

Abstract
Immunological tolerance theory in chronic lymphocytic leukaemia (CLL): we suggest that B cells that express B-cell receptors (BCR) that recognize their own BCR epitopes are viewed by immune system as ‘dangerous cells’. BCR autonomous signalling may induce constant receptor editing and mistakes in allelic exclusion. The fact that whole BCR recognizes a self-antigen or foreing antigen may be irrelevant in early B cell development. In early B cells, autonomous signalling induced by recognition of the BCR’s own epitopes simulates an antigen-antibody engagement. In the bone marrow this interaction is viewed as recognition of self-molecules and induces receptor editing. In mature B cells autonomous signalling by the BCR may promote ‘reversible anergy’ and also may correct self-reactivity induced by the somatic hypermutation mechanisms in mutated CLL B cells. However, in unmutated CLL B cells, BCR autonomous signalling in addition to self-antigen recognition augments B cell activation, proliferation and genomic instability. We suggest that CLL originates from a coordinated normal immunologic tolerance mechanism to destroy self-reactive B cells. Additional genetic damage induced by tolerance mechanisms may immortalize self-reactive B cells and transform them into a leukemia.
Keywords: anergy, chronic lymphocytic leukaemia, clonal selection theory, immunological tolerance, receptor editing
Introduction
Much evidence has accumulated supporting the idea that neoplasms, including those of a lymphoproliferative nature, are influenced by their environment.1 This evidence has come from the study of a range of conditions such as inflammation, bacterial and viral infections, endemic diseases and autoantigen stimulation of the immune system.1 Immunological tolerance mechanisms, in a way similar to other stimuli, also induce the persistence of B-cell receptor changes that induce genetic instability and finally result in molecular aberrations that promote the development of a neoplasm.2–8
Here, we review tolerance mechanisms to prevent self-reactivity in chronic lymphocytic leukaemia (CLL) B lymphocytes and propose a hypothesis in which tolerance mechanisms play a key role in CLL development.
The objective in writing this review was to present a hypothesis about the generation of CLL in terms of the clonal selection approach. Using the same method as Burnet,1 we try to express CLL development as a logical and linear development of the forbidden clone concept formed within the framework of clonal selection theory.
B-cell development occurs initially in the bone marrow
Stem cells differentiate into pro-B lymphocytes. These pro-B lymphocytes undergo a rearrangement of their immunoglobulin heavy chain genes and are known as pre-B lymphocytes. Subsequent rearrangement of the light chain enables the cells to express surface IgM and they becomes immature transitional B lymphocytes. When these lymphocytes leave the bone marrow they are called naive B lymphocytes. During this process several tolerance mechanisms control the survival of self-reactive B-lymphocytes.
It is interesting to note that the B-lymphocyte differentiation from pro-B to pre-B to early B to mature B lymphocyte is marked by the expression of the B-cell receptor (BCR) on the cell surface and by constant ‘education’ to avoid autoimmunity. Despite the fact that unmutated CLL (U-CLL) and mutated CLL (M-CLL) lymphocytes receive the same strategies (receptor editing and anergy) to correct their self-reactive BCR, it appears that the U-CLL subset is resistant to immunological tolerance, but the M-CLL subset is not.9–11 Finally, germinal centre exclusion12–15 may be the censor checkpoint which divides CLL cells into M-CLL and U-CLL (germinal centre excluded B cells).
The essence of the ‘forbidden clone’ concept
On differentiation, a stem cell becomes a lymphocyte carrying a specific pattern of BCR. If accessible, antigenic determinants are present soon after differentiation. Any normal lymphocyte capable of reacting with them will be eliminated. If, however, the stem line has at some point undergone a somatic mutation increasing resistance, the differentiated lymphocyte can, on specific stimulation, initiate a forbidden clone.5
Hypothesis
CLL B lymphocytes fulfil the immunological tolerance rules proposed by Burnet and can be classified as ‘malignant’ forbidden clones, which suggests that CLL B lymphocytes may be generated by immunological tolerance mechanisms.
CLL B lymphocytes qualify as a malignant forbidden clone
The cause of CLL is unknown. Several cellular origins of CLL have been proposed, supporting the idea that CLL may be generated at different levels of maturation and activation. In particular, marginal zone B cells,16 human B1 cells,17 CD5+ B cells,18 regulatory B cells19,20 and memory B cells21 have been suggested as the cellular origin of CLL. Despite extensive searching it is not yet known whether there is an equivalent normal cell from which the CLL lymphocyte arises,22–26 and it is not certain at what stage of lymphocyte maturation the cell arises, because roughly equal numbers seem to come from pre- and post-germinal centre B lymphocytes.27 However, BCR of CLL lymphocytes demonstrate highly selected immunoglobulin heavy chain variable (IGHV) gene usage, or even very similar entire antigen-binding sites, coded by both heavy-chain28–35 and light-chain genes.36,37 These findings argue in favour of a limited set of autoantigens promoting the division of precursor cells and clonal evolution.38–42 Immunoglobulin genes are rearranged with 40–50% of cases unmutated (> 98% homology with germline) and 50–60% showing somatic hypermutation (SHM). IGHV gene usage in CLL is highly selective and often associated with autoantibody reactivity.43–45 Importantly, U-CLL and M-CLL lymphocytes derive from self-reactive B lymphocytes, despite expressing different antibody reactivity.9
Burnet's rules about immunological tolerance and their application in CLL B-cell development
Taking advantage of the comparison of CLL development data with a linear normal B-lymphocyte differentiation we can observe that at all stages of B-cell development there is evidence that CLL cells may be induced or promoted by the persistent action of a tolerance mechanism.
Burnet's immunological tolerance rules about generation of self BCR and autoantigens indicate that:
Since an immune pattern is generated by a random process, a mechanism must exist whereby the ‘self-reactive’ cells that may emerge can be eliminated or functionally inhibited.5 More than one mechanism may be needed to establish and maintain this intrinsic immunological tolerance toward self components.5,46–58
The accessibility, amount and physiological character of the [autoantigen] plays an essential part in determining both to what extent a potentially pathogenic clone is stimulated to proliferate and what opportunity it has to damage cells and tissues in the body.5
These rules are reflected in CLL development because on differentiation the CLL stem cell becomes a lymphocyte carrying a specific pattern of BCR.59 CLL haemopoietic stem cells generate aberrantly increased amounts of pro-B lymphocytes that have an intrinsic propensity to generate clonal CLL-like B lymphocytes with normal karyotypes.59 In pro-B lymphocytes, DH-JH joining precedes VH-DJH rearrangement, followed by VL-JL joining in the late-stage pre-B lymphocytes. Pro-B lymphocytes derived from patients with CLL are also altered because they continue to rearrange heavy chains and some of them fail to induce allelic exclusion.60 Intriguingly, CLL B lymphocytes express BCR that recognize their own BCR and also have an autonomous signalling capacity,61 in a way similar to the pre-B-cell receptor expressed in pre-B lymphocytes.62,63 These BCR are continuously accessible to antigenic determinants present in their own BCR.61 This persistent stimulus induces an early positive selection and promotes their differentiation.62 We suggest that this early B-cell stage BCR (whether self-reactive or not) can be viewed as ‘dangerous BCR’ because CLL B cells try to delete or correct it. These new B lymphocytes, capable of reacting with their own BCR (internal self antigen), will be eliminated or silenced.63,64 However, this forbidden B lymphocyte is resistant to tolerance mechanisms such as receptor editing because its heavy and light chains,64 frequently used by its BCR, are similar to heavy chains that cannot be silenced by receptor editing.31,36,37,50 For this reason, CLL B lymphocytes are in constant receptor editing65 and some of them have a lack of allelic exclusion.60 This suggests that CLL B lymphocytes retain the BCR autonomous signalling capacity acquired during the pre-B-cell development in both U-CLL and M-CLL.61 This is also supported by the fact that CLL B lymphocytes recognize galectin-1, a ligand of pre-B BCR in pre-B cells.66,67 CLL B lymphocytes also fail to inhibit the autonomous signalling capacity of the BCR induced by the production of conventional light chain.61,64
Burnet's immunological tolerance rule about ‘points of decision’ such as anergy indicates that:
There are many genetically determined anomalies in the functioning of the immune system. It is reasonable to suggest that it functions by shifting the ‘point of decision’, determining whether a lymphocyte reacting with its corresponding determinant will be stimulated to proliferation and functional activity or will be destroyed or functionally inhibited.5
Chronic lymphocytic leukaemia was considered to be a tumour of circulating B lymphocytes, variably stimulated and anergized following exposure to antigen in lymphoid tissue.68 This is in concordance with our hypothesis that CLL B lymphocytes are persistently under check by a tolerance mechanism because they are chronically exposed to self antigens (or their own BCR).
