
Interleukin-1 plays a key role in ischemic/hypoxic brain injury. This study investigated whether microglia induce interleukin-1 release during ischemia/hypoxia.
-
(4)
P2X4 receptor and P2X7 receptor mediated interleukin-1α and interleukin-1β release from microglia resulted in hippocampal ischemic/hypoxic injury.
-
(5)
Astrocytes were activated, but did not synthesize or release interleukin-1α and interleukin-1β.
INTRODUCTION
The inflammatory response plays an important role in ischemic/hypoxic brain injury[1,2]. Following ischemic/hypoxic injury, increased release of adenosine triphosphate leads to microglial activation. Activated microglia release interleukin, interferon, tumor necrosis factor, prostaglandin and reactive oxygen species[3]. The above factors, especially interleukin, have strong toxic effects on neuronal cells. They mediate the inflammatory reaction and aggravate ischemic/hypoxic injury which results in cell death. Interleukin-1 is an important inflammatory cytokine and exerts a crucial effect on the initial stage of the inflammatory reaction[4,5]. Interleukin-1 is composed of interleukin-1α and interleukin-1β and exerts biological effects via interleukin-1 receptors. Interleukin-1α is mostly membrane-associated and exerts a biological activity, but interleukin-1β mainly exists in the circulation. Interleukin-1 receptor antagonists[6] are specific inhibitors of interleukin-1, and can suppress the activities of interleukin-1α and interleukin-1β, thus having a protective effect against ischemic/hypoxic injury[7,8,9].
Normal brain tissue expresses interleukin-1, but this low level of expression cannot induce neuronal death. Neurons, microglia and vascular endothelial cells can synthesize interleukin-1. A large number of interleukin-1β can be detected during ischemic/hypoxic injury. Interleukin-1β mediated cytokine release from microglia, promoted adhesion molecule expression in endothelial cells, stimulated the production of interleukin-6, interleukin-8 and tumor necrosis factor in monocytes and macrophages, strengthened leukocyte infiltration, released inflammatory mediators, and aggravated local inflammatory reaction. In addition, following brain ischemia, increased exogenous interleukin-1β obviously aggravated brain injury[10]. Touzani et al[11] injected interleukin-1β into the cerebral ventricles of rats with middle cerebral artery occlusion and found that the injury was aggravated, and a remarkable increase in infarct volume was observed. Following cerebral ischemia, interleukin-1 receptor expression in the cerebral cortex was significantly increased at 6 hours, which coincided with interleukin-1β levels. An interleukin-1 receptor antagonist used as a preventative factor can inhibit interleukin-1β activity, and lessen neuronal injury in the ischemic region[12,8,13]. Caso et al[14] reported that following ischemic/hypoxic injury, an interleukin-1 receptor antagonist inhibited interleukin-1β expression. The interleukin-1 receptor antagonist lessened cerebral edema, reduced infarct area, decreased leukocyte infiltration, and blocked interleukin-1β-induced neural cell injury[15]. To further confirm this observation, interleukin-1β gene knock-out mice display significantly reduced infarct area following cerebral ischemia[16,17,18,19].
Our previous studies confirmed that interleukin-1β regulated the opening and closing of L-type Ca2+ channels, decreased L-type Ca2+ current, suppressed Ca2+ overload, reversed glutamate cytotoxicity, and exerted a neuroprotective effect. Nesic et al[20] and Weng et al[21] confirmed that interleukin-1β can protect injured spinal nerves. These studies demonstrated that interleukin-1β expression increased during ischemic/hypoxic injury, but whether interleukin-1β contributed to or relieved brain injury required further elucidation. Moreover, few studies addressing the effects of interleukin-1α in ischemic brain injury have been published, with the results remaining controversial.
Interleukin-1α and interleukin-1β have been shown to participate in ischemic/hypoxic brain injury, but their manner of release remains unclear[22]. Under the stimulation of lipopolysaccharide, the P2X7 receptor induced interleukin-1β release in macrophages or microglia[23,24,25]. Therefore, during ischemic/hypoxic injury, P2X7 receptor mediated interleukin-1β release from microglia or astrocytes is involved in neuronal injury. The concentration of adenosine triphosphate, an endogenous ligand of the P2X7 receptor, has been shown to increase in extracellular fluid during cerebral ischemia[26]. Seven subtypes of adenosine receptors for adenosine triphosphate, P2X1-7 receptors, have been identified. Of these, P2X4 and P2X7 expression is abundant in the central nervous system[27,28,29]. Previous studies have demonstrated that the P2X4 receptor participated in the inflammatory reaction after traumatic brain injury, and P2X4 receptor expression was upregulated during brain ischemia[30,31]. Thus, the P2X4 receptor possibly mediates interleukin-1 release, together with the P2X7 receptor. Furthermore, these receptors may participate in the starting and development of ischemic/hypoxic brain injury.
In the central nervous system, microglia are strongly associated with interleukin-1 during ischemic/hypoxic brain injury[32,33]. Recent studies showed that activated astrocytes take on an active role in the inflammatory reaction following traumatic brain injury[34,35]. Whether microglia or astrocytes induce interleukin-1 release requires further investigations. This study investigated the effects of interleukin-1α and interleukin-1β after hypoxic brain injury in hippocampal slices subjected to oxygen and glucose deprivation. The source of interleukin-1α and 1β during hypoxia, and the precise mechanisms of their release were also investigated, so as to provide firm experimental evidence for future treatment of ischemic brain injury.
RESULTS
Effect of interleukin-1 on neuronal injury in hippocampal slice cultures
At 75 minutes following oxygen-glucose deprivation, the morphology of nerve cells in the control group was normal. Lightly stained basophilic granules were visible in the cytoplasm. The boundary between nerve cell membranes and surrounding regions was unclear. In the oxygen-glucose deprivation group, nerve cells displayed hypoxic injury, cell atrophy, acidophilic granules in the cytoplasm, and a clear boundary between nerve cell membranes and surrounding regions was observed. Some nerve cells exhibited vacuole-like changes. In the oxygen-glucose deprivation + interleukin-1 receptor antagonist group, nerve cells displayed normal morphology, lightly stained basophilic granules were observed in the cytoplasm and an unclear boundary between nerve cell membranes and surrounding regions was evident. No significant difference between this group and controls was seen (Figure 1).
