

Abstract
Studies have shown that glycolysis increases during seizures, and that the glycolytic metabolite lactic acid can be used as an energy source. However, how lactic acid provides energy for seizures and how it can participate in the termination of seizures remains unclear. We reviewed possible mechanisms of glycolysis involved in seizure onset. Results showed that lactic acid was involved in seizure onset and provided energy at early stages. As seizures progress, lactic acid reduces the pH of tissue and induces metabolic acidosis, which terminates the seizure. The specific mechanism of lactic acid-induced acidosis involves several aspects, which include lactic acid-induced inhibition of the glycolytic enzyme 6-diphosphate kinase-1, inhibition of the N-methyl-D-aspartate receptor, activation of the acid-sensitive 1A ion channel, strengthening of the receptive mechanism of the inhibitory neurotransmitter γ-minobutyric acid, and changes in the intra- and extracellular environment.
Keywords: neural regeneration, reviews, epilepsy, energy metabolism, glycolysis, epileptogenesis, termination, ATP, aerobic metabolism, 6-diphosphate kinase-1, N-methyl-D-aspartate receptor, acid-sensitive 1A ion channel, γ-aminobutyric acid, intra- and extracellular environment, voltage-gated Na+ and Ca2+, adenosine receptors, ATP receptor, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
Glycolysis increases during seizures, and the glycolytic metabolite lactic acid is used as an energy source during seizures.
-
(2)
The abnormal synchronized discharge of a large number of neurons leads to great consumption of brain bio-energy, so the body has to accelerate the bio-energy of ATP to maintain energy consumption by seizures through aerobic metabolism and anaerobic glycolysis.
-
(3)
During seizures, the aerobic metabolism pathway is inhibited, and the glycolytic pathway enhances the energy shortage caused by insufficient aerobic metabolism. The glycolytic metabolite lactic acid may be involved in supplying energy during seizures.
-
(4)
The main mechanisms of seizure termination include energy consumption and metabolic acidosis. Lactic acid induces acidosis by several possible pathways: lactic acid inhibits the activity of 6 diphosphate kinase-1 and N-methyl-D-aspartate receptors, activates the acid-sensitive 1A ion channel, and strengthens the receptive mechanism of the inhibitory neurotransmitter γ-aminobutyric acid.
INTRODUCTION
Epilepsy, second only to cerebrovascular disease, is one of the most common nervous system diseases[1]. Caused by a variety of factors, epilepsy is a chronic recurrent transient cerebral dysfunction syndrome characterized by the abnormal firing of neurons, and frequent seizures that bring about progressive damage to the brain. Epilepsy afflicts more than 50 million people worldwide, and more than 20 million of those affected continue to have seizures despite treatment with current antiepileptic drugs or surgery[2]. Recurring behavioral seizures are not merely disruptive; they are accompanied by long-term co-morbidities such as memory, cognitive and affective dysfunction. Epileptic seizures, a clinical feature of epilepsy, are an abnormal process in which different causes act on some part of the cerebral cortex of the brain, resulting in an excessive or hypersynchronous discharge of nerve cell groups, with corresponding appearance of clinical performances.
To date, how to clarify the pathogenesis of epilepsy remains a challenge in the field of neuroscience. Epilepsy affects the brain at the molecular, cellular, and neural network level, leading to a rapid cascade of reactions[3]. Seizures involve a series of complex processes, such as nerve cell membrane depolarization caused by an instant change in ion channel dynamics, the rapid transfer of post-synaptic membrane action potentials caused by an excitatory and inhibitory neurotransmitter imbalance and the synchronized depolarization of a large number of neurons. The most important aspect of seizures is the fluctuating change in energy and bio-energy. Due to the unpredictability of epilepsy and the limitations of energetics research methods, most studies have focused on the change in glucose consumption of brain tissue, phosphocreatine/ATP and other energy related molecules before and after seizures using magnetic resonance spectroscopy, and functional MRI[4]. Few studies have investigated the effect of the continuous change in energy on the initiation, development and termination process of epilepsy, or the main energy source of the process during abnormal firing of neurons.
Similar to earthquakes, seizures are a non-linear, slowly accumulating-quick release, continuous energy change process, when neural network energy reaches a critical state after the outbreak of the release, and then reaches a new equilibrium[5]. The entire system relies on a continuous and stable energy supply. During seizures, ion channels open, resulting in an imbalance in excitatory and inhibitory neurotransmitters, which causes increased neuronal excitability and increased energy consumption[2]. In addition, glucose metabolism increases, along with blood flow and oxygen consumption, while the local deoxyhemoglobin concentration and blood oxygen levels decrease signals as detected by functional MRI[6]. These findings indicate that energy consumption increases during seizures. The abnormal synchronized discharge of a large number of neurons leads to a great consumption of bio-energy in the brain. Thus, the body has to increase ATP production to maintain energy consumption caused by seizures through aerobic metabolism and anaerobic glycolysis[7]. A ketogenic diet is a high-fat, low-protein, low carbohydrate diet that has been employed as a treatment for medically intractable epilepsy for 86 years. The “classic” ketogenic diet is based upon consumption of long-chain saturated triglycerides in a 3:1–4:1 ketogenic diet ratio of fats to carbohydrates + protein (by weight)[8]. The vast majority of calories (> 90%) are derived from fat. Despite nearly a century of use, the mechanisms underlying its clinical efficacy remain unknown. A remarkable feature of the ketogenic diet is that ingestion of even a small amount of carbohydrate by patients who have achieved seizure control on the diet can rapidly reduce the diet effectiveness and result in seizure recurrence[9]. This clinical observation suggests that glycolysis and carbohydrate metabolism may promote seizure susceptibility and that inhibition or reduction of glycolysis may have anticonvulsant effects.
Thus, glycolysis, an important in vivo bypass for energy supply, can relieve the energy shortage caused by the energy damage of aerobic metabolism. The glycolytic metabolite lactic acid is involved in seizure onset and provides energy in the early periods of the seizure. As seizures progress, lactic acid can terminate the seizures.
In this paper, we reviewed the possible mechanism where glycolysis is involved in seizure onset to investigate the role of glycolysis in energy metabolism in seizures and to identify how lactic acid participates and terminates seizures. We hope to further understand the association between anaerobic metabolism and the pathogenesis of epilepsy.