The low expression of BCR is the hallmark of CLL and anergic B lymphocytes. Despite their initial resistance to receptor editing, these CLL B lymphocytes are forced to engage with the environment as a test of self-reactivity (negative selection),67,69 becoming ‘edited, anergized CLL B lymphocytes’.10,70,71 However, there is a difference between BCR signalling in U-CLL and M-CLL.10 The molecular signature of anergy has been detected in both U-CLL and M-CLL.10,70,71 In Fig. 1 we provide a schematic representation that summarizes several central tolerance mechanisms involved in the development of CLL B cells and monoclonal B-cell lymphocytosis.
Figure 1.

Tolerance mechanisms are involved in chronic lymphocytic leukaemia (CLL). Stem cells from patients with CLL produce an increased amount of pro-B cells. These pro-B cells undergo tolerance in bone marrow. Pro-B lymphocytes derived from patients with CLL continue to rearrange heavy chains and some of them fail to induce allelic exclusion. Some pro-B cells mature into pre-B cells that express B-cell receptors (BCR) that recognize their own BCR and also have an autonomous signalling capacity. These BCR (self-reactive or not) with autonomous signalling capacity induce constant receptor editing, a mechanism involved in inducing immunological tolerance. Despite active receptor editing, B cells that express BCR with autonomous signalling capacity are viewed by the immune system as ‘dangerous cells’ and suffer different degrees of ‘reversible anergy’. These mechanisms are common in monoclonal B-cell lymphocytosis, unmutated CLL (U-CLL) B cells and mutated CLL (M-CLL) B cells. However, M-CLL B cells may be expressing a profound level of anergy compared with U-CLL B cells. Importantly, M-CLL can correct their initial self-reactive BCR during central tolerance mechanisms in bone marrow. U-CLL B cells remain self-reactive B cells.
Burnet's immunological tolerance rules about ‘resistance to elimination’ indicate that:
Changes due to somatic mutation or to physiological factors may render a newly differentiated lymphocyte (the antigen-reactive cell) non-susceptible to elimination or inhibition by the ‘censorship’ mechanisms concerned. As in virtually every statement one can make in an immunological context, the word ‘non-susceptible’ is not an absolute but simply refers to a range of susceptibility below the level, determined by genetic factors, of the normal newly differentiated lymphocyte.5
When, through somatic mutation, a lymphocyte line develops that has an undue resistance to elimination by antigenic contact, and reacts specifically with an accessible body component functioning as antigenic determinant, it is potentially capable of initiating a forbidden clone of directly or indirectly pathogenic cells. As such, they will have all the essential characteristics of a conditioned malignancy.5
Self-reactive B lymphocytes decrease from 75% in immature B lymphocytes in the bone marrow to 20% in the mature naive B-lymphocyte population in healthy individuals.47 Lymphocytes require very low affinity binding to self antigens to remain viable in the periphery. Interestingly, aberrations in PTPN22 over-expression in CLL represent a protective mechanism that allows autoantigen-activated CLL cells to escape from negative selection and tolerance mechanisms.72 Importantly, decreased BCR signalling is sufficient to alter the removal of developing self-reactive B cells,73,56 on the one hand, and PTPN22 alterations might induce a failure in central tolerance in healthy donors,73 on the other. One of the most intriguing phenomena related to immunological tolerance is that sometimes a mechanism created to protect against self-reactivity leads to damage and promotes malignancy (‘horror autotoxicus’).3,6–8
Several lines of evidence suggest that normal cells that become CLL lymphocytes progress through various stages before becoming overtly leukaemic. Healthy individuals may show monoclonal or oligoclonal B-cell expansions with the characteristic phenotype of CLL in about 3·5% of tested subjects > 40 years old, and the estimated rate of progression of monoclonal B-cell lymphocytosis to CLL requiring treatment is 1·1% per year.74–76 This implies that B CLL lymphocytes (forbidden clone) undergo at some point somatic mutations or molecular aberrations as microRNA disturbances,11,77,78 mutations in MyD88,79 NOTCH-180–82 and SF3B183,84 or trisomy 12,85,86 13q deletion,87 11q deletion,87 and 17p deletion,87 increasing their resistance to being eliminated. Interestingly, karyotype evolution has been reported during the long-term follow up of patients with untreated early-stage CLL.76,88 We suggest that genetic programmes are progressively disrupted during B-cell development and tolerance checkpoints. Combinations of disrupted genetic programmes originate different ways to make a CLL-like clone. These new ‘edited anergized’ B CLL lymphocytes continue with their differentiation and can, on specific stimulation, initiate a process of forbidden clone proliferation.
Burnet's immunological tolerance rule about ‘autoantibodies’ indicates that:
It is necessary for survival that neither lymphocytes nor antibodies which are reactive to more than a minimal degree with any accessible body component should exist in the body.5
Within germinal centres, naive B lymphocytes undergo activation, proliferation, SHM, isotype switching and subsequent positive and negative selection by antigen. Somatic mutations are introduced at a high rate in the germinal centres, and are typically involved in single nucleotide exchanges.
An important peripheral checkpoint is entry into the T-lymphocyte-dependent, long-term memory compartment. Lymphocytes that express antibodies encoded by the VH4-34 gene are self-reactive B cells.12,13 B lymphocytes that express self-reactive BCR encoded by the IGHV4-34 are excluded from T-lymphocyte-dependent IgG memory and plasma cell populations, suggesting that these autoreactive lymphocytes fail to cross a developmental checkpoint following activation in normal individuals.12,13 This implies that negative selection of autoreactive cells occurs at the transition of naive to germinal centre cells. The comparison of the methylation patterns between B-cell populations and CLL B cells leads to the conclusion that malignant U-CLL cells are related to pre-germinal (germinal-centre-excluded?) B cells, whereas M-CLL lymphocytes probably stem from germinal-centre-experienced B cells 24 (memory B cells?).
Remarkably, 79·3% of U-CLL antibodies are polyreactive and some of them recognize apoptotic cells, IgG, thyroglobulin, DNA, actin, cardiolipin and other nuclear antigens.9,38–41 These features of U-CLL are in concordance with the negative selection at the transition from naive to germinal centre cells and suggest that U-CLL lymphocytes are excluded from germinal centres. In Fig. 2 we provide a schematic overview of the genesis of the U-CLL B-cell subset and M-CLL subset.
Figure 2.
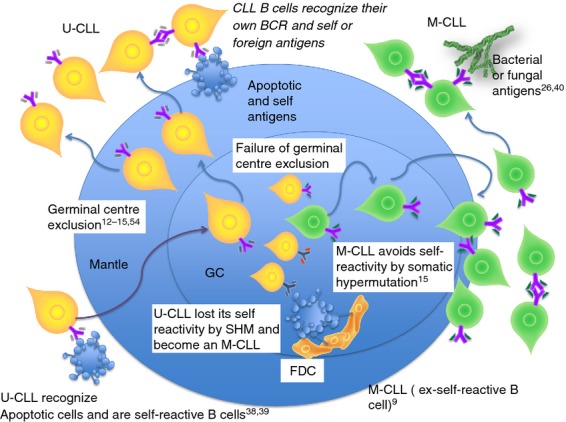
Tolerance mechanisms in the germinal centre are involved in chronic lymphocytic leukaemia (CLL). Germinal centre exclusion of self-reactive B cells maintains the unmutated CLL (U-CLL) subset without somatic mutations in their heavy and light chains. However, if a failure of germinal centre exclusion occurs, somatic hypermutation may correct the initial self-reactive B-cell receptor (BCR). Newly generated ex-self-mutated CLL may recognize their own BCR to avoid apoptosis and/or recognize new antigens (self or foreign).