Figure 1.
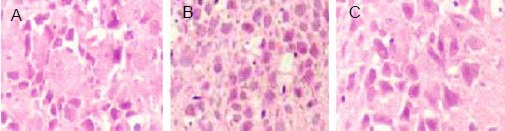
Morphology of neurons in rat hippocampal slices following oxygen-glucose deprivation (light microscope, × 200).
Hematoxylin-eosin staining showed lightly stained basophilic granules in the cytoplasm and unclear boundaries between neuronal membranes and surrounding regions in the control (A) and oxygen-glucose deprivation + interleukin-1 receptor antagonist groups (C). Neuronal atrophy, clear boundaries between neuronal cells and surrounding regions, acidophilic granules in the cytoplasm, and vacuole-like changes in some neuronal cells were observed in the oxygen-glucose deprivation group (B).
At 75 minutes after oxygen-glucose deprivation, the release of lactate dehydrogenase significantly increased compared with the control group (P < 0.01). Interleukin-1 receptor antagonist could significantly inhibit the release of lactate dehydrogenase mediated by oxygen-glucose deprivation (P < 0.01), but a significant elevation was still detected when compared with the control group (P < 0.01; Figure 2).
Figure 2.
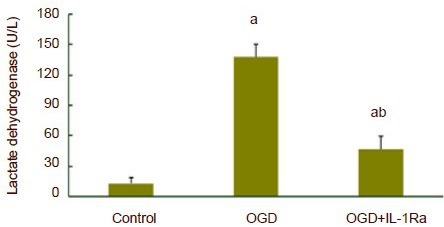
Release of lactate dehydrogenase in hippocampal slices subjected to oxygen-glucose deprivation (OGD).
At 75 minutes after OGD, the release of lactate dehydrogenase significantly increased compared with the control group. Interleukin-1 receptor antagonist (IL-1Ra) could significantly inhibit the release of lactate dehydrogenase. aP < 0.01, vs. control group; bP < 0.01, vs. OGD group. Data are expressed as mean ± SD. Experiments were repeated six times. One-way analysis of variance and least significant difference t-test were used for statistical analysis.
Release of interleukin-1α and interleukin-1β in hippocampal slices subjected to oxygen-glucose deprivation
At 75 minutes after oxygen-glucose deprivation, interleukin-1α or interleukin-1β concentration significantly increased in artificial cerebrospinal fluid (P < 0.01). The P2X7 receptor antagonist, A438079, did not affect interleukin-1α concentration. However, oxygen-glucose deprivation-induced interleukin-1α release was completely inhibited after P2X4 receptor antibody was added to artificial cerebrospinal fluid (P < 0.01; Figure 3). A438079 could completely suppress oxygen-glucose deprivation-induced interleukin-1β release (P < 0.01), but P2X4 receptor antibody did not affect interleukin-1β release (Figure 4).
Figure 3.
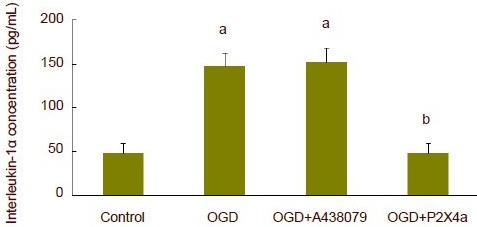
Release of interleukin-1α in hippocampal slices subjected to oxygen-glucose deprivation (OGD).
At 75 minutes after OGD, interleukin-1β concentration significantly increased. P2X4 receptor antibody could completely inhibit OGD-induced interleukin-1α release. aP < 0.01, vs. control group; bP < 0.01, vs. OGD group. Data are expressed as mean ± SD. Experiments were repeated six times. One-way analysis of variance and least significant difference t-test were used for statistical analysis, P2X4a: P2X4 receptor antibody.
Figure 4.
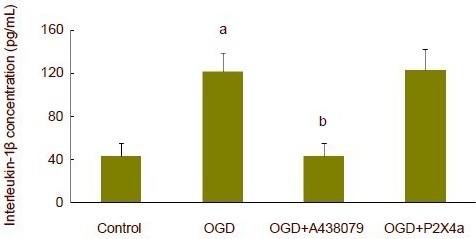
Release of interleukin-1β in hippocampal slices subjected to oxygen-glucose deprivation (OGD).
At 75 minutes after OGD, interleukin-1β concentration significantly increased, and P2X7 receptor antagonist A438079 could completely inhibit the release of OGD-induced interleukin-1β. aP < 0.01, vs. control group; bP < 0.01, vs. OGD group. Data are expressed as mean ± SD. Experiments were repeated six times. One-way analysis of variance and least significant difference t-test were used for statistical analysis. P2X4a: P2X4 receptor antibody.
Effects of exogenous interleukin-1α and interleukin-1β in hippocampal slices
A significant increase in lactate dehydrogenase release was observed at 75 minutes following incubation in artificial cerebrospinal fluid with the addition of exogenous interleukin-1α or interleukin-1β (P < 0.01). A further increase in lactate dehydrogenase release was observed following the combined addition of interleukin-1α and interleukin-1β compared with the addition of interleukin-1α alone or interleukin-1β alone (P < 0.01; Figure 5).
Figure 5.
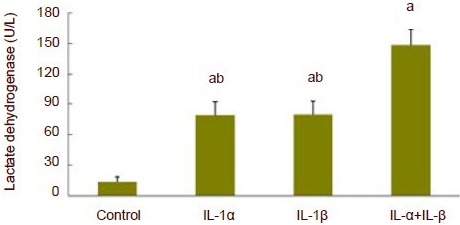
Release of lactate dehydrogenase in hippocampal slices following addition of exogenous interleukin-1α (IL-1α) and interleukin-1β (IL-1β). At 75 minutes following incubation of hippocampal slices in artificial cerebrospinal fluid supplemented with exogenous interleukin-1α (IL-α) or interleukin-1β (IL-β) under normal oxygen conditions, a significant increase in lactate dehydrogenase release was observed. A further increase in lactate dehydrogenase release was observed following the combined addition of IL-1α and IL-1β compared with the addition of IL-1α alone or IL-1β alone. aP < 0.01, vs. control group; bP < 0.01, vs. IL-1α + IL-1β group. Data are expressed as mean ± SD. Experiments were repeated six times. One-way analysis of variance and least significant difference t-test were used for statistical analysis.