ROLE OF GLYCOLYSIS IN ENERGY METABOLISM
Glycolysis is the process by which glucose decomposes into pyruvate in the cytoplasm, and is accompanied by the generation of a small amount of ATP. This process is carried out in the cytoplasm, without oxygen consumption, with the basic steps for each reaction being catalyzed by specific enzymes. Pyruvate deoxidizes to lactate under anoxic conditions, and pyruvate can be further oxidized into acetyl-CoA, which enters the citric acid cycle and produces CO2 and H2O under aerobic conditions. The overall reaction is: glucose + 2 ATP + 2 ADP + 2 phosphate + 2 nicotinamide adenine dinucleotide --> 2 pyruvate + 4 ATP + 2 nicotinamide adenine dinucleotide + 2 hydrogen ions (H+) + 2 H2O. The outcome of this process is that two ATPs are consumed and four ATPs are generated. Therefore, two more ATPs are generated (Figure 1).
Figure 1.
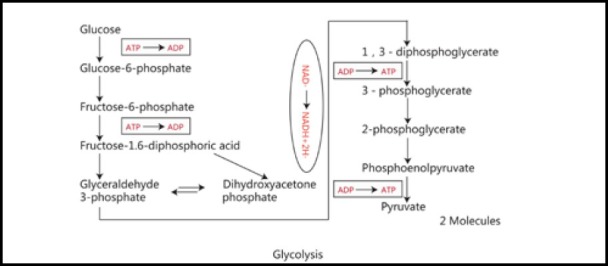
Process of glycolysis.
Glycolysis is the process by which glucose decomposes into pyruvate in the cytoplasm, accompanied by the generation of a small amount of ATP.
Regulation of glycolysis
Hypoxia is the most decisive factor for the initiation of glycolysis. Under stable conditions (when the oxygen level within the cell exceeds the needs of biological energy), aerobic metabolism occurs in cells. Under hypoxic conditions, mitochondrial activity in most cells reduces, and glycolysis becomes the main pathway to generate ATP[10]. The regulation of glycolysis is related to the key rate- limiting enzymes in glycolysis. These enzymes include hexokinase, 6 diphosphate kinase-1 and pyruvate kinase. The activity of these enzymes are regulated by multiple factors such as hypoxia[10], ATP levels[11], insulin, metabolites[11,12] (both substrates and product), AMP, 2,6-diphosphate, 1,6-diphosphate, and citric acid.
ENHANCED GLYCOLYSIS DURING SEIZURES
Characteristics of aerobic energy supply during seizures
During a seizure, abnormal discharge occurs in a large number of neurons, followed by rapid and repeated depolarization of the neuronal cell membrane, and an increase in cerebral blood flow, and metabolic rate of glucose and oxygen consumption[13]. Therefore, the brain is driven into a high metabolic state, making brain and cerebral cortex ATP levels decrease. As a result, energy supply to the brain becomes temporarily insufficient[14]. Therefore, energy supply is limited in epileptic seizures. Under these conditions, in order to maintain energy supply during a seizure, the body needs to speed up the production of ATP through aerobic metabolism (Krebs cycle) and glycolysis. In seizures, there is a significant increase in oxygen demand, which induces the brain into a relatively hypoxic environment; this will inevitably cause the decline of aerobic metabolism. Studies have found that the activity of key enzymes of the tricarboxylic acid cycle such as aconitase, malate dehydrogenase and succinate dehydrogenase decrease in epileptic seizures[15,16], and mitochondrial oxidative shock, which leads to the dysfunction of the electron transport chain (respiratory chain) and the reduction of ATP generation, eventually brings about a barrier to energy supply to the brain[17]. In addition, studies have shown that energy consumption by the central nervous system increases in epileptic seizures, but that barriers to aerobic metabolism impair the energy supply of aerobic metabolism[15,16,17].
Glycolysis increases in seizures
The aerobic pathway does not supply sufficient energy for seizures, and provides the condition in which glycolysis becomes the way of supplying energy for seizures. Studies have confirmed that glycolysis increases during seizures. For example, the activity of some key enzymes involved in glycolytic metabolism such as phosphofructokinase and glucose kinase increase[18]; the glycolytic metabolic rate increases five times; ATP decreases by 15%; phosphocreatine decreases by 44%, while the metabolite of glycolysis lactic acid increases by 87% in the rat cerebral cortex[14]. These findings indicate that ATP levels in the cerebral cortex decrease and that energy supply is relatively insufficient. The increase in anaerobic glycolysis is involved in energy supply during seizure stress. In addition, proton magnetic resonance spectroscopy of seizure rats showed that lactic acid gradually increased, but peaked during and after seizure, which further suggested that anaerobic metabolism gradually increases during seizures[19]. This study shows that in seizures, when the energy provided by the aerobic metabolic pathway is relatively insufficient, the glycolytic pathway is needed to supply energy to the brain[19].
Lactic acid may be involved in supplying energy for seizures
Tetramethylammonium-selective microelectrodes and diffusion-weighted magnetic resonance imaging show that pilocarpine application leads to a rise in lactate, the lactate/pyruvate ratio and glutamate levels within 100 minutes, with a subsequent decrease to control values 140 minutes later[20]. The highest glucose levels occurred 40 minutes after pilocarpine injection. This result shows that the source of energy during seizures may not be the direct decomposition of glucose. Dalsgaard and Secher[21] showed that the increase in glucose and oxygen consumption are disproportionate during a seizure, which further illustrates the important role of the glycolytic pathway in seizures, and that lactic acid may be the major source of energy.
During seizures, abnormal discharge occurs in a large number of neurons, which is followed by rapid and repeated depolarization that extends to the neuronal cell membrane. In addition, cerebral blood flow and the cerebral metabolic rate of glucose and oxygen rapidly increases[22]. Therefore, the brain is driven into a high metabolic state, resulting in the consumption of great amounts of energy and a reduction in ATP levels in the cerebral cortex. As a result, the energy supply to the brain becomes temporarily insufficient, which induces status epilepticus and the destruction of brain function. Studies have shown brain glycogen, an energy material stored in astrocytes that is usually synthesized during sleep, decomposes during rest or in the physical state and is maintained at relatively low levels[23]. Once neuronal activity and energy consumption increase, as occurs during seizures, the metabolic rate of the brain rapidly increases, causing a great deal of brain glucose consumption. Thus, other energy supplies are needed. Glycogen stores in astrocytes are degraded by glycogen phosphorylase, and glycolysis occurs within a short time to allow ATP to supplement the energy required for seizures[24]. A study has shown that brain glycogen can decompose into lactic acid, which is an important energy source during epilepsy[21]. Moreover, lactic acid can provide energy through the astrocyte-neuron lactate shuttle pathway[25] and by the gap junction-mediated metabolic cycle pathway[26] under the transient depletion of glucose. Therefore, lactic acid may be involved in energy supply during primary stages of seizures.