Mutated CLL lymphocytes appear to have undergone the normal process of SHM and antigen selection in the germinal centres before transformation and might be considered ex-self-reactive B cells.9 Although receptor editing65–67 and anergy10,70,71 may play a role in the tolerance of M-CLL cases, the current hypothesis is that M-CLL avoids self reactivity by SHM.78 This implies a failure of germinal centre exclusion of self-reactive B cells,12–15 or the generation of new self-reactive B cells during SHM.52,53 In the first scenario a self-reactive B cell (U-CLL) loses its self-reactivity when passing through the germinal centre reaction by SHM.89 This is important because point mutations can both correct self-reactive BCR and abolish autonomous signalling of CLL BCR. In the second scenario, new self-reactive B cells could avoid self recognition by anergy and heavy-chain or light-chain editing. Both contexts result in an ex-self-reactive M-CLL cell. Figure 3 summarizes the different processes of M-CLL subset genesis taking into consideration the origin of B cells (non-self B cells, ex-self B cells or self B cells that produce natural autoantibodies).
Figure 3.

Immunological consequences of developing B-cell receptors (BCR) that recognize their own BCR epitopes. Non-self-reactive B cells, ex-self-reactive B cells and B cells that encode natural autoantibodies may also be an origin of mutated chronic lymphocytic leukaemia (M-CLL) cells if these cells previously acquire BCRs that recognize their own BCR in the bone marrow. When they enter a normal immune response they may acquire self reactivity during the somatic hypermutation process. However, persistent receptor editing and anergy induced by their ‘special BCR’ with autonomous signalling capacity may correct their recently acquired self-reactive BCR during somatic hypermutation within the germinal centre.
Tolerance mechanisms involved in the development of memory B cells
B cells expressing self-reactive antibodies and antibodies with broad specificity for bacteria are removed from the repertoire in the transition from naive to IgM+ memory B cells.15 However, rare IgM+ memory B cells that produce antibodies with low levels of self reactivity acquire this reactivity by SHM.15 In a similar way, autoreactivity in human IgG+ memory B cells is mostly created by SHM.54 The fact that CLL B cells undergo isotype switching with and without acquiring somatic mutations suggests that germinal centre chekpoints may be disrupted in CLL patients.30–35,40 Some BCR of M-CLL B cells could bind carbohydrate determinants of bacterial capsules, viral coats and certain autoantigens.25,32–34,42,54 These facts indirectly support a failure in tolerance mechanisms within germinal centre checkpoints.32–34,40,42,54
M-CLL B lymphocytes might also be generated during a normal immune response against some pathogens without any stigma of self reactivity. Recently, a new subset of M-CLL lymphocytes expressing BCR highly specific for a major antigenic determinant of yeasts and filamentous fungi has been reported.26 Importantly, this subset represents > 0·3% of CLL and is not associated with germline self reactivity.26
Burnet's immunological tolerance rule about ‘self-reactive T cells and autoantibodies’ indicates that:
In all probability, the clinical manifestations of autoimmune disease are due to aggressive T lymphocytes producing damage to target cells; to the deposition of antigen–antibody complexes elsewhere; and to a directly damaging action of an antibody, but probably only when the autoantigen is present in the circulating blood either in cells or as a soluble component.5
Indeed, although the BCR of CLL B cells may be non-self-reactive, BCR of CLL B lymphocytes may be viewed for the immune system as a self antigen in an early developmental stage because these ‘autonomous signalling BCR’ recognize their own BCR internal epitopes.61 It is improbable that an immune attack against this epitope would induce an autoimmune disease.5 In theory, however, an attack against their self BCR may increase their own BCR stimulation; induce chronic activation of mechanisms of tolerance as receptor editing; and promote BCR surface elimination with the consequent low surface IgM detected in CLL B lymphocytes.
The immunodeficiency of CLL
All patients with CLL have a degree of immunodeficiency,90,91 which may be a direct consequence of leukaemic cell action. The CLL lymphocytes themselves can induce profound immunosuppression by direct contact or through cytokines such as transforming growth factor-β and interleukin-10 (IL-10).92,20 Direct contact with CLL lymphocytes can induce synapse inhibition in previously healthy T lymphocytes,93,94 and probably may also promote the induction of regulatory T cells.95–97 Remarkably, CLL B lymphocytes may kill plasma cells during cell–cell contact.98 Over-expression of CAV1 in CLL B lymphocytes from lymph nodes increases the capacity to interact with T lymphocytes and promotes an immunosuppressive environment.99 Once T-cell functions are blocked or disturbed100,101 the rest of the immune system also fails.102 Decreased immunoglobulin synthesis develops in about 85% of patients and > 50% suffer recurrent infections. Surprisingly, the innate immune system also has defects in natural killer cells,103 phagocytic cells104 and complement system.105 Finally, low levels of immunoglobulin and complement may decrease the clearance of self antigens (apoptotic blebs), with the subsequently increased risk of autoimmunity and CLL disease progression.2,106,107
The role of transcription factors in CLL development
B-cell ontogeny-determining transcription factors may also support the hypothesis about several cellular origins of CLL B cells. As mentioned before, B-cell differentiation is typically viewed as a linear process as defined by regulated expression of specific sets of transcription factors and BCR expression.
PAX5
Haemopoietic stem cells (HSC) are capable of developing into all the blood cell types. B cells are continually generated from HSC in the bone marrow. Initial commitment to the B-cell lineage requires activation of a series of transcriptional factors. At the nuclear level, the transcription factors PU.1, Ikaros, E2A, early B-cell factor (EBF) and PAX-5 play major roles in committing progenitor cells to the B-cell lineage.108 However, after lineage commitment has been established, it is the composition of the BCR that controls further development, as mentioned above. Throughout B-cell development, precursor B cells produce CD19+ pro-B cells that are irreversibly committed to becoming B cells, due to expression of PAX-5. PAX-5 is a paired-box transcription factor which, among the progeny of HSC, is expressed exclusively in cells of the B-cell lineage. CD79A and PAX-5 appear at the time of heavy chain gene rearrangement. Importantly, PAX-5 is expressed at higher levels in U-CLL B cells, when it is compared with normal B cells and M-CLL B cells.109 Interestingly, PAX-5 mRNA decreases in plasma B cells, suggesting that the lower level of PAX-5 in M-CLL cells may be a sign of developmental block before M-CLL cells become plasma B cells.