In double immunofluorescence labeling, hippocampal slices subjected to oxygen-glucose deprivation were assigned to eight groups. Using double labeling immunofluorescence, monoclonal antibodies of OX42 and glial fibrillary acidic protein were used to label microglia and astrocytes, respectively. At 75 minutes after oxygen-glucose deprivation, staining for these two markers was visible. P2X4 receptor and P2X7 receptor were successfully stained in astrocytes and microglia, suggesting that microglia and astrocytes were activated. Interleukin-1α and interleukin-1β were seen in OX 42-positive cells, suggesting that microglia could synthesize interleukin-1α and interleukin-1β. These interleukins could not be immunostained in glial fibrillary acidic protein-positive cells, suggesting that astrocytes were activated during oxygen-glucose deprivation, but could not synthesize or secrete interleukin-1α and interleukin-1β (Figure 6).
Figure 6.
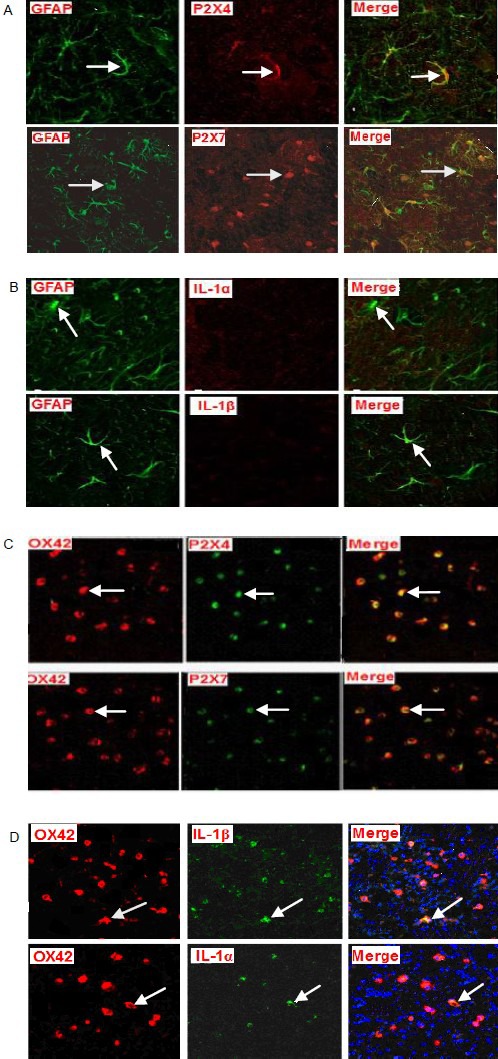
Double fluorescence staining for the P2X4 receptor, P2X7 receptor, interleukin (IL)-1α and IL-1β expression in astrocytes and microglia in hippocampal slices subjected to oxygen-glucose deprivation (confocal laser scanning microscope, × 100).
At 75 minutes after oxygen-glucose deprivation, in green astrocytes (Cy2-stained; glial fibrillary acidic protein, GFAP), P2X4 and P2X7 receptors were stained red with Cy3. After merging the two images, yellow staining appeared (arrows; A) indicating colocalization. IL-1α and IL-1β were not stained with Cy3. After merging of these images, no colocalization was observed (arrows; B). In red microglia (Cy3 stained; OX42), P2X4 receptor, P2X7 receptor, IL-1α and IL-1β all stained green with Cy2, and after merging of the images yellow double staining appeared (arrows; C, D), indicating colocalization.
DISCUSSION
During cerebral ischemia and hypoxia, necrosis and apoptosis, two types of neuronal death, can occur[36,37,38,39,40]. Whether it is necrosis or apoptosis depends on the character and intensity of various nociceptive stimuli, and also ischemic time. Apoptosis frequently occurs after slight ischemia or postischemic reperfusion[41,42,43]. Apoptosis is a naturally occurring programmed and targeted cause of cellular death, showing cell shrinkage, with the presence of intact cell membrane and apoptotic bodies, which are phagocytozed. Cellular contents are contained within the cell membrane, and do not induce an inflammatory reaction. Apoptotic cells are often scattered throughout a sample[40]. Necrosis is a form of cell injury that results in the premature death of cells in living tissue, displaying cell swelling, broken cell membranes, leakage of cellular contents, slow changes in nuclei, insufficient DNA degradation, and local severe inflammatory response. Necrotic cells are widely distributed throughout ischemic tissue. Hematoxylin-eosin staining in the oxygen-glucose deprivation group showed nerve cell atrophy and a clear boundary between cell membranes.
Interleukin-1 receptor antagonist could block this change in the oxygen-glucose deprivation group, suggesting that apoptotic hippocampal nerve cells were found after ischemia/hypoxia, and the occurrence of apoptosis was mediated by interleukin-1, which was consistent with previously published results[40]. Lactate dehydrogenase is known to catalyze the conversion of pyruvic acid to lactic acid[44]. In the oxygen-glucose deprivation group, expression of lactate dehydrogenase significantly increased, and the addition of interleukin-1 receptor antagonist could partially inhibit the increased expression of oxygen-glucose deprivation-mediated lactate dehydrogenase, which was consistent with the results from the hematoxylin-eosin staining studies. These findings indicated that this study successfully established a hippocampal slice model of hypoxia, and the hippocampal nerve cells experienced anaerobic glycolysis, showing hypoxic injury. Simultaneously, these findings suggested that interleukin-1 may promote the effects on hypoxic injury in rat hippocampal neurons. Following oxygen-glucose deprivation, even with the addition of interleukin-1 receptor antagonist, the concentrations of lactate dehydrogenase remain higher than the control group. The above-mentioned results suggested that during cerebral ischemia/hypoxia, other factors, besides the interleukin-1-mediated inflammatory reaction, also participated in neuronal injury.