Mechanism of glycolysis activation during seizures
During seizures, brain hypoxia increases the transcriptional level of the glycolytic metabolic molecule hypoxia inducible factor, so as to increase the activity of glycolytic metabolic enzymes and activate the glycolytic production of energy[10]. Low ATP levels and the glycolytic substrate 2,6-diphosphate fructose in the brain enhances the activity of PFKs by regulating the balance between the dimer and tetramer of 6-phosphate fructose kinase-1, thereby increasing the energy generated by glycolysis[11]. In addition, the decrease of blood glucose in brain tissue in seizures[14] stimulates insulin secretion, and insulin can induce in vivo synthesis of glucokinase, 6 diphosphate kinase-1, and pyruvate kinase, thereby promoting the activity of these enzymes, and the energy generated by glycolysis.
GLYCOLYSIS METABOLITES MAY BE INVOLVED IN THE TERMINATION OF SEIZURES
Multiple mechanisms may terminate seizures. There are two mechanisms involved in the termination of seizures: the energy depletion mechanism and the metabolic acidosis mechanism.
Mechanism of energy consumption and seizure termination
The energy depletion hypothesis is based on the fact that neurons abnormally discharge during seizures, resulting in energy depletion, which may lead to termination of seizures. This hypothesis is evidenced by the fact that seizures can reduce oxygen, glucose, and metabolic substrates required for neurotransmission[27,28,29,30]. In vitro recordings show that electrographic seizure-like activity decreases when extracellular glucose is reduced in hippocampal slices maintained in low magnesium[31]. The effect of hypoglycemia on seizure-like discharges in vitro was statistically significant and slow. 50% fewer seizure-like discharges occurred in the 24-minute period following application of low glucose artificial cerebrospinal fluid when compared to the frequency of discharges in 30 minutes prior to application. Moreover, low glucose reduced the amplitude of the seizure-like discharge by 25%. These effects on the frequency and amplitude of seizure-like discharges were reversed by restoration of normal glucose levels. Some anti-epileptic treatments, such as the ketogenic diet, 1,6-diphosphate, and 2-deoxy-glucose have an effect on the inhibition of glycolysis. Their mechanism of action may involve a reduction in the production of energy during seizures, resulting in lowered neuron excitability. Thus the seizures are terminated. Previous studies have shown that the ketogenic diet is very effective in a variety of seizure types[32]. The seizure frequency of most epilepsy patients was reduced by more than 50%[33,34]; and it had an antiepileptic effect on animal models of acute and chronic epilepsy[35,36]. The most common antiepileptic mechanisms are the norepinephrine hypothesis[37,38,39,40,41,42] and the γ-aminobutyric acid hypothesis[43,44,45]. Recent studies have found that reductions in neuronal excitability inhibit epileptic seizures by lowering nicotinamide adenine dinucleotide-dependent gene regulation[46]. It was also found that 2-deoxy D-glucose has acute and chronic antiepileptic effects in many epilepsy models[47,48]. 2-Deoxy D-glucose generates-6-phosphate 2-deoxy D-glucose, which is a competitive inhibitor of glucose that cannot be converted to fructose-6-phosphate by phosphorylate glucose isomerase, thus inhibiting glycolysis. Its acute antiepileptic mechanism remains unclear, but its chronic antiepileptic mechanism involves decreasing the expression of brain-derived neurotrophic factor and tyrosine kinase B, and decreasing neuronal excitability, thereby inhibiting seizures[47]. 1,6- Diphosphate has antiepileptic effects in many epilepsy models[49,50], and inhibits glycolysis by transferring glucose to the pentose phosphate pathway. Its antiepileptic mechanism parallels that of 2-deoxy D-glucose[51], that is, by changing the nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide ratio. The corresponding genetic changes down- regulates the expression of brain-derived neurotrophic factor and tyrosine kinase B, thereby reducing the excitability of nerve cells. In addition, the antiepileptic mechanism of fructose-1,6-diphosphate involves the activation of phospholipcase C, which increases the activity of the nitrogen-activated extracellular signal protein kinase/extracellular signal activated protein kinase signaling pathway[52]. However, the role of these factors remains unclear. While some studies suggest that depleting oxygen, glutamate and ATP can interrupt seizure-like activity, other works suggest reduced levels of these factors might initiate and worsen seizures[31,53]. Moreover, it is well established that energy deprivation via either hypoxia or hypoglycemia-such as that produced by an insulin overdose-often results in coma and neuronal death, and is sometimes associated with onset of seizure activity rather than seizure control. Furthermore, in status epilepticus, seizures may last for several hours, indicating that prolonged seizures do not exhaust the fuel that sustains them and prolonged seizures cannot be terminated by energy depletion.
Mechanism of metabolic acidosis and seizure termination
Metabolic acidosis may be another mechanism by which seizures are terminated. During the epileptic discharge process, the accumulation of various metabolites that are generated by neuron misfiring during seizures leads to acidosis, and this may be involved in the process of seizure termination. Anaerobic glycolysis is known to increase seizures, and its metabolite lactic acid terminates seizures[54] (Figure 2).
Figure 2.
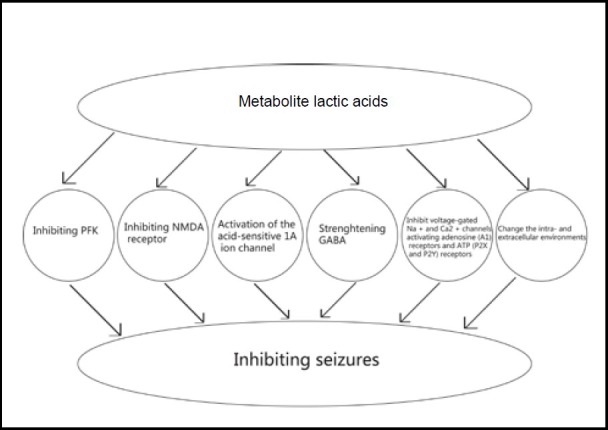
Mechanism of metabolite acidosis and seizure termination.
The glycolytic metabolite lactic acid can decrease the pH of tissue via the following ways: (1) inhibiting the activity of glycolytic enzyme 6 diphosphate kinase-1; (2) inhibiting the N-methyl-D-aspartate receptor; (3) activating the acid-sensitive 1A ion channel; (4) strengthening the receptive mechanism of the inhibitory neurotransmitter γ-aminobutyric acid; (5) changing intra- and extracellular environments and (6) other mechanisms such as directly suppressing the activity of voltage-gated Na+ and Ca2+ channels, and activating adenosine receptors and ATP receptors.