Helix-loop-helix transcription factors E2A and EBF
E2A codes two transcription factors E12 and E47, members of the basic helix-loop-helix family, and its induction is crucial from the earliest stages of B-cell lineage development. E12 is a better activator of EBF and PAX-5 and E47 play a greater role in driving TdT and RAG, implicating this transcription factor in the process of chromatin remodelling of the immunoglobulin heavy chain locus that permits accessibility by the recombinase machinery.108,110 Interestingly, in CLL B cells E2A is elevated at the mRNA and protein levels compared with normal B-cell subsets.111 This finding is consistent with the circumstance that CLL B cells have constant BCR rearrangement and (auto)antigen-driven receptor editing65 to avoid self reactivity or autonomous BCR signalling.61 Moreover, receptor editing in marginal zone B-cell development is regulated by E2A.112 E2A proteins are required to regulate secondary gene rearrangement in B cells that express an autoreactive BCR, due in part to activated RAG expression and also because E2A proteins are required to promote developmental progression of autoreactive B cells.112 Moreover, this circumstance may explain the increased ability to activate secondary immunoglobulin light chain gene rearrangement65 in CLL. However, this increased receptor editing is not enough to induce IgL gene rearrangement that will permit surface immunoglobulin expression without autonomous signalling.61 Importantly, E2A binds to a large subset of genes involved in pre-BCR signalling,113 supporting the notion that links autonomous BCR signalling61 in CLL B cells with normal pre-B cells.62–64
Early B-cell factor-1 (EBF1) is a helix-loop-helix like transcription factor. EBF1 is expressed at all stages of differentiation except in plasma cells and may be critical in the progression of B cells past the early pro-B cell stage. Significantly low levels of EBF1 in both M-CLL and U-CLL cells compared with CD5+ or conventional B cells have been reported.18 EBF1 is also a repressor of the transcription factor TCF7.113 TCF7 is known to be a T-cell-specific transcription factor required for T-cell development. Importantly, high expression of TCF7 is associated with prolonged survival times and M-CLL.114 Importantly, the low expression of EBF1 may lead to reduced levels of numerous B-cell signalling factors, contributing to an anergy signature of CLL cases.18 Taken together it is possible that the low levels of EBF1 and high expression of TCF7 contribute to the diminished response of BCR to stimulus and are associated with prolonged survival of patients with M-CLL.
Transcription factors control pro-B and pre-B cell development
PU.1 is an ETS family loop-helix-loop (winged helix) transcription factor. PU.1 regulates a number of B-cell-specific genes including CD79a, joining chain, µ chain, κ chain, λ chain, RAG and TdT.115,116 Significantly, PU.1 is poorly expressed in CLL B cells compared with CD5+ B cells from normal blood, control tonsils and non-CLL B-cell lymphomas.117 Interestingly, in pro-B cells specifically deleted for PU.1 these genes continue to be expressed.118,119 We suggest that low levels of PU.1 may contribute to the increased BCR rearrangements65 and lack of allelic exclusion60 detected in CLL B cells. PU.1 directly activates the expression of EBF1.120 Importantly, low levels of EBF1 detected in CLL B cells are in concordance with the low levels of PU.1 observed in CLL B cells. Such low levels of both PU.1 and EBF1 in B cells do not compromise their ability to establish an immune response upon BCR stimulation.119 The recent finding that PU.1 is a potent tumour suppressor in classical Hodgkin's lymphoma cells121 suggests that low expression of PU.1 in CLL B cells might also play a role in the progression of disease.
LEF-1 is specifically expressed in pro-B and pre-B cells.122,123 Surprisingly, monoclonal B-cell lymphocytosis and both M-CLL and U-CLL B cells express LEF-1.124,125 Interestingly, pro-B, pre-B and CLL B cells share the expression of LEF-1 as a pro-survival factor. We speculate that some CLL B cells derive from pro-B cells taking into consideration that some CLL B cells lack allelic exclusion60 and express LEF-1.124,125 In the same line of thinking some CLL B cells derive from pre-B cells because they share with pre-B cells the expression of LEF-1124,125 and an autonomous BCR signalling similar to pre-BCR.62,64
ID-1 and ID-2 have a helix-loop-helix domain, but lack a DNA binding domain. Hence, they can function as a dominant negative factor, inhibiting the function of helix-loop-helix transcription factors such E2A. ID-1 is expressed only in pro-B cells. However, pre-B cells in adults, showed highly up-regulated expression of ID-2, in the absence of changes in expression of E2A126 in bone marrow. Over-expression of ID-2 in follicular lymphoma cells may regulate proliferation or contribute to the maturation arrest in these tumour cells.127 In M-CLL B cells ID-2 expression is high but may be down-regulated after stimulation in a similar manner to normal B cells.109 However, in U-CLL B cells, high expression of PAX-5 and lower levels of ID2109 are associated with constitutive expression of activation-induced cytidine deaminase (AID). Importantly, U-CLL B cells are frequently self-reactive and more prone to acquire more genetic lesions than M-CLL B cells. Interestingly, AID is required for the establishment of both central and peripheral B-cell tolerance.128,129 Taken together it is possible that low expression of ID-2 contributes to constitutive expression of AID130,131 and reflects the self-reactive nature of U-CLL B cells.
FOXP1 is a forkhead transcription factor involved in the control of V/D/J recombination of the genes encoding immunoglobulin heavy chain.
FOXP1 deficiency is associated with a block in the transition from pro-B to pre-B cells.132 FOXP1 also regulates recombination-activating genes (RAG1 and RAG2) in B cells and in cancer cells.133 In human B cells, FOXP1 shows the opposite expression pattern to BCL-6, suggesting that FOXP1 regulates the transition from resting follicular B cell to activated germinal centre B cell.134 Intriguingly, FOXP1 is down-regulated in some autoreactive human naive B cells refractory to antigenic stimulation (anergic B cells).58 However, FOXP1 expression is higher in some high-risk U-CLL B cells, especially in those which also have SF3B1 mutations83 and deletions in chromosome 11q.84 We speculate that this deregulation in some transcription factors such as FOXP1 may induce some resistance to U-CLL B cells to becoming anergic B cells. High expression of FOXP1 may also explain germinal centre exclusion and the lack of SHM in U-CLL B cells.58,134
The chromatin regulator Aiolos and the transcriptional co-activator OBF-1 have been implicated in the control of genes involved in pre-BCR function.135 They also play a key role in the transition from pre-B to immature B cells.136 OBF-1 has comparable levels of mRNA and protein in CLL B cells and normal blood CD5+ B cells.117 However, Aiolos is over-expressed in CLL B cells in comparison with healthy donors. Remarkably, Aiolos is also involved in cell survival.137 In addition, OCT-2 and OBF1 transcription factors are expressed in CLL B cells at comparable levels to CD5+ B cells.117 SPI-B is also expressed in both CLL B cells and normal B cells.138
Transcription factors associated with marginal zone, follicular B cells and human B1 B cells
NOTCH-2 is preferentially expressed in mature B cells and supports marginal zone B-cell development and type 2 transitional B cells.139
Although the precursor of CLL remains elusive, marginal zone B cells have been suggested to have one normal counterpart,16 because they display an activated B-cell phenotype, and express either mutated or unmutated IGHV genes that can be biased in usage and produce poly/autoreactive antibodies. Interestingly, the transcription factor NOTCH-2 is constitutively expressed in both U-CLL and M-CLL B cells but not in normal B cells.80 The expression of NOTCH-2 in CLL is in agreement with marginal zone B cells as CLL B-cell precursors.
Nonetheless, marginal zone B cells may in several cases be the precursors of CLL B cells; some CLL also may derive from follicular B cells. c-Myb is critical for B-cell development and maintenance of follicular B cells 140 and is expressed also in normal pre-B cells. Interestingly, only a few CLL B cells express the c-Myb transcription factor.141 This finding argues in favour of multiple origins for CLL B-cell development theory. The expression pattern of Myb in B cells inversely correlates with that of miR-150.142 Interestingly, miR-150 is highly expressed in the majority of CLL tumour cells, but not in the proliferation centres.143 M-CLL (clinically indolent) has significantly higher levels of miR-150 than U-CLL (clinically aggressive).11,77 The mantle zone is rich in VH4-34 self-reactive B cells excluded from germinal centres.3,12,13 Remarkably, in the mantle zone, the expression of miR-150 is high and of c-Myb is weak144 in a similar way to stereotypical subset 4 (IGHV4-34/IGKV2-30 mutated, good prognosis).11 However, subset 1 (IGHV1/5/7-IGKV1(D)39, unmutated, bad prognosis) down-regulates miR-150 in a similar manner to germinal centres and proliferation centres. Notably, miR-150 null mice possess a higher proportion of B1 cells and show a significant increase in the production of ‘natural’ antibodies.142 This is in keeping with the lower levels of miR-150, increased expression of ZAP-70, increased proliferative response to BCR stimulation and the suggestion that human B1 B cells or marginal zone B cells16,17 may be a possible origin of the U-CLL subsets.