This study verified that the addition of exogenous interleukin-1α and interleukin-1β could directly induce nerve injury, which confirmed that interleukin-1α and interleukin-1β exert a crucial effect on ischemia/hypoxia-induced nerve injury. Furthermore, the augmentation of the release of lactate dehydrogenase seen following the combined addition of interleukin-1α and interleukin-1β was greater than that using interleukin-1α or interleukin-1β alone. This finding suggests that the mechanisms of action of interleukin-1α and interleukin-1β are different, and their combination shows a synergistic effect on lactate dehydrogenase release. The precise mechanism remains unclear and requires further investigations.
During cerebral ischemia/hypoxia, damaged or necrotic nerve cells significantly release adenosine triphosphate, so that the intracellular and extracellular concentrations of adenosine triphosphate are altered[26]. Elevated adenosine triphosphate can lead to cerebral injury[45,46]. Thus, during hypoxia in hippocampal slices, interleukin-1α- and interleukin-1β-mediated nerve injury may possibly be associated with adenosine triphosphate. Adenosine triphosphate is an endogenous ligand of P2X4 and P2X7.
Adenosine triphosphate receptors have seven subtypes, of these, P2X4 and P2X7 receptors are highly expressed in the central nervous system[28,29]. A selective antagonist of the P2X7 receptor is A438079[47]. The P2X4 receptor does not have a specific antagonist. A previous study showed that P2X4 receptor antibody could effectively suppress the P2X4-gating current[48]. Therefore, P2X4 receptor antibody and A438079, respectively, blocked the P2X4 and P2X7 receptors to observe their effects in oxygen-glucose deprivation-mediated interleukin-1α and interleukin-1β expression. Results revealed that oxygen-glucose deprivation-mediated release of interleukin-1α and interleukin-1β can only be completely suppressed by P2X4 receptor antibody and A438079. These findings suggested that during ischemia/hypoxia, interleukin-1α is mediated by P2X4, but interleukin-1β is mediated by P2X7. The pathway and mechanism of high expression of interleukin-1α and interleukin-1β are not identical.
After P2X4 and P2X7 receptors were blocked, the hypoxia-mediated release of interleukin-1α and interleukin-1β was inhibited, but their physiological release was not affected (as shown in the control group), indicating that the mechanisms of physiological release and hypoxia-induced release are different. Physiological release of interleukin-1α and interleukin-1β was not mediated by the interaction of adenosine triphosphate and its P2X receptor. On the basis of previous studies, this study further confirmed that interleukin-1α and interleukin-1β were involved in hippocampal neuronal injury following oxygen-glucose deprivation via P2X4 and P2X7 receptors, respectively.
The hippocampal cell type that releases interleukin-1α and interleukin-1β remains unclear. A previous study reported that P2X7 receptor mediated interleukin-1β release occurs in macrophages or microglia after stimulation with lipopolysaccharide[49]. It remains unclear whether the mechanism of interleukin-1β synthesis and release during cerebral ischemia/hypoxia is similar to the mechanism of interleukin-1β synthesis and release in response to stimulation with lipopolysaccharide. Microglia and astrocytes are activated and involved in neuronal injury during ischemic/hypoxic brain injury[34,35]. Microglia are considered as “macrophages of the central nervous system”, and play a key role in the innate immune[50,51] and adaptive immune response[52]. Normally, microglia are in the resting state, and mainly nourish and support neurons. Simultaneously, microglia are the principal immune cells in the brain and function in immune defense. During cerebral ischemia/hypoxia, microglia are activated and proliferate, with the presence of reduced number of shortened processes, resulting in microglial pinocytosis and phagocytosis. Thus, microglia can secrete various cytokines, and express various specific proteins, such as CD68 and CD116. Appropriate microglial activation contributes to injury repair, but excessive activation will release interleukin, interferon and tumor necrosis factor, thereby aggravating injury[53,54,55].
To explore which kind of cells released interleukin-1α and interleukin-1β and mediated neuronal injury during ischemia/hypoxia, this study investigated the expression of P2X4 receptor, P2X7 receptor, interleukin-1α and interleukin-1β in astrocytes and microglia using double immunofluorescence labeling. Results demonstrated that at 75 minutes following oxygen-glucose deprivation, astrocytes and microglia were activated, and P2X4 and P2X7 receptors were expressed in both astrocytes and microglia. Interleukin-1α and interleukin-1β, however, were only expressed in microglia. These results verified that interleukin-1α and interleukin-1β were synthesized and released by microglia during cerebral hypoxia, and discovered the source of significant increase in interleukin-1α and interleukin-1β expression under hypoxia.
Following hypoxia, astrocytes were activated, but there was no immunostaining for interleukin-1α and interleukin-1β under these conditions, even though staining for the P2X receptor subtypes was present. These observations may be the case because (1) astrocytes did not synthesize interleukin-1α and interleukin-1β during hypoxia, but mediated the inflammatory reaction of ischemic/hypoxic brain injury by synthesizing other inflammatory factors. (2) Astrocytes synthesized interleukin-1α and interleukin-1β during hypoxia, but interleukin-1α and interleukin-1β were fully released from astrocytes at 75 minutes following hypoxia. Thus, expression of interleukin-1α and interleukin-1β was not detectable in astrocytes. (3) Astrocytes were activated in an early stage of cerebral hypoxia, but astrocytes did not synthesize interleukin-1α and interleukin-1β. The synthesis of interleukin-1α and interleukin-1β began at 75 minutes following oxygen-glucose deprivation, so the expression of interleukin-1α and interleukin-1β was not detected at the sampling time points. Nevertheless, results from previous studies demonstrated that interleukin-1β expression peaked at 60 minutes following cerebral ischemia/hypoxia, which differs from the latter two presumptions[56].
The inflammatory reaction is a major characteristic of continuous damage following cerebral ischemia/hypoxia, and is involved in the whole process of ischemic/hypoxic brain injury, especially in ischemia/reperfusion brain injury. Therefore, studying the pathogenesis of the inflammatory reaction and pathways used by inflammatory molecules allows further understanding for future lessening and prevention of ischemic/hypoxic brain injury.
Studies confirmed that P2X4 and P2X7 receptors, respectively, mediated the release of interleukin-1α and interleukin-1β from microglia during hypoxia. Astrocytes were activated during cerebral hypoxia, but did not express interleukin-1α and interleukin-1β, which further revealed the mechanisms of action of interleukin-1α and interleukin-1β in ischemic/hypoxic brain injury. This observation provides important theoretical evidence for the treatment of ischemic cerebrovascular disease, such as stroke. However, the way that the P2X4 and P2X7 receptors control the release of interleukin-1α and interleukin-1β from microglia and how P2X4 and P2X7 activation mediates neuronal injury and death remain poorly understood and require further investigations.