Lactic acid terminates seizures by inhibiting 6-diphosphate kinase-1
6-Diphosphate kinase-1 is a key regulatory glycolytic enzyme characterized by allosteric kinetics, a complex oligomeric structure with multiple modes of regulation. This enzyme is regulated by a variety of ligands including its substrates, reaction products and various other cellular metabolites such as adenosine monophosphate, fructose-2,6-bisphosphate, glucose 1,6-bisphosphate, and citrate. ATP, has a dual effect. At concentrations of up to 1 mmol/L it acts as an enzyme activator, but at higher concentrations it acts as an inhibitor. However, fructose 2,6-bisphosphate reverses the inhibitory effects of ATP. Below, we discuss how lactate down-regulates 6-diphosphate kinase-1 activity.
Glycolysis produces large amounts of lactic acid, causing the pH to decrease, resulting in extracellular acidosis. It was found when the pH is comparatively low (pH less than 7.5)[54], lactic acid can act on 6-diphosphate kinase-1 through allosteric regulation, making it separate from the tetrameric structure with strong metabolic activity to the dimer structure with low metabolic activity. There are many studies that show that fructose 2,6-bisphosphate is a potent activator of 6-diphosphate kinase-1[55]. 6-Diphosphate kinase-1 associates with f-actin, which induces the same effect as fructose 2,6-bisphosphate, such as stabilization of the tetrameric conformation of the enzyme[56,57]. Meanwhile, 6- diphosphate kinase-1 is phosphorylated by protein kinase A upon adrenergic stimulation leading to a more active tetrameric-stable enzyme[58,59]. Fructose- 2,6-bisphosphate and f-actin present distinct binding sites and protein kinase A promotes a covalent modification of the enzyme, counteracting the effect of lactate on 6-diphosphate kinase-1 activity. These results corroborate that lactate is acting on 6-diphosphate kinase-1 to dissociate the tetrameric structure of the enzyme, which inhibits its catalytic activity. Therefore lactate can inhibit 6-diphosphate kinase-1 activity which is counteracted by several physiological signals such as an increase in fructose-2,6-bisphosphate, the action of protein kinase A and the association of the enzyme with f-actin, which is mediated by many other signals[60,61]. Moreover, lactate inhibits 6-diphosphate kinase-1 by dissociating the enzyme active tetramers into less active dimers. Glycolysis is inhibited, and finally seizures are terminated because of the shortage of energy supply.
Lactic acid terminates seizures by inhibiting N-methyl-D-aspartate receptor
Studies show that lowering the pH to 6.7 by either means increases the interval between seizure-like events, and acidification slows the rate of seizure propagation in [Mg2+ ]-induced epileptiform bursting in hippocampal slices and entorhinal cortex preparation, which is presumably by activation of N-methyl-D-aspartate receptors, which show blockade of N-methyl-D-aspartate currents by protons. This may be an important component of the anticonvulsant action of extracellular acidosis[62]. Protons inhibit N-methyl-D-aspartate receptors by decreasing the opening frequency of 30- 50 pS N-methyl-D-aspartate channels, and reducing the relative proportion of longer bursts. N-methyl-D-aspartate receptor antagonists attenuate the acid effect on epileptiform activity in brain slices[63]. Therefore, a decrease in pH can inhibit N-methyl-D-aspartate receptor activity, and seizure activity is terminated.
Lactic acid terminates seizures by activating acid-sensitive 1A ion channel
Acid-sensitive 1A ion channels (ASICs) are proton-gated members of the degenerin/epithelial Na+ channel family[64]. There are three ASICs, ASIC1a, -2a, and -2b, which are widely expressed in the central nervous system[65,66,67]. In neurons of the central nervous system, ASIC1a is sensitive to the pH of the extracellular environment and excitability change of nerve cell. Generally, ASIC1a is activated in response to pH values between 7.2 and 5.0, and extracellular acidosis activates acid-sensitive 1a ion channels to initiate neuron firing. The current generated following ASIC-induced acidosis inhibits the epileptic discharge[68]. The detailed mechanism involves ASICs triggering inhibitory neuron activity to terminate seizures. In addition, ASICs are also expressed in excitatory pyramidal neurons, which may contribute to reduced activity, possibly through blockade of depolarization. Moreover, ASICs are also expressed in many different brain regions and it is possible that activation of inhibitory neurons in other regions may also contribute to seizure termination through increasing the inhibitory neurotransmitter γ-aminobutyric acid[66,67,68].
Lactic acid terminates seizures by strengthening γ-aminobutyric acid transaminase
Glutamic acid and γ-aminobutyric acid which are the most important excitable and inhibitory neurotransmitters in the central nervous system are widely distributed in various brain areas[69,70,71]. H+ ions can effect γ-aminobutyric acid transaminase receptor function via three ways. First, the decrease in external pH may affect protonating weak acidic and basic groups contained within the receptor protein, which may modulate receptor function. Moreover, protonation and subsequent determination of pKa values may allow the broad identification of key amino acid residues that are involved in receptor function. Second, previous studies have demonstrated that changing external pH can differentially influence native γ-aminobutyric acid transaminase receptor function[72]. Third, glutamate in the brain can be decarboxylated to generate γ-aminobutyric acid, which is catalyzed by glutanic acid decarboxylase with pyridoxal phosphate as the coenzyme[73]. Meanwhile, γ-aminobutyric acid transaminase can catalyze the transamination between gamma-aminobutyric acid transaminase and alkone glutaric acid to generate glutamate. The change in pH values in the body can modulate the enzyme activity of glutanic acid decarboxylase and γ-aminobutyric acid transaminase. The optimal pH values of the two enzymes are different, 6.5 for glutanic acid decarboxylase and 8.2 for γ-aminobutyric acid transaminase. Seizures and lactic acids reduce the extracellular pH from 7.35 to 6.8[74,75,76,77], which increases glutanic acid decarboxylase activity, resulting in the decreased activity of γ-aminobutyric acid transaminase, an increase in the inhibitory neurotransmitter γ-aminobutyric acid. These alterations inhibit seizures[78]. (Figure 3), (Figure 4), (Figure 5) and (Figure 6).
Figure 3.

The metabolite lactic acid terminates seizures by inhibiting 6 diphosphate kinase-1(PFK).
PKA: protein kinase A.
Figure 4.
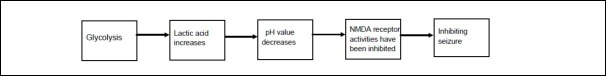
The metabolite lactic acid terminates seizures by inhibiting N-methyl-D-aspartate (NMDA) receptors.