Transcription factors control receptor editing, germinal centre formation and exclusion, class switch recombination and SHM
The transcription factor IRF4 regulates immunoglobulin class switch recombination and plasma cell differentiation. Transient expression of IRF4 induces the expression of germinal centre genes including BCL-6 and AID.145 However, sustained and higher concentration of IRF4 promotes the generation of plasma cells.145 IRF4 mutations in CLL induce higher IRF4 mRNA expression.146 Surprisingly, patients with IRF4 mutations have a paradoxically good prognosis because they express high risk factors such as U-CLL B cells and trisomy 12.146 We speculate that sustained IRF4 in self-reactive B cells may induce germinal centre exclusion and block the differentiation to plasma cells in U-CLL B cells. Importantly, increased IRF4 transcripts are associated with increased receptor editing in self-reactive pre-B cells.55 Along the same lines, expression of MUM1/IRF4 correlates with M-CLL, post-germinal centre origin and a more favourable clinical course.147 This suggests that only ex-self-reactive or re-educated M-CLL B cells may form germinal centres and suffer SHM.2,13,15,54
The differentiation of mature B cells into antibody-secreting plasma cells and memory B cells is controlled by the presence or absence of transcription factors, including BLIMP-1, BCL-6 and IRF-4.148,149
Chronic lymphocytic leukaemia cells are fixed in a transition stage between immature and mature resting naive B cells, which continue to express PAX-5 and do not up-regulate BLIMP-1.138 BCL-6 is a central transcription factor that promotes the germinal centre phenotype, and while doing so, prevents plasmablast differentiation. However, in CLL B cells BCL-6 is expressed at very low levels compared with germinal centre B cells although CLL B cells have higher BCL-6 levels than memory B cells.150 Taken together, this suggests that some tolerance mechanism prevents the transition of self-reactive B cells into autoantibody-producing plasma B cells.
The increased expression of PAX-5 in U-CLL130 supports the notion that U-CLL cells are immature self-reactive B cells excluded from germinal centres. In M-CLL a low level of PAX-5130 may represent the induction of the plasma cell/memory B-cell transcription programme, which leads to the down-regulation of BCL-6, but this process is not complete. We speculate that interruption of this transition from activated-IgH mutated B cells to plasma B cells represents a mechanism of control for potentially dangerous autoantibody-secreting B cells or the transition of an ex-self-reactive B cell to a ‘fixed’ memory B cell. Importantly, BCL-6 mutations are associated with M-CLL.
Chromosomal abnormalities in M-CLL B cells and their relationship with tolerance
Deletion at 13q
The commonest abnormalities include del 13q in > 50% of patients and are usually associated with good prognosis.87 Deletion of 13q14.3 contains two micro-RNAs, miRs15a and 16-1.78 Interestingly the miR15/16 cluster controls the production of BCL-2, an anti-apoptotic factor produced at high levels in CLL B cells. This suggests that miRNAs can act as tumour suppressors. Deletions at 13q often have leukaemic CLL clones with mutated IGHV genes.
MyD88
Whole genome sequencing demonstrated that recurrent mutations in some genes are more frequent in patients with M-CLL. Two types of mutations in MyD88 have been described in CLL; an activating mutation (MyD88 L265P) and an inactivating mutation (MyD88 E52DEL).88
Interestingly, IRAK.4/MyD88 complexes play a major role in the establishment of central B-cell tolerance in the bone marrow by counterselecting developing autoreactive B cells.57 Defects in receptor editing contribute to persistence of self-reactive B cells in patients with MyD88 deficiency.57 We speculate that patients with CLL clones with an inactivating mutation (MyD88 E52DEL) have self-reactive B cells that are inactivated by anergy or SHM and not by receptor editing. This is in accordance with the fact that M-CLL are ex-self-reactive B cells. However, in M-CLL with the activating mutation (MyD88 L265P), receptor editing may be increased with the subsequent abolition of self reactivity.
Another important consequence in M-CLL with activating mutation (MyD88 L265P) is the function of Toll-like receptors (TLR). Most self-reactive B cells are normally counterselected during early B-cell development and TLR7, TLR8 and TLR9 may prevent the recruitment of developing self-reactive B cells in healthy individuals. TLR9 promotes tolerance by restricting the survival of anergic B cells with specificity for DNA.151 Activating MyD88 mutation enhances the response of TLR to their ligands.88 Even more, IL-10 production by B cells stimulated by contact with apoptotic cells results from the engagement with TLR9.152 Interestingly human circulating CD27+ B cells respond to DNA-bearing apoptotic cells secreting IL-10.84
These facts suggest a role for MyD88 mutations in the development of M-CLL and their similarities with IL-10 regulatory B cells. Interestingly, TLR9 stimulation can also induce apoptosis in M-CLL and proliferation in U-CLL.153 This may be explained by the fact that TLR9 stimulation after BCR triggering can also increase BCR-initiated activation. The synergic stimulation of TLR9 and BCR might be present only in U-CLL because M-CLL cases involve mainly anergic B cells. It is possible that signalling through mitogen-activated protein kinase and nuclear factor-κB, which are downstream of both TLR9 and the BCR, promote proliferation in U-CLL B cells; however, in M-CLL B cells promote apoptosis and IL-10 secretion.
Importantly, TLR9 stimulation induces CD86 expression preferentially in M-CLL.35 The interaction of CD86 with CD28 provides important co-stimulatory signals for T cells activated through CD3/T-cell receptor. Furthermore, stimulation of TLR9 in concert with the secretion of various chemokines and IL-10 from M-CLL cells contribute to the impaired synapses, T-cell exhaustion and generation of an increased amount of regulatory T cells in patients with CLL. In Fig. 4 we provide a schematic model of M-CLL subset genesis and the role of TLR, MyD88 mutations, del13q, microRNAs and immunological tolerance.
Figure 4.

Mutated chronic lymphocytic leukaemia (M-CLL) lymphocytes have molecular and genetic disturbances that promote survival and tolerance. Deletion of 13q14.3 contains two micro-RNAs, miRs15a and 16-1, involved in the control and the production of BCL-2, an anti-apoptotic factor produced at high levels in CLL B cells. Activating MyD88 mutation enhances the response of Toll-like receptor (TLR) to their ligands. Immunosuppressive cytokines such as interleukin-10 (IL-10) may be induced in M-CLL B cells by the engagement of TLR9 with apoptotic cells. TLR9 stimulation induces CD86 expression in M-CLL. The interaction of CD86 with CD28 provides co-stimulatory signals for T cells activated through the T-cell receptor. These interactions may promote survival of M-CLL B cells but also promote tolerance and B-cell maturation arrest. Interactions of B-cell receptors (BCR) that recognize their own BCR epitopes may promote survival and prevent apoptosis.
Chromosomal abnormalities in U-CLL B cells and their relationship with tolerance
Trisomy 12 and NOTCH-1 mutations
Trisomy 12 is found in about 15% of CLL patients. Interestingly, the NOTCH-1 mutations are associated with trisomy 12 and U-CLL cases.80–82,88 These mutations in NOTCH-1 result in the accumulation of an active protein isoform in CLL B cells, and these patients appear to have a worse clinical course.
NOTCH-1 is expressed throughout normal B-cell development and in leukaemic B-cell lineage cells and may regulate human B-cell development. The NOTCH ligand Delta is expressed in the bone marrow B lineage cells and the NOTCH ligand Jagged-1 is expressed in bone marrow stromal cells.154 The NOTCH-1–Delta interaction promotes the differentiation of B lymphocytes and co-engagement of NOTCH and the BCR results in increased activation of the mitogen-activated protein kinase pathway.155 It is possible that NOTCH-1 ligands also contribute to the development of CLL.