In summary, interleukin-1α and interleukin-1β played a key role in hypoxic injury in hippocampal slices. The P2X4 receptor and P2X7 receptor, respectively, mediated the release of interleukin-1α and interleukin-1β from microglia. In hypoxic hippocampal slices, astrocytes were activated, but did not synthesize or release interleukin-1α or interleukin-1β. The above mentioned results provide further evidence for developing and applying targeted drugs for the prevention and treatment of neuronal injury following ischemia/hypoxia.
MATERIALS AND METHODS
Design
A randomized, controlled in vitro study.
Time and setting
Experiments were performed at the Clinical Skill Center, Affiliated Hospital of Guangdong Medical College, China from June 2011 to March 2012.
Materials
A total of 15 healthy, clean, male, Sprague-Dawley rats aged 7 or 8 weeks old and weighing 150–200 g were provided by the Experimental Animal Center, Guangdong Medical College, China with license No. A2010067, SCXK (Yue) 2008-0008. Rats were housed in a quiet room with ventilation and air filtration system at 20°C and 50% humidity, and allowed free access to food and water. Bedding and drinking water were replaced daily.
All protocols were in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China[57].
Methods
Preparation of rat hippocampal slices
Rats were decapitated under anesthesia. Hippocampi were separated and washed with artificial cerebrospinal fluid (Shanghai Chemical Agent Company, Shanghai, China) at 0–4°C. Two minutes later, hippocampi were sliced into 400-μm thick coronal sections on a vibratome (Leica, Wetzlar, Germany) and incubated at 35°C in artificial cerebrospinal fluid supplemented with NaCl 126.5 mmol/L, KCI 2.4 mmol/L, KH2 PO4 0.5 mmol/L, CaCl2 1.1 mmol/L, MgCl2 0.83 mmol/L, NaHCO3 27.5 mmol/L, Na2 SO4 and glucose 11.8 mmol/L, pH 7.3, and maintained in 95% O2 + 5% CO2. In glucose-free artificial cerebrospinal fluid, glucose was replaced by 10 mmol/L sucrose (Shanghai Chemical Agent Company).
Oxygen-glucose deprivation tests
Hippocampal slices were randomly assigned into three groups. In the control group, slices were incubated in artificial cerebrospinal fluid in 95% O2 + 5% CO2. In the oxygen-glucose deprivation group[58,59], slices were incubated in artificial cerebrospinal fluid in 95% O2 + 5% CO2 for 20 minutes, and then in glucose-free artificial cerebrospinal fluid in 95% N2 + 5% CO2 for 75 minutes. In the oxygen-glucose deprivation + interleukin-1 receptor antagonist group, sections were incubated in artificial cerebrospinal fluid containing interleukin-1 receptor antagonist (1 mg/mL; R&D, Minneapolis, MN, USA) in 95% O2 + 5% CO2, and then in glucose-free artificial cerebrospinal fluid containing interleukin-1 receptor antagonist (1 mg/mL) in 95% N2 + 5% CO2 for 75 minutes.
Lactate dehydrogenase detection kit (Daiichi Pure Chemicals Co., Ltd., Tokyo, Japan) was used to measure the concentration of lactate dehydrogenase in artificial cerebrospinal fluid, which determined whether hippocampal slices subjected to oxygen-glucose deprivation successfully displayed hypoxic injury. Hematoxylin-eosin staining was utilized to observe morphological changes in neurons. Lactate dehydrogenase detection (lactate dehydrogenase colorimetry) occurred as follows. Standards (10 μL) and samples of artificial cerebrospinal fluid (10 μL) were added in microwell plates. Substrate buffer (reagent A) was mixed with beta-nicotinamide adenine dinucleotide (reagent B) at a ratio of 5:1, and 60 μL of the mixture was added in each well. This mixture was placed in a water bath at 37°C for 15 minutes. 2,4-dinitrophenylhydrazine (reagent C; 50 μL) was mixed with standard preparations or specimens in a water bath at 37°C for 15 minutes. Stop solution (150 μL) was added at room temperature for 3 minutes. Specimens were measured at 440 nm. Subsequently, stock solution of sodium propionate standard preparation was separately diluted into 2.5, 2, 1.5, 1, 0.5, 0.25, 0.125 and 0 (blank well) μmol/mL.
Absorbance values of each well were measured at 440 nm. All experiments were performed at 35°C and repeated six times in six separate cultures.
Detection of interleukin-1α and interleukin-1β release in hippocampal slices subjected to oxygen-glucose deprivation
In the oxygen-glucose deprivation + A438079 group, slices were incubated in artificial cerebrospinal fluid containing A438079 (10 μmol/L) in 95% O2 + 5% CO2 for 20 minutes, and then in glucose-free artificial cerebrospinal fluid containing A438079 (10 μmol/L) in 95% N2 + 5% CO2 for 75 minutes. In the oxygen-glucose deprivation + P2X4 receptor antibody group, slices were incubated in artificial cerebrospinal fluid containing rabbit P2X4 receptor monoclonal antibody (1 μg/mL; Guangzhou Qiyun Biotechnology Co., Ltd., Guangzhou, Guangdong Province, China) in 95% O2 + 5% CO2 for 20 minutes, and then in glucose-free artificial cerebrospinal fluid containing rabbit P2X4 receptor monoclonal antibody (1 μg/mL) in 95% N2 + 5% CO2 for 75 minutes.