Figure 5.
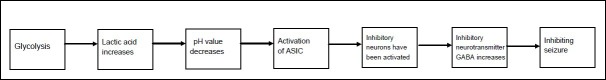
The metabolite lactic acid terminates seizures by activating acid-sensitive 1A ion channels.
ASIC: activation of acid-sensitive 1A.
Figure 6.

The metabolite lactic acid terminates seizures by strengthening γ-aminobutyric acid.
GAD: Glutanic acid decarboxylase; GABA: γ-aminobutyric acid.
A recent study found that the ketogenic diet may also increase the ratio of nicotinamide adenine dinucleotide/ nicotinamide adenine dinucleotide and enhance glyceraldehyde-3-phosphate dehydrogenase enzyme activity. These changes subject γ-aminobutyric acid to endogenous phosphorylation and activate γ-aminobutyric acid α receptors, which play a role in the antiepileptic effect[69].
Metabolite lactic acid terminates seizures by changing the intra- and extracellular environment
As determined previously, in a low [Mg2+]-induced in vitro model of epilepsy, a pH of 6.7 terminated seizure-like burst firing. This decrease in epileptiform activity began within minutes of lowering the pH[64], and caused lactic acid production. This observation may be due to a decrease in N-methyl-D-aspartate receptor function and a loss of synaptic long-term potentiation. A decrease in pH to 7.1 produced milder synaptic impairment with continued loss of long-term potentiation[79]. Moreover, inhibition of carbonic anhydrase, which alters extracellular pH, had an anticonvulsant effect, and the carbonic anhydrase inhibitor acetozolamide had an anticonvulsant effect[80]. Studies that compared wild type mice with knockout mice deficient in carbonic anhydrase were severely acidotic and were resistant to seizures induced by flurothyl gas[81].
Intracellular acidification also contributes to the termination of seizure discharge. Spontaneous interictal spiking is induced by the application of bicuculline in the piriform cortex in in vitro whole brain preparations. This was associated with periodic abrupt alkanization of the extracellular space followed by return of baseline pH[82]. These studies provide evidence of intracellular acidification. The increase in neuronal excitability and the presence of discharges following each spike were induced by application of ammonium chloride in the perfusing medium, which prevents intracellular acidification. Researchers hypothesized that the intracellular acidification reduced excitability through reducing gap junction function. In addition, octanol, 4-aminopyridine which is a gap junction blocker, abolished spontaneous interictal spiking[83]. Moreover, similar research from our lab using the gap junction blocker carbenoxolone revealed a decrease in epileptiform discharge and negated the dependence of burst firing[84].
The possible mechanism may be that gap junction opening is dependent on pH[85] and that the drop in pH can lead to the closure of gap junctions, thus disrupting synchronization of the network of neurons and thereby inhibiting seizures.
The altering of chloride homeostasis in the extracellular milieu may disrupt epileptic activity. There are studies that show that furosemide applied to an epileptic initiation site, either in vitro or in vivo, terminates seizure discharge but does not interfere with excitatory synaptic transmission[86]. The mechanism of furosemide appears to be mediated by changes in chloride concentration. Furosemide disrupted the synchronization of action potential firing between neurons without affecting synaptic activity through the changing of the extracellular environment[87].
Lactic acid is known to lead to a drop in pH. And the decreasing pH can terminate seizures through changing the intra- and extracellular environments.
Other mechanisms of lactic acid-induced termination of seizures
Lactic acid brings about an increase in H+ ions in the extracellular environment and a reduction of pH, which leads to acidosis. Furthermore, the reduction in pH can also inhibit voltage-gated Na+ and Ca2+ channels[78], thereby suppressing seizures. Extracellular fluid acidosis raises the adenosine density, activating adenosine receptors and ATP receptors, and reduced the activity of cranial slices epileptiform[88]. Recent studies have found that 1,6-diphosphate plays an anticonvulsant role by increasing glutathione levels in astrocytes[89,90].
DISCUSSION
Seizures involve energy storage in the brain being exhausted when neurons discharge abnormally. During this process, glycolysis becomes an important source of energy supply. Enhancement of glycolysis can relieve the energy shortage caused by the energy damage to aerobic metabolism. The glycolytic metabolite lactic acid is involved in seizures and provides energy in the early period of the seizure. As seizures progress, lactic acid can decrease the pH of tissue through the following possible ways: (1) inhibiting the active mechanism of the key glycolytic enzyme 6-diphosphate kinase-1; as seizures progress, lactic acid increases, which leads to the decrease in pH. Following this, 6-diphosphate kinase-1 is phosphorylated by protein kinase A and 6-diphosphate kinase-1 activity is inhibited and the seizures are terminated. (2) inhibiting the active mechanism of N-methyl-D-aspartate receptor; lactic acid increases which leads to a decrease in pH. N-methyl-D-aspartate receptor activity is inhibited, and seizures are terminated. (3) activating the acid-sensitive 1A Ion channel; lactic acid increases, which leads to a decrease in pH. The acid-sensitive 1A ion channel is activated and the inhibitory neurons are activated. Seizures are terminated through increasing inhibitory neurotransmitter γ-aminobutyric acid. (4) strengthening the receptive mechanism of the inhibitory neurotransmitter γ-aminobutyric acid; lactic acid increases, which leads to a decrease in pH, and glutamic acid decarboxylase is activated and the inhibitory neurotransmitter γ-aminobutyric acid increases. Thus, seizures are terminated. (5) changing the intra- and extracellular environment. Lactic acid can lead to a drop in pH. Decreasing pH can terminate seizures through changing the intra- and extracellular environments. (6) other mechanisms such as directly suppressing the channel activity of voltage-gated Na+ and Ca2+, and activating adenosine receptors and ATP receptors. The understanding of specific roles of glycolysis during different types of seizures and periods will help investigation on epileptic pathogenesis, and also provide brand-new treatment strategies for antiepileptic therapies.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 30971534 and 125 Project of the Third Xiangya Hospital, China.
Conflicts of interest: None declared.