Deletion at 11q and SF3B1 mutations
The approximately 20% of patients who exhibit this deletion often present beyond Binet A stage and follow an aggressive disease course with bulky adenopathy and diminished survival.87 These patients have an increased proportion of U-CLL clones. Del 11q22-q23 in most cases affects the ataxia telangiectasia mutated (ATM) gene, the deficiency of which causes genomic instability. Interestingly, mutations in the SF3B1 gene are associated with del(11).83,84 The SF3B1 gene regulates the alternative splicing programme of genes controlling cell cycle progression and apoptosis.83,84
We suggest that once an autoreactive lymphocyte acquires a chromosomal abnormality induced by the tolerance mechanisms, it may be considered a malignancy of self-reactive B cells. It is possible that the criteria to differentiate a benign proliferation of self-reactive B cells as monoclonal B-cell lymphocytosis76 from leukaemia may be the presence of chromosomal or genetic abnormalities. In Fig. 5 we provide a schematic model of U-CLL subset genesis and the role of TLR, NOTCH and FOXP1 mutations, del11q, and immunological tolerance mechanisms that promote chromosomal instability and progression.
Figure 5.

Unmutated chronic lymphocytic leukaemia (U-CLL) lymphocytes have molecular and genetic disturbances that promote survival, proliferation and genomic instability. Deletion of 11q and acquisition of aberrations in SF3B1, Notch-1 and FOXP1 causes genomic instability with the risk of acquiring further genetic damage and progression. Co-engagement of Notch ligands and self-B-cell receptor (BCR) activation results in increased proliferation and survival in U-CLL. These interactions may overcome the low level of anergy that the U-CLL subset undergoes. Signalling through self-reactive BCR, Toll-like receptor 9 (TLR9) with apoptotic cells and interactions of BCR with their own BCR epitopes promote proliferation of U-CLL B cells. In cells with increased levels of FOXP1, augmented expression of RAG enzymes may also promote new genetic damage by persistent V/D/J recombination or receptor editing.
In summary, we suggest that B cells that express BCR that recognize their own BCR epitopes are viewed by the immune system as ‘dangerous cells’. We speculate that BCR autonomous signalling in early B-cell development may induce constant receptor editing (in heavy and light chains) a mechanism involved in immunological tolerance. This constant activation of receptor editing may increase risks of mistakes in allelic exclusion. It may also induce genomic instability and increase the risk of other molecular and genetic disturbances. We advocate that autonomous signalling by the BCR is the clue to understanding the concept of receptor editing and allelic exclusion in CLL B cells. The fact that the whole BCR recognizes a self antigen or foreign antigen may be irrelevant in early B-cell development. In early B cells, autonomous signalling induced by recognition of the BCR's own epitopes simulates an antigen–antibody engagement. In the bone marrow this interaction is viewed as recognition of self molecules and induces receptor editing. This receptor editing may correct germline autoreactivity in the M-CLL subset but fails to induce tolerance in the U-CLL subset. In mature B cells, autonomous signalling by the BCR may promote ‘reversible anergy’ and also may correct self reactivity induced by the somatic hypermutation mechanism in M-CLL. However, in U-CLL, BCR autonomous signalling in addition to self antigen recognition augments B-cell activation, proliferation and genomic instability.
Therapy approaches at the immunological level
B-cell receptor inhibitors, such as ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor, have demonstrated significant activity against CLL.156–158 Recently, the effect of ibrutinib in potentially dangerous CLL clones with high sIgM and CXCR4 expression levels was proposed as a critical factor in therapeutic success.68 CLL was considered to be a tumour of circulating B lymphocytes, variably stimulated and anergized following exposure to antigen in lymphoid tissue.68 This is in concordance with our hypothesis that CLL B lymphocytes are persistently under check by a tolerance mechanism because they are chronically exposed to self antigens. In this context, inhibition of BCR signalling is very important because it can control autonomous CLL lymphocyte BCR signalling capacity and self antigen BCR activation. A consequence of this BCR inhibition may be the inhibition of tolerance mechanisms such as receptor editing and reduction of the risk of acquiring of new molecular aberrations and clonal evolution.
Conclusion
We suggest that CLL originates from a coordinated normal immunological tolerance mechanism to destroy self-reactive B cells. CLL is a malignancy of lymphocytes that acquire a self BCR with autonomous BCR signalling that induces them to proliferate and mature independently of their maturation stage. Additional genetic damage induced by tolerance mechanisms may immortalize them and transform a self-reactive B cell into a leukaemia. The result of tolerogenic mechanisms and genetic aberrations is the survival of CLL B-cell clones with similar markers and homogeneous gene expression signatures despite the different stages of maturation at which the initial damage occurs. BCR signalling inhibitors may correct the immunological disturbances blocking the stimuli that induce chronic activation or tolerance mechanisms in CLL B lymphocytes.
Disclosures
The authors declare no conflict of interest.
References
- 1.Herreros B, Sanchez-Agulere A, Piris MA. Lymphoma microenvironment. In: Magrath IT, Bathia K, Boffetta P, Dearden C, Diehl V, Gascoyne RD, Muller-Hermelink HK, Potter M, Rohatiner A, editors. The Lymphoid Neoplams. 3rd edn. London, UK: Oxford University Press; 2010. p. 1552. [Google Scholar]
- 2.García-Muñoz R, Galiacho VR, Llorente L. Immunological aspects in chronic lymphocytic leukemia (CLL) development. Ann Hematol. 2012;91:981–96. doi: 10.1007/s00277-012-1460-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.García-Muñoz R, Panizo C, Bendandi M, Llorente L. Autoimmunity and lymphoma: is mantle cell lymphoma a mistake of the receptor editing mechanism? Leuk Res. 2009;33:1437–9. doi: 10.1016/j.leukres.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Luning Prak ET, Monestier M, Eisenberg RA. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci. 2011;1217:96–121. doi: 10.1111/j.1749-6632.2010.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnet M. Auto-Immunity and Auto-Immune Disease. 1st edn. Letchworth, UK: The Garden City Press Limited; 1972. Tolerance and paralysis; p. 59. In: Sir Macfarlane Burnet. [Google Scholar]
- 6.Wang JH, Alt FW, Gostissa M, et al. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–90. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobrin C, Cha SC, Qin H, et al. Molecular analysis of light-chain switch and acute lymphoblastic leukemia transformation in two follicular lymphomas: implications for lymphomagenesis. Leuk Lymphoma. 2006;47:1523–34. doi: 10.1080/10428190600612909. [DOI] [PubMed] [Google Scholar]
- 8.Yonetani N, Ueda C, Akasaka T, Nishikori M, Uchiyama T, Ohno H. Heterogeneous breakpoints on the immunoglobulin genes are involved in fusion 5' region of BCL-2 in B cell tumors. Jpn J Cancer Res. 2001;92:933–40. doi: 10.1111/j.1349-7006.2001.tb01183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, Chiorazzi N, Meffre E. Unmutated and mutated chronic lyphocytic leukemia derive from self reactive B cell precursors despite expressing antibody reactivity. J Clin Invest. 2005;115:1636–43. doi: 10.1172/JCI24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109:4424–31. doi: 10.1182/blood-2006-11-056648. [DOI] [PubMed] [Google Scholar]