Interleukin-1α and interleukin-1β concentration were measured using enzyme-linked immunosorbent assay[60]. Artificial cerebrospinal fluid was centrifuged at 1 000 r/min at room temperature for 20 minutes, and the supernatant was obtained. A 96-well assay plate was divided into blank wells, standard wells and pending test sample wells. Sample diluent (100 μL) was added in the blank wells. Standard preparations (100 μL) and pending test samples (100 μL) were separately added to other wells. Samples were covered with a film and maintained at 37°C for 120 minutes. After removal of the liquid, specimens were dried in air. Detection solution A working fluid (100 μL) was added in each well and the sample was covered with a film for incubation at 37°C for 60 minutes. After removal of the liquid, specimens were dried in air. Liquid A (400 μL/well) was added for 2 minutes. The plate was washed three times with PBS pH 7.4. Detection solution B working fluid (100 μL) was added and the sample was covered with a film for incubation at 37°C for 60 minutes. After removal of the liquid, the specimens were dried in air. Liquid B (350 μL/well) was added for 2 minutes, followed by drying in air. Substrate solution (90 μL) was added to each well in order. The assay plate was covered with a film and maintained at 37°C in the dark for coloration. Stop solution (50 μL) was then added in each well in order to terminate the reaction.
Absorbance values of each well were measured at 450 nm using a microplate reader (Shanghai Kang Medical Technology Development Co., Ltd., Shanghai, China). A standard curve was drawn using curve expert 1.3 curve software (Daniel G. Hyams, Hixson, TN, USA) where standard substance concentration was on the Y-axis and absorbance was on the X-axis. Thus, interleukin-1β concentrations were obtained according to the standard curve. The detection procedure of interleukin-1α was identical to that of interleukin-1β. Experiments were repeated six times.
Determination of the effects of exogenous interleukin-1α and interleukin-1β in hippocampal slices subjected to nerve injury
Hippocampal slices were assigned to four groups. In the control group, slices were incubated in artificial cerebrospinal fluid in 95% O2 + 5% CO2 for 75 minutes. In the exogenous interleukin-1α group, exogenous interleukin-1β group and exogenous interleukin-1α + interleukin-1β group, slices were incubated in artificial cerebrospinal fluid containing exogenous interleukin-1α (100 ng/mL) and/or interleukin-1β (100 ng/mL) in 95% O2 + 5% CO2 for 75 minutes. The concentrations of lactate dehydrogenase were measured.
The measurement method was identical to that used in oxygen-glucose deprivation studies. Experiments were repeated six times.
Immunofluorescence for the expression of the P2X4 receptor, P2X7 receptor, interleukin-1α and interleukin-1β in astrocytes and microglia
Hippocampal slices were incubated in glucose-free artificial cerebrospinal fluid in 95% N2 + 5% CO2 for 75 minutes. The expression of the P2X4 and P2X7 receptor and interleukin-1α and interleukin-1β in astrocytes and microglia were determined using immunofluorescence. Following oxygen-glucose deprivation, hippocampal slices were divided into eight groups. Precise fluorescent labeling is as follows:
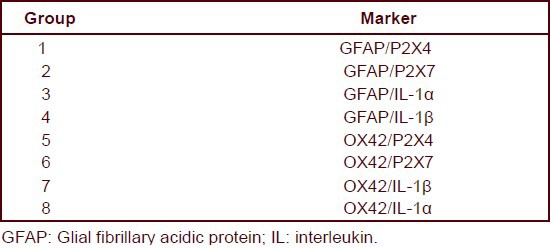
Hippocampal slices in groups 1–4 were washed with Tris buffered saline (0.05 mol/L, pH 7.6), blocked with 5% fetal calf serum, incubated with rat glial fibrillary acidic protein monoclonal antibody (1:1 000; Guangzhou Qiyun Biotechnology Co., Ltd.), and then with secondary antibody targeted against Cy2-labeled goat anti-rat IgG (1:400; Guangzhou Qiyun Biotechnology Co., Ltd.) for fluorescence staining. Subsequently, slices in groups 1 and 2 were incubated in primary antibody rabbit P2X4 and P2X7 receptor monoclonal antibody (P2X4: 1:500, P2X7: 1:400; Guangzhou Qiyun Biotechnology Co., Ltd.). Slices in groups 3 and 4 were incubated with primary antibody hamster interleukin-1 monoclonal antibody (interleukin-1α: 1:200, interleukin-1β: 1:200; Guangzhou Qiyun Biotechnology Co., Ltd.). Subsequently, slices were treated with secondary antibody Cy3-labeled goat anti-rabbit IgG (1:400; Guangzhou Qiyun Biotechnology Co., Ltd.) or secondary antibody Cy3-labeled goat anti-hamster IgG (1:200) for fluorescence staining.
Hippocampal slices in groups 5–8 were incubated in rat OX42 monoclonal antibody (1:2 000; Guangzhou Qiyun Biotechnology Co., Ltd.), and then in the secondary antibody Cy3-labeled goat anti-rat IgG (1:500; Guangzhou Qiyun Biotechnology Co., Ltd.) for fluorescence staining. Subsequently, hippocampal slices in groups 5 and 6 were incubated with primary antibody targeted towards the rabbit P2X4 receptor and P2X7 receptor monoclonal antibody (P2X4: 1:500, P2X7: 1:500). Hippocampal slices in groups 7 and 8 were incubated with primary antibody hamster interleukin-1 monoclonal antibody (interleukin-1α: 1:200; interleukin-1β: 1:200). Secondary antibody Cy2-labeled goat anti-rabbit (1:400; Guangzhou Qiyun Biotechnology Co., Ltd.) or secondary antibody Cy2-labeled goat anti-hamster IgG (1:200; Guangzhou Qiyun Biotechnology Co., Ltd.) were used for fluorescence staining.
Double fluorescence detection was performed using confocal laser scanning microscope (Olympus, Tokyo, Japan). A red signal was revealed by immunofluorescence for Cy3 at an excitation wavelength of 543 nm, and a green signal was revealed by immunofluorescence for Cy2 at an excitation wavelength of 488 nm.
Statistical analysis
Data were expressed as mean ± SD. Data were analyzed using SPSS 16.0 software (SPSS, Chicago, IL, USA). Intergroup differences were compared by one-way analysis of variance. Intergroup paired comparison was performed using least significant difference t-test. A value of P < 0.05 was considered statistically significant.
Acknowledgments:
We thank Guangdong Medical College and Affiliated Hospital of Guangdong Medical College in China for technical support; and the staff of the Library of Guangdong Medical College in China for their help.
Footnotes
Conflicts of interest: None declared.
Funding: This study was supported by the Natural Science Foundation of Guangdong Province, No. S2011010004096.