(Reviewed by Diwakarla S, Frenchman B, Di ZL, Liu SY)
(Edited by Wang J, Su LL, Li CH, Song LP)
REFERENCES
- 1.Scharfman HE. The neurobiology of epilepsy. Curr Neurol Neurosci Rep. 2007;7(4):348–354. doi: 10.1007/s11910-007-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birbeck G, Chomba E, Atadzhanov M, et al. The social and economic impact of epilepsy in Zambia: a cross-sectional study. Lancet Neurol. 2007;6(1):39–44. doi: 10.1016/S1474-4422(06)70629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wasterlain CG, Fujikawa DG, Penix L, et al. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34(Suppl 1):S37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- 4.Williamson A, Patrylo PR, Pan J, et al. Correlations between granule cell physiology and bioenergetics in human temporal lobe epilepsy. Brain. 2005;128(Pt 5):1199–1208. doi: 10.1093/brain/awh444. [DOI] [PubMed] [Google Scholar]
- 5.Osorio I, Frei MG, Sornette D, et al. Epileptic seizures: Quakes of the brain? Phys Rev E Stat Nonlin Soft Matter Phys. 2010;82(2 Pt 1):021919. doi: 10.1103/PhysRevE.82.021919. [DOI] [PubMed] [Google Scholar]
- 6.Federico P, Abbott DF, Briellmann RS, et al. Functional MRI of the pre-ictal state. Brain. 2005;128(Pt 8):1811–1817. doi: 10.1093/brain/awh533. [DOI] [PubMed] [Google Scholar]
- 7.Wasterlain CG, Thompson KW, Suchomelova L, et al. Brain energy metabolism during experimental neonatal seizures. Neurochem Res. 2010;35(12):2193–2198. doi: 10.1007/s11064-010-0339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kossoff EH, McGrogan JR. Worldwide use of the ketogenic diet. Epilepsia. 2005;46(2):280–289. doi: 10.1111/j.0013-9580.2005.42704.x. [DOI] [PubMed] [Google Scholar]
- 9.Huttenlocher PR. Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res. 1976;10(5):536–540. doi: 10.1203/00006450-197605000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405(1):1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 11.Zancan P, Marinho-Carvalho MM, Faber-Barata J, et al. ATP and fructose-2, 6-bisphosphate regulate skeletal muscle 6-phosphofructo-1-kinase by altering its quaternary structure. IUBMB Life. 2008;60(8):526–533. doi: 10.1002/iub.58. [DOI] [PubMed] [Google Scholar]
- 12.Uyeda K. Phosphofructokinase. Adv Enzymol Relat Areas Mol Biol. 1979;48:193–244. doi: 10.1002/9780470122938.ch4. [DOI] [PubMed] [Google Scholar]
- 13.Wasterlain CG, Fujikawa DG, Penix L, et al. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34(Suppl 1):37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- 14.Wasterlain CG, Thompson KW, Suchomelova L, et al. Brain energy metabolism during experimental neonatal seizures. Neurochem Res. 2010;35(12):2193–2198. doi: 10.1007/s11064-010-0339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folbergrová J, Jesina P, Drahota Z, et al. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Exp Neurol. 2007;204(2):597–609. doi: 10.1016/j.expneurol.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Acharya MM, Katyare SS. Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp Neurol. 2005;192(1):79–88. doi: 10.1016/j.expneurol.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Waldbaum S, Patel M. Mitochondria, oxidative stress and temporal lobe epilepsy. Epilepsy Res. 2010;88(1):23–45. doi: 10.1016/j.eplepsyres.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sacktor B, Wilson JE, Tiekert CG. Regulation of glycolysis in brain, in situ, during convulsions. J Biol Chem. 1966;241(21):5071–5075. [PubMed] [Google Scholar]
- 19.Neppl R, Nguyen CM, Bowen W, et al. In vivo detection of postictal perturbations of cerebral metabolism by use of proton MR spectroscopy: preliminary results in a canine model of prolonged generalized seizures. AJNR Am J Neuroradiol. 2001;22(10):1933–1943. [PMC free article] [PubMed] [Google Scholar]
- 20.Slais K, Vorisek I, Zoremba N, et al. Brain metabolism and diffusion in the rat cerebral cortex during pilocarpine- induced status epilepticus. Exp Neurol. 2008;209(1):145–154. doi: 10.1016/j.expneurol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Dalsgaard MK, Secher NH. The brain at work: a cerebral metabolic manifestation of central fatigue? J Neurosci Res. 2007;85(15):3334–3339. doi: 10.1002/jnr.21274. [DOI] [PubMed] [Google Scholar]
- 22.Cavus I, Kasoff WS, Cassaday MP, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57(2):226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- 23.Oz G, Seaquist ER, Kumar A, et al. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol Endocrinol Metab. 2007;292(3):E946–951. doi: 10.1152/ajpendo.00424.2006. [DOI] [PubMed] [Google Scholar]
- 24.Wiesinger H, Hamprecht B, Dringen R. Metabolic pathways for glucose in astrocytes. Glia. 1997;21(1):22–34. doi: 10.1002/(sici)1098-1136(199709)21:1<22::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Pellerin L, Bouzier-Sore AK, Aubert A, et al. Activity- dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55(12):1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 26.Rouach N, Koulakoff A, Abudara V, et al. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322(5907):1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- 27.Freund TF, Buzsaki G, Prohaska OJ, et al. Simultaneous recording of local electrical activity, partial oxygen tension and temperature in the rat hippocampus with a chamber- type microelectrode. Effects of anaesthesia, ischemia and epilepsy. Neuroscience. 1989;28(3):539–549. doi: 10.1016/0306-4522(89)90003-1. [DOI] [PubMed] [Google Scholar]
- 28.Chapman AG, Meldrum BS, Siesjo BK. Cerebral metabolic changes during prolonged epileptic seizures in rats. J Neurochem. 1977;28(5):1025–1035. doi: 10.1111/j.1471-4159.1977.tb10665.x. [DOI] [PubMed] [Google Scholar]
- 29.Kloiber O, Bockhorst K, Hoehn-Berlage M, et al. Effect of hypoxia on bicuculline seizures of rat: NMR spectroscopy and bioluminescence imaging. NMR Biomed. 1993;6(5):333–338. doi: 10.1002/nbm.1940060509. [DOI] [PubMed] [Google Scholar]
- 30.Yamada K, Ji JJ, Yuan H, et al. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292(5521):1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- 31.Kirchner A, Veliskova J, Velisek L. Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. Eur J Neurosci. 2006;23(6):1512–1522. doi: 10.1111/j.1460-9568.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- 32.Bainbridge JL, Gidal BE, Ryan M. The ketogenic diet. Central nervous system practice and research network of the american college of clinical pharmacy. Pharmacotherapy. 1999;19(6):782–786. doi: 10.1592/phco.19.9.782.31535. [DOI] [PubMed] [Google Scholar]