- 11.Papakonstantinou N, Ntoufa S, Chartomatsidou E, et al. Differential microRNA profiles and their functional implications in different immunogenetic subsets of chronic lymphocytic leukemia. Mol Med. 2013;19:115–23. doi: 10.2119/molmed.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappione A, 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005;115:3205–16. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pugh-Bernard AE, Silverman GJ, Cappione AJ, Villano ME, Ryan DH, Insel RA, Sanz I. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001;108:1061–70. doi: 10.1172/JCI12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matejuk A, Beardall M, Xu Y, Tian Q, Phillips D, Alabyev B, Mannoor K, Chen C. Exclusion of natural autoantibody-producing B cells from IgG memory B cell compartment during T cell-dependent immune response. J Immunol. 2009;182:7634–43. doi: 10.4049/jimmunol.0801562. [DOI] [PubMed] [Google Scholar]
- 15.Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzeig MC, Wardemann H. A checkpoint for autoreactivity in human IgM+ memory B cell development. J Exp Med. 2006;203:393–400. doi: 10.1084/jem.20052033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117:1781–91. doi: 10.1182/blood-2010-07-155663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70−. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seifert M, Sellmann L, Bioehdorn J, Wein F, Stilgenbauer S, Dürig J, Küppers R. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209:2183–98. doi: 10.1084/jem.20120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103:4389–95. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 20.DiLillo DJ, Weinberg JB, Yoshizaki A, et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia. 2013;27:170–82. doi: 10.1038/leu.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194:1625–38. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caligaris-Cappio F. B-chronic lymphocytic leukemia: a malignancy of anti-self B cells. Blood. 1996;87:2615–20. [PubMed] [Google Scholar]
- 23.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194:1639–47. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 25.Hoogeboom R, Wormhoudt TA, Schipperus MR, Langerak AW, Dunn-Walters DK, Guikema JE, Bende RJ, van Noesel CJ. A novel chronic lymphocytic leukemia subset expressing mutated IGHV3-7 encoded rheumatoid factor B cell receptors that are functionally proficient. Leukemia. 2013;27:738–40. doi: 10.1038/leu.2012.238. [DOI] [PubMed] [Google Scholar]
- 26.Hoogeboom R, van Kessel KP, Hochstenbach F, et al. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med. 2013;210:59–70. doi: 10.1084/jem.20121801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamblin T, Hamblin A. Chronic lymphocytic leukemia. In: Saba HI, Mufti GJ, editors. Advances in Malignant Hematology. 1st edn. Oxford: Blackwell Science Inc; 2011. p. 211. [Google Scholar]
- 28.Agathangelidis A, Darzentas N, Hadzidimitriou A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119:4467–75. doi: 10.1182/blood-2011-11-393694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forconi F, Potter KN, Wheatley I, et al. The normal IGHV1-69 derived B cell repertoire contains stereotypic patterns characteristic of unmutated CLL. Blood. 2010;115:71–7. doi: 10.1182/blood-2009-06-225813. [DOI] [PubMed] [Google Scholar]
- 30.Oscier DG, Thompsett A, Zhu D, Stevenson FK. Differential rates of somatic hypermutation in VH genes among subsets of chronic lymphocytic leukemia defined by chromosomal abnormalities. Blood. 1997;89:4153–60. [PubMed] [Google Scholar]
- 31.Ghia P, Stamatopoulos K, Belessi C, et al. Geographic patterns and pathogenetic implications of IGHV gene usage in chronic lymphocytic leukemia: the lesson of the IGHV3-21 gene. Blood. 2005;105:1678–85. doi: 10.1182/blood-2004-07-2606. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto S, Dono M, Wakai M, et al. Somatic diversification and selection of immunoglobulin heavy and light chain variable region genes in IgG+ CD5+ chronic lymphocytic leukmemia B cells. J Exp Med. 1995;181:1507–17. doi: 10.1084/jem.181.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potter KN, Mockridge CL, Neville L, et al. Structural and functional features of the B cell receptor in IgG-positive chronic lymphocytic leukemia. Clin Cancer Res. 2006;12:1672–9. doi: 10.1158/1078-0432.CCR-05-2164. [DOI] [PubMed] [Google Scholar]
- 34.Ghiotto F, Fais F, Valetto A, et al. Remarably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113:1008–16. doi: 10.1172/JCI19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ntoufa S, Vardi A, Papakonstantinou N, et al. Distinct innate immunity pathways to activation and tolerance in subgroups of chronic lymphocytic leukemia with distinct immunoglobulin receptors. Mol Med. 2012;18:1281–91. doi: 10.2119/molmed.2011.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tobin G, Thunberg U, Johnson A, et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted Vλ2-14 gene use and homologous CDR3s: implication recognition of a common antigen epitope. Blood. 2003;101:4952–7. doi: 10.1182/blood-2002-11-3485. [DOI] [PubMed] [Google Scholar]
- 37.Stamatopoulos K, Belessi C, Hadzidimitriou A, et al. Immunoglobulin light chain repertoire in chronic lymphocytic leukemia. Blood. 2005;106:3575–83. doi: 10.1182/blood-2005-04-1511. [DOI] [PubMed] [Google Scholar]
- 38.Chu CC, Catera R, Hatzi K, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112:5122–9. doi: 10.1182/blood-2008-06-162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catera R, Silverman GJ, Hatzi K, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med. 2008;14:665–74. doi: 10.2119/2008-00102.Catera. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lanemo Myhrinder A, Hellqvist E, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111:3838–48. doi: 10.1182/blood-2007-11-125450. [DOI] [PubMed] [Google Scholar]
- 41.Sthoeger ZM, Wakai M, Tse DB, et al. Production of autoantibodies by CD5-expressing B lymphocytes from patients with chronic lymphocytic leukemia. J Exp Med. 1989;169:255–68. doi: 10.1084/jem.169.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efremov DG, Ivanovski M, Batista FD, Pozzato G, Burrone OR. IgM-producing chronic lymphocytic leukemia cells undergo immunoglobulin isotype-switching without acquiring somatic mutations. J Clin Invest. 1996;98:290–8. doi: 10.1172/JCI118792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54. [PubMed] [Google Scholar]
- 44.Damle RN, Wasil T, Fais F, et al. IgV gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7. [PubMed] [Google Scholar]
- 45.Muller-Hermelink HK, Montserrat E, Catovsky D, Campo E, Harris NL, Stain H. Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Clasification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 180–2. [Google Scholar]
- 46.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 47.Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenszweig MC. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol. 2004;5:645–50. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Zemlin M, Wang YH, et al. Contribution of VH gene replacement to the primary B cell repertoire. Immunity. 2003;19:21–31. doi: 10.1016/s1074-7613(03)00170-5. [DOI] [PubMed] [Google Scholar]
- 50.Wardemann H, Hammersen J, Nussenzeig MC. Human autoantibody silencing by immunoglobulin light chains. J Exp Med. 2004;200:191–9. doi: 10.1084/jem.20040818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duty JA, Szodoray P, Zheng NY, et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med. 2009;206:139–51. doi: 10.1084/jem.20080611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan TD, Brink R. Affinity-based selection and the germinal center response. Immunol Rev. 2012;247:11–23. doi: 10.1111/j.1600-065X.2012.01118.x. [DOI] [PubMed] [Google Scholar]
- 53.Weistein JS, Hernandez SG, Craft J. T cells that promote B-cell maturation in systemic autoimmunity. Immunol Rev. 2012;247:160–71. doi: 10.1111/j.1600-065X.2012.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussensweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26:205–13. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cadera EJ, Wan G, Amin RH, Nolla H, Lenardo MJ, Schlissel MS. NF-κB activity marks cells engaged in receptor editing. J Exp Med. 2009;206:1803–16. doi: 10.1084/jem.20082815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20:632–8. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Isnardi I, Ng YS, Srdanovic I, et al. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity. 2008;29:746–57. doi: 10.1016/j.immuni.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isnardi I, Ng YS, Menard L, et al. Complement receptor 2/CD21-human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026–36. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20:246–59. doi: 10.1016/j.ccr.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 60.Rassenti LZ, Kipps TJ. Lack of allelic exclusion in B cell chronic lymphocytic leukemia. J Exp Med. 1997;185:1435–45. doi: 10.1084/jem.185.8.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dühren-von Minden M, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signaling. Nature. 2012;489:309–12. doi: 10.1038/nature11309. [DOI] [PubMed] [Google Scholar]