Ethical approval: The study gained full ethical approval from the Animal Ethics Committee of Guangdong Medical College in China.
(Reviewed by Apricòk, Hindle A, Feng JC, Wang YL)
(Edited by Wang J, Qiu Y, Li CH, Song LP)
REFERENCES
- 1.Mu S, Ouyang L, Liu B, et al. Relationship between inflammatory reaction and ischemic injury of caudate-putamen in rats: inflammatory reaction and brain ischemia. Anat Sci Int. 2011;86(2):86–97. doi: 10.1007/s12565-010-0091-5. [DOI] [PubMed] [Google Scholar]
- 2.Sun M, Zhao Y, Gu Y, et al. Anti-inflammatory mechanism of taurine against ischemic stroke is related to down-regulation of PARP and NF-κB. Amino Acids. 2012;42(5):1735–1747. doi: 10.1007/s00726-011-0885-3. [DOI] [PubMed] [Google Scholar]
- 3.Lalancette-Hébert M, Swarup V, Beaulieu JM, et al. Galectin-3 is required for resident microglia activation and proliferation in response toischemic injury. J Neurosci. 2012;32(30):10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuen CM, Chiu CA, Chang LT, et al. Level and value of interleukin-18 after acute ischemic stroke. Circ J. 2007;71(11):1691–1696. doi: 10.1253/circj.71.1691. [DOI] [PubMed] [Google Scholar]
- 5.Yamasaki Y, Matsuura N, Shozuhara H, et al. interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26(4):676–680. doi: 10.1161/01.str.26.4.676. [DOI] [PubMed] [Google Scholar]
- 6.Nicklin MJ, Barton JL, Nguyen M, et al. A sequence-based map of the nine genes of the human interleukin-1 cluster. Genomics. 2002;79(5):718–725. doi: 10.1006/geno.2002.6751. [DOI] [PubMed] [Google Scholar]
- 7.Pinteaux E, Rothwell NJ, Boutin H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia. 2006;53(5):551–556. doi: 10.1002/glia.20308. [DOI] [PubMed] [Google Scholar]
- 8.Girard S, Sébire H, Brochu ME, et al. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav Immun. 2012;26(8):1331–1339. doi: 10.1016/j.bbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrier A, Olaya N, Chiappini F, et al. Ischemic preconditioning modulates the expression of several genes, leading to the overproduction of IL-1Ra, iNOS, and Bcl-2 in a human model of liver ischemia-reperfusion. FASEB J. 2005;19(12):1617–1626. doi: 10.1096/fj.04-3445com. [DOI] [PubMed] [Google Scholar]
- 10.Schielke GP, Yang GY, Silverstein FS, et al. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J Cereb Blood Flow Meteb. 1999;19(10):1099–1108. doi: 10.1097/00004647-199910000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Touzani O, Boutin H, LeFeuvre R, et al. Interleukin-1 influences ischemic brain damage in the mouse independently of the interleukin-1 type I receptor. J Neurosci. 2002;22(1):38–43. doi: 10.1523/JNEUROSCI.22-01-00038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradillo JM, Denes A, Greenhalgh AD, et al. Delayed administration of interleukin-1 receptor antagonist reduces ischemic brain damage and inflammation in comorbid rats. J Cereb Blood Flow Metab. 2012;32(9):1810–1819. doi: 10.1038/jcbfm.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith CJ, Emsley HC, Udeh CT, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 2012;58(3):384–389. doi: 10.1016/j.cyto.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Caso JR, Moro MA, Lorenzo P, et al. Involvement of IL-1beta in acute stress-induced worsening of cerebral ischaemia in rats. Eur Neuropsychopharmacol. 2007;17(9):600–607. doi: 10.1016/j.euroneuro.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Savitz SI, Erhardt JA, Anthony JV, et al. The novel beta-blocker, carvedilol, provides neuroprotection in transient focal stroke. J Cereb Blood Flow Metab. 2000;20(8):1197–204. doi: 10.1097/00004647-200008000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Touzani O, Boutin H, Chuquet J, et al. Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J Neuroimmunol. 1999;100(1-2):203–215. doi: 10.1016/s0165-5728(99)00202-7. [DOI] [PubMed] [Google Scholar]
- 17.Betz AL, Schielke GP, Yang GY. Interleukin-1 in cerebral ischemia. Keio J Med. 1996;45(3):230–237. doi: 10.2302/kjm.45.230. [DOI] [PubMed] [Google Scholar]
- 18.Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J Cereb Blood Flow Metab. 1998;18(8):833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Hara H, Fink K, Endres M, et al. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997;17(4):370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Nesic O, Xu GY, McAdoo D, et al. IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma. 2001;18(9):947–956. doi: 10.1089/089771501750451857. [DOI] [PubMed] [Google Scholar]
- 21.Weng Y, Khatri B, Hong G, et al. Protective effect of interleukin-1beta on motor neurons after sciatic nerve injury in rats. J Huazhong Univ Sci Technolog Med Sci. 2004;24(1):71–74. doi: 10.1007/BF02830711. [DOI] [PubMed] [Google Scholar]
- 22.Thornton P, McColl BW, Greenhalgh A, et al. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. 2010;115(17):3632–3639. doi: 10.1182/blood-2009-11-252643. [DOI] [PubMed] [Google Scholar]
- 23.Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87(2):659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- 24.Burnstock G, Kennedy C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol. 1985;16(5):433–440. doi: 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- 25.Boeynaems JM, Communi D, Gonzalez NS, et al. Overview of the P2 receptors. Semin Thromb Hemost. 2005;31(2):139–149. doi: 10.1055/s-2005-869519. [DOI] [PubMed] [Google Scholar]
- 26.Abbracchio MP, Ceruti S. Roles of P2 receptors in glial cells: focus on astrocytes. Purinergic Signal. 2006;2(4):595–604. doi: 10.1007/s11302-006-9016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gum RJ, Wakefield B, Jarvis MF. P2X receptor antagonists for pain management: examination of binding and physicochemical properties. Purinergic Signal. 2012;8(Suppl 1):41–56. doi: 10.1007/s11302-011-9272-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vavra V, Bhattacharya A, Zemkova H. Facilitation of glutamate and GABA release by P2X receptor activation in supraoptic neurons from freshly isolated rat brain slices. Neuroscience. 2011;188:1–12. doi: 10.1016/j.neuroscience.2011.04.067. [DOI] [PubMed] [Google Scholar]