- 33.Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59(2):293–315. doi: 10.1016/j.brainresrev.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freeman JM, Vining EP, Pillas DJ, et al. The efficacy of the ketogenic diet-1998: a prospective evaluation of intervention in 150 children. Pediatrics. 1998;102(6):1358–1363. doi: 10.1542/peds.102.6.1358. [DOI] [PubMed] [Google Scholar]
- 35.Bough KJ, Wetherington J, Hassel B, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60(2):223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 36.Bough KJ, Gudi K, Han FT, et al. An anticonvulsant profile of the ketogenic diet in the rat. Epilepsy Res. 2002;50(3):313–325. doi: 10.1016/s0920-1211(02)00086-4. [DOI] [PubMed] [Google Scholar]
- 37.Yan QS, Jobe PC, Dailey JW. Noradrenergic mechanisms for the anticonvulsant effects of desipramine and yohimbine in genetically epilepsy-prone rats: studies with microdialysis. Brain Res. 1993;610(1):24–31. doi: 10.1016/0006-8993(93)91212-b. [DOI] [PubMed] [Google Scholar]
- 38.Weinshenker D, Szot P. The role of catecholamines in seizure susceptibility: new results using genetically engineered mice. Pharmacol Ther. 2002;94(3):213–233. doi: 10.1016/s0163-7258(02)00218-8. [DOI] [PubMed] [Google Scholar]
- 39.Szot P, Weinshenker D, Rho JM, et al. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Brain Res Dev Brain Res. 2001;129(2):211–214. doi: 10.1016/s0165-3806(01)00213-9. [DOI] [PubMed] [Google Scholar]
- 40.Richichi C, Lin EJ, Stefanin D, et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24(12):3051–3059. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vezzani A, Sperk G. Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides. 2004;38(4):245–252. doi: 10.1016/j.npep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Schlifke I, Kuteeva E, Hokfelt T, et al. Galanin expressed in the excitatory fibers attenuates synaptic strength and generalized seizures in the piriform cortex of mice. Exp Neurol. 2006;200(2):398–406. doi: 10.1016/j.expneurol.2006.02.124. [DOI] [PubMed] [Google Scholar]
- 43.Cheng CM, Hicks K, Wang J, et al. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J Neurosci Res. 2004;77(2):270–276. doi: 10.1002/jnr.20144. [DOI] [PubMed] [Google Scholar]
- 44.Yudkoff M, Daikhin Y, Nissim I, et al. Effects of ketone bodies on astrocyte amino acid metabolism. J Neurochem. 1997;69(2):682–692. doi: 10.1046/j.1471-4159.1997.69020682.x. [DOI] [PubMed] [Google Scholar]
- 45.Bough K. Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):91–93. doi: 10.1111/j.1528-1167.2008.01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang YZ, McNamara JO. Inhibiting glycolysis to reduce seizures: how it might work. Nat Neurosci. 2006;9(11):1351–1352. doi: 10.1038/nn1106-1351. [DOI] [PubMed] [Google Scholar]
- 47.Stafstrom CE, Ockuly JC, Murphree L, et al. Anticonvulsant and antiepileptic actions of 2-deoxy-D- glucose in epilepsy models. Ann Neurol. 2009;65(4):435–447. doi: 10.1002/ana.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stafstrom CE, Roopra A, Sutula TP. Seizure suppression via glycolysis inhibition with 2-deoxy-D-glucose (2DG) Epilepsia. 2008;49(Suppl 8):97–100. doi: 10.1111/j.1528-1167.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- 49.Garriga-Canut M, Schoenike B, Qazi R, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9(11):1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 50.Gasior M, Yankura J, Hartman AL, et al. Anticonvulsant and proconvulsant actions of 2-deoxy-D-glucose. Epilepsia. 2010;51(8):1385–1394. doi: 10.1111/j.1528-1167.2010.02593.x. [DOI] [PubMed] [Google Scholar]
- 51.Lian XY, Khan FA, Stringer JL. Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci. 2007;27(14):12007–12011. doi: 10.1523/JNEUROSCI.3163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fahlman CS, Bickler PE, Sullivan B, et al. Activation of the neuroprotective ERK signaling pathway by fructose-1,6-bisphosphate during hypoxia involves intracellular Ca2+ and phospholipase C. Brain Res. 2002;958(1):43–51. doi: 10.1016/s0006-8993(02)03433-9. [DOI] [PubMed] [Google Scholar]
- 53.Haglund MM, Schwartzkroin PA. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63(2):225–239. doi: 10.1152/jn.1990.63.2.225. [DOI] [PubMed] [Google Scholar]
- 54.Costa Leite T, Da Silva D, Guimarães Coelho R, et al. Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J. 2007;408(1):123–130. doi: 10.1042/BJ20070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kristensen M, Albertsen J, Rentsch M, et al. Lactate and force production in skeletal muscle. J Physiol. 2005;562(Pt 2):521–526. doi: 10.1113/jphysiol.2004.078014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liou RS, Anderson S. Activation of rabbit muscle phosphofructokinase by F-actin and reconstituted thin filaments. Biochemistry. 1980;19(12):2684–2688. doi: 10.1021/bi00553a022. [DOI] [PubMed] [Google Scholar]
- 57.Clarke F, Stephan P, Morton DJ, et al. Actin: Structure and Function in Muscle and Non-Muscle Cells. Sydney: 2nd International Congress of Biochemistry, Australia; 1983. The role of actin and associated structural proteins in the organization of glycolytic enzymes. [Google Scholar]
- 58.Silva AP, Alves GG, Araujo AH, et al. Effects of insulin and actin on phosphofructokinase activity and cellular distribution in skeletal muscle. Ann Acad Bras Cienc. 2004;76(3):541–548. doi: 10.1590/s0001-37652004000300008. [DOI] [PubMed] [Google Scholar]
- 59.Gomes Alves G, Sola-Penna M. Epinephrine modulates cellular distribution of muscle phosphofructokinase. Mol Genet Metab. 2003;78(4):302–306. doi: 10.1016/s1096-7192(03)00037-4. [DOI] [PubMed] [Google Scholar]