- 29.Lorca RA, Rozas C, Loyola S, et al. Zinc enhances long-term potentiation through P2X receptor modulation in the hippocampal CA1 region. Eur J Neurosci. 2011;33(7):1175–1185. doi: 10.1111/j.1460-9568.2010.07589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li F, Wang L, Li JW, et al. Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC Neurosci. 2011;12:111. doi: 10.1186/1471-2202-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wixey JA, Reinebrant HE, Carty ML, et al. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature ratbrain. J Neuroimmunol. 2009;212(1-2):35–43. doi: 10.1016/j.jneuroim.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 32.Ferreira R, Xapelli S, Santos T, et al. Neuropeptide Y modulation of interleukin-1{beta} (IL-1{beta})-induced nitric oxide production in microglia. J Biol Chem. 2010;285(53):41921–41934. doi: 10.1074/jbc.M110.164020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianco F, Pravettoni E, Colombo A, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005;174(11):7268–7277. doi: 10.4049/jimmunol.174.11.7268. [DOI] [PubMed] [Google Scholar]
- 34.Franke H, Schepper C, Illes P, et al. Involvement of P2X and P2Y receptors in microglial activation in vivo. Purinergic Signal. 2007;3(4):435–445. doi: 10.1007/s11302-007-9082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasielewski B, Jensen A, Roth-Härer A, et al. Neuroglial activation and Cx43 expression are reduced upon transplantation of human umbilical cord blood cells after perinatal hypoxic-ischemic injury. Brain Res. 2012;1487:39–53. doi: 10.1016/j.brainres.2012.05.066. [DOI] [PubMed] [Google Scholar]
- 36.Wang JY, Xia Q, Chu KT, et al. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011;70(4):314–322. doi: 10.1097/NEN.0b013e31821352bd. [DOI] [PubMed] [Google Scholar]
- 37.Arsava EM, Gurer G, Gursoy-Ozdemir Y, et al. A new model of transient focal cerebral ischemia for inducing selective neuronal necrosis. Brain Res Bull. 2009;78(4-5):226–231. doi: 10.1016/j.brainresbull.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Gürer G, Gursoy-Ozdemir Y, Erdemli E, et al. Astrocytes are more resistant to focal cerebral ischemia than neurons and die by a delayed necrosis. Brain Pathol. 2009;19(4):630–641. doi: 10.1111/j.1750-3639.2008.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bi GR, Zhang HM, Bai LJ, et al. Effect of adrenomedullin on neuron apoptosis, infarction volume and expression of Egr-1 mRNA after focal ischemia-reperfusion in rats. Neurosci Bull. 2006;22(6):323–330. [PubMed] [Google Scholar]
- 40.Zhou H, Ma Y, Zhou Y, et al. Effects of magnesium sulfate on neuron apoptosis and expression of caspase-3, bax and bcl-2 after cerebral ischemia-reperfusion injury. Chin Med J (Engl) 2003;116(10):1532–1534. [PubMed] [Google Scholar]
- 41.Lu Y, Zhang J, Ma B, et al. Glycine attenuates cerebral ischemia/reperfusion injury by inhibiting neuronal apoptosis in mice. Neurochem Int. 2012;61(5):649–658. doi: 10.1016/j.neuint.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Zhao JX, Tian YX, Xiao HL, et al. Effects of electroacupuncture on hippocampal and cortical apoptosis in a mouse model ofcerebral ischemia-reperfusion injury. J Tradit Chin Med. 2011;31(4):349–355. doi: 10.1016/s0254-6272(12)60017-x. [DOI] [PubMed] [Google Scholar]
- 43.Xing B, Chen H, Zhang M, et al. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke. 2008;39(8):2362–2369. doi: 10.1161/STROKEAHA.107.507939. [DOI] [PubMed] [Google Scholar]
- 44.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 45.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 46.Mohr C, Brady JD, Rossi DJ. Young age and low temperature, but not female gender delay ATP loss and glutamate release, and protect Purkinje cells during simulated ischemia in cerebellar slices. Neuropharmacology. 2010;58(2):392–403. doi: 10.1016/j.neuropharm.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark AK, Staniland AA, Marchand F, et al. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30(2):573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu DM, Adams DJ. Ionic selectivity of native ATP-activated (P2X) receptorchannels in dissociated neurones from rat parasympathetic ganglia. J Physiol. 2001;534(Pt 2):423–435. doi: 10.1111/j.1469-7793.2001.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takenouchi T, Iwamaru Y, Sugama S, et al. The activation of P2X7 receptor induces cathepsin D-dependent production of a 20-kDa form of IL-1β under acidic extracellular pH in LPS-primed microglial cells. J Neurochem. 2011;117(4):712–723. doi: 10.1111/j.1471-4159.2011.07240.x. [DOI] [PubMed] [Google Scholar]
- 50.van Horssen J, Singh S, van der Pol S, et al. Clusters of activated microglia in normal-appearing white matter show signs of innateimmune activation. J Neuroinflammation. 2012;9:156. doi: 10.1186/1742-2094-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147(4):867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 52.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 53.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 54.Garden GA, Möller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1(2):127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 55.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 56.Zhang BL, Liu HL, Jing YM, et al. Role of P2X receptors in synthesis and release of IL-1β during oxygen-glucose deprivation in rat hippocampus. Zhonghua Mazuixue Zazhi. 2010;30(8):1012–1015. [Google Scholar]
- 57.The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]
- 58.Lossi L, Alasia S, Salio C, et al. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog Neurobiol. 2009;88(4):221–245. doi: 10.1016/j.pneurobio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 59.Sun HS, French RJ, Feng ZP. A method for identifying viable and damaged neurons in adult mouse brain slices. Acta Histochem. 2009;111(6):531–537. doi: 10.1016/j.acthis.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Cardenas JM, Marshall P, Henderson B, et al. Human interleukin 2. Quantitation by a sensitive radioimmunoassay. J Immunol Methods. 1986;89(2):181–189. doi: 10.1016/0022-1759(86)90356-x. [DOI] [PubMed] [Google Scholar]