- 60.Zancan P, Sola-Penna M. Regulation of human erythrocyte metabolism by insulin: cellular distribution of 6-phosphofructo-1-kinase and its implication for red blood cell function. Mol Genet Metab. 2005;86(3):401–411. doi: 10.1016/j.ymgme.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 61.Zancan P, Rosas AO, Marcondes MC, et al. Clotrimazole inhibits and modulates heterologous association of the key glycolytic enzyme 6-phosphofructo-1-kinase. Biochem Pharmacol. 2007;73(10):1520–1527. doi: 10.1016/j.bcp.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 62.Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345(6273):347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- 63.Velisek L, Dreier JP, Stanton PK, et al. Lowering of extracellular pH suppresses low-Mg2+ -induces seizures in combined entorhinal cortex-hippocampal slices. Exp Brain Res. 1994;101(1):44–52. doi: 10.1007/BF00243215. [DOI] [PubMed] [Google Scholar]
- 64.McKhann GM., 2nd Seizure termination by acidosis depends on ASIC1a. Neurosurgery. 2008;63(4):N10. doi: 10.1227/01.NEU.0000339453.48345.4C. [DOI] [PubMed] [Google Scholar]
- 65.Waldmann R, Champigny G, Bassilana F, et al. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 66.García-Añoveros J, Derfler B, Neville-Golden J, et al. BNaC1 and BNaC2 constitute a new family of human neuronal sodium channels related to degenerins and epithelial sodium channels. Proc Natl Acad Sci U S A. 1997;94(4):1459–1464. doi: 10.1073/pnas.94.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coryell MW, Ziemann AE, Westmoreland PJ, et al. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry. 2007;62(10):1140–1148. doi: 10.1016/j.biopsych.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Ziemann AE, Schnizler MK, Albert GW, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11(7):816–822. doi: 10.1038/nn.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin SH, Gong SS, Yan KS, et al. Effects of neuroglobin gene transfer in vivo on hearing response properties of neurons in the inferior colliculus in mice after administration of sodium salicylate. Sheng Li Xue Bao. 2005;57(4):529–536. [PubMed] [Google Scholar]
- 70.Faingold CL. Role of GABA abnormalities in the inferior colliculus pathophysiology-audiogenic seizures. Hear Res. 2002;168(1-2):223–237. doi: 10.1016/s0378-5955(02)00373-8. [DOI] [PubMed] [Google Scholar]
- 71.Storozhuk VM, Khorevin VI, Razumna NN, et al. The effects of activation of glutamate ionotropic connections of neurons in the sensorimotor cortex in a conditioned reflex. Neurosci Behav Physiol. 2003;33(5):479–488. doi: 10.1023/a:1023415317960. [DOI] [PubMed] [Google Scholar]
- 72.Chen JC, Chesler M. Modulation of extracellular pH by glutamate and GABA in rat hippocampal slices. J Neurophysiol. 1992;67(1):29–36. doi: 10.1152/jn.1992.67.1.29. [DOI] [PubMed] [Google Scholar]
- 73.Sloviter RS, Dichter MA, Rachinsky TL, et al. Basal expression and induction of glutamate decarboxylase and GABA in excitatory granule cells of the rat and monkey hippocampal dentate gyrus. J Comp Neurol. 1996;373(4):593–618. doi: 10.1002/(SICI)1096-9861(19960930)373:4<593::AID-CNE8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 74.Somjen GG. Acidification of interstitial fluid in hippocampal formation caused by seizures and by spreading depression. Brain Res. 1984;311(1):186–188. doi: 10.1016/0006-8993(84)91416-1. [DOI] [PubMed] [Google Scholar]
- 75.Wang RI, Sonnenschein RR. PH of cerebral cortex during induced convulsions. J Neurophysiol. 1955;18(2):130–137. doi: 10.1152/jn.1955.18.2.130. [DOI] [PubMed] [Google Scholar]
- 76.Wemmie JA, Chen J, Askwith CC, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34(3):463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- 77.Xiong ZG, Zhu XM, Chu XP, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 78.Somjen GG, Tombaugh GC. pH modulation of neuronal excitability and central nervous system functions. In: Kaila K, Ransom BR, editors. pH and Brain Function. New York: Wiley-Leiss Inc, USA; 1998. [Google Scholar]
- 79.Velisek L. Extracellular acidosis and high levels of carbon dioxide suppress synaptic transmission and prevent the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Hippocampus. 1998;8(1):24–32. doi: 10.1002/(SICI)1098-1063(1998)8:1<24::AID-HIPO3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 80.Thiry A, Dogne JM, Supuran CT, et al. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem. 2007;7(9):855–864. doi: 10.2174/156802607780636726. [DOI] [PubMed] [Google Scholar]
- 81.Velisek L, Moshe SL, Xu SG, et al. Reduced susceptibility to seizures in carbonic anhydrase II deficient mutant mice. Epilepsy Res. 1993;14(2):115–121. doi: 10.1016/0920-1211(93)90016-z. [DOI] [PubMed] [Google Scholar]
- 82.de Curtis M, Manfridi A, Biella G. Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. J Neurosci. 1998;18(18):7543–7551. doi: 10.1523/JNEUROSCI.18-18-07543.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jahromi SS, Wentlandt K, Piran S, et al. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol. 2002;88(4):1893–1902. doi: 10.1152/jn.2002.88.4.1893. [DOI] [PubMed] [Google Scholar]
- 84.Valiunas V. Biophysical properties of connexin-45 gap junction hemichannels studied in vertebrate cells. J Gen Physiol. 2002;119(2):147–164. doi: 10.1085/jgp.119.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hochman DW, Baraban SC, Owens JW, et al. Dissociation of synchronization and excitability in furosemide blockade of epileptiform activity. Science. 1995;270(5233):99–102. doi: 10.1126/science.270.5233.99. [DOI] [PubMed] [Google Scholar]
- 86.Hochman DW, Schwartzkroin PA. Chloride-cotransport blockade desynchronizes neuronal discharge in the “epileptic” hippocampal slice. J Neurophysiol. 2000;83(1):406–417. doi: 10.1152/jn.2000.83.1.406. [DOI] [PubMed] [Google Scholar]
- 87.Dulla CG, Dobelis P, Pearson T, et al. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron. 2005;48(6):1011–1023. doi: 10.1016/j.neuron.2005.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stringer JL, Xu K. Possible mechanisms for the anticonvulsant activity of fructose-1,6-diphosphate. Epilepsia. 2008;49(Suppl 8):101–103. doi: 10.1111/j.1528-1167.2008.01849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abe K, Nakanishi K, Saito H. The possible role of endogenous glutathione as an anticonvulsant in mice. Brain Res. 2000;854(1-2):235–238. doi: 10.1016/s0006-8993(99)02269-6. [DOI] [PubMed] [Google Scholar]
- 90.Vexler ZS, Wong A, Francisco C, et al. Fructose- 1,6-bisphosphate preserves intracellular glutathione and protects cortical neurons against oxidative stress. Brain Res. 2003;960(1-2):90–98. doi: 10.1016/s0006-8993(02)03777-0. [DOI] [PubMed] [Google Scholar]