- 62.Köhler F, Hug E, Eschbach C, Meixlsperger S, Hobeika E, Kofer J, Wardemann H, Jumaa H. Autoreactive B cell receptors mimic autonomous pre-B cell receptor signaling and induce proliferation of early B cells. Immunity. 2008;29:912–21. doi: 10.1016/j.immuni.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 63.van Loo PF, Dingjan GM, Maas A, Hendriks RW. Surrogate-light-chain silencing is not critical for the limitation of pre-B cell expansion but is for the termination of constitutive signaling. Immunity. 2007;27:468–80. doi: 10.1016/j.immuni.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 64.Meixlsperger S, Köhler F, Wossning T, Reppel M, Müschen M, Jumaa H. Conventional light chains inhibit the autonomous signaling capacity of the B cell receptor. Immunity. 2007;26:323–33. doi: 10.1016/j.immuni.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 65.Hadzidimitriou A, Darzentas N, Murray F, et al. Evidence for the significant role of immunoglobulin light chains in antigen recognition and selection in chronic lymphocytic leukemia. Blood. 2009;113:403–11. doi: 10.1182/blood-2008-07-166868. [DOI] [PubMed] [Google Scholar]
- 66.Gauthier L, Rossi B, Roux F, Termine E, Schiff C. Galectin-1 is a stromal cell ligand of the pre-B cell receptor (BCR) implicated in synapse formation between pre-B and stromal cells and in pre-BCR triggering. Proc Natl Acad Sci USA. 2002;99:13014–9. doi: 10.1073/pnas.202323999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Croci DO, Morande PE, Dergan-Dylon S, et al. Nurse-like cells control the activity of chronic lymphocytic leukemia B cells via galectin-1. Leukemia. 2013;27:1413–6. doi: 10.1038/leu.2012.315. [DOI] [PubMed] [Google Scholar]
- 68.Coelho V, Krysov S, Steele A, et al. Identification in CLL of circulating intraclonal subgrups with varying B cell receptor expression and function. Blood. 2013;122:2664–72. doi: 10.1182/blood-2013-02-485425. [DOI] [PubMed] [Google Scholar]
- 69.Klippel JH, Crofford LJ, Stone JH, Weyland CM, editors. Primer on the Rheumatic Diseases. 12th edn. Atlanta, GA, USA: Arthritis Foundation; 2001. [Google Scholar]
- 70.Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, Caligaris-Cappio F, Ghia P. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood. 2008;112:188–95. doi: 10.1182/blood-2007-09-111344. [DOI] [PubMed] [Google Scholar]
- 71.Apollonio B, Scielzo C, Bertilaccio MT, et al. Targeting B cell anergy in chronic lymphocytic leukemia. Blood. 2013;121:3879–88. doi: 10.1182/blood-2012-12-474718. [DOI] [PubMed] [Google Scholar]
- 72.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119:6278–87. doi: 10.1182/blood-2012-01-403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–44. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rawstron AC, Bennet FL, O'Connor SJM, et al. Monoclonal B cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359:575–83. doi: 10.1056/NEJMoa075290. [DOI] [PubMed] [Google Scholar]
- 75.Ladgren O, Albitar M, Wanlong M, et al. B cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360:659–67. doi: 10.1056/NEJMoa0806122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Henriques A, Rodriguez-Caballero A, Nieto WG, et al. Combined patterns of IGHV repertoire and cytogenetic/molecular alterations in monoclonal B cell lymphocytosis versus chronic lymphocytic leukemia. PLoS One. 2013;8:e67751. doi: 10.1371/journal.pone.0067751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109:4944–51. doi: 10.1182/blood-2006-12-062398. [DOI] [PubMed] [Google Scholar]
- 78.Calin GA, Liu CG, Sevignani C, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004;101:11755–60. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–65. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- 81.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–9. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 84.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, Pekarsky Y, Croce CM. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119:329–31. doi: 10.1182/blood-2011-10-386144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balatti V, Lerner S, Rizzotto L, et al. Trisomy 12 CLLs progress through NOTCH1 mutations. Leukemia. 2013;27:740–3. doi: 10.1038/leu.2012.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 88.Shanafelt TD, Witzing TE, Fink SR, et al. Prospective evolution during long-term follow-up of patients with untreated early stage chronic lymphocytic leukemia. J Clin Oncol. 2006;24:4634–41. doi: 10.1200/JCO.2006.06.9492. [DOI] [PubMed] [Google Scholar]
- 89.Murrray F, Darzentas N, Hadzidimitrioy A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111:1524–33. doi: 10.1182/blood-2007-07-099564. [DOI] [PubMed] [Google Scholar]
- 90.Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukemia. Br Med Bull. 2008;87:49–62. doi: 10.1093/bmb/ldn034. [DOI] [PubMed] [Google Scholar]
- 91.García-Muñoz R, García DK, Roldan-Galiacho V, Merchante-Andreu M, Campeny-Najara A, Rabasa P. Therapy-related acute myeloid leukemia in a patient with chronic lymphocytic leukemia treated with Rituximab-Bendamustine. Ann Hematol. 2014;93:699–702. doi: 10.1007/s00277-013-1844-8. [DOI] [PubMed] [Google Scholar]
- 92.Lotz M, Ranheim E, Kipps TJ. Transforming growth factor β as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J Exp Med. 1994;179:999–1004. doi: 10.1084/jem.179.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Le Dieu R, Blum W, Byrd JC, Gribben JG. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118:2427–37. doi: 10.1172/JCI35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120:1412–21. doi: 10.1182/blood-2012-02-411678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weiss L, Melchardt T, Eagle A, Grabmer C, Greil R, Tinhofer I. Regulatory T cells predict the time to initial treatment in early stage chronic lymphocytic leukemia. Cancer. 2009;27:e5–6. doi: 10.1002/cncr.25752. [DOI] [PubMed] [Google Scholar]
- 96.D'Arena G, D'Auria F, Simeon V, et al. A shorter time to the first treatment may be predicted by the absolute number of regulatory T cells in patients with Rai stage 0 chronic lymphocytic leukemia. Am J Hematol. 2012;87:628–31. doi: 10.1002/ajh.23170. [DOI] [PubMed] [Google Scholar]
- 97.Lad DP, Varma S, Varma N, Sachdeva MU, Bose P, Malhotra P. Regulatory T cells in B cell chronic lymphocytic leukemia: their role in disease progression and autoimmune cytopenias. Leuk Lymphoma. 2013;54:1012. doi: 10.3109/10428194.2012.728287. [DOI] [PubMed] [Google Scholar]
- 98.Sampalo A, Brieva JH. Humoral immunodeficiency in chronic lymphocytic leukemia: role of CD95/95L in tumoral damage and escape. Leuk Lymphoma. 2002;43:881–4. doi: 10.1080/10428190290017033. [DOI] [PubMed] [Google Scholar]
- 99.Gilling CE, Mittal AK, Chaturvedi NK, et al. Lymph node induced immune tolerance in chronic lymphocytic leukaemia: a role for caveolin-1. Br J Haematol. 2012;158:216–31. doi: 10.1111/j.1365-2141.2012.09148.x. [DOI] [PubMed] [Google Scholar]
- 100.Riches JC, Davies JK, McClanahan F, Fatah R, Igbal S, Agrawal S, Ramsay AG, Gribben JG. T cells from CLL patients exhibit features of T cell exhaustion but retain capacity for cytokine production. Blood. 2013;121:1612–21. doi: 10.1182/blood-2012-09-457531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C. Expansion of a CD8+PD-1+ replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res. 2012;18:678–87. doi: 10.1158/1078-0432.CCR-11-2630. [DOI] [PubMed] [Google Scholar]
- 102.Dearden C. Disease-specific complications of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2008;2008:450–6. doi: 10.1182/asheducation-2008.1.450. [DOI] [PubMed] [Google Scholar]
- 103.Kay NE, Zarling JM. Impaired natural killer activity in patients with chronic lymphocytic leukemia is associated with deficiency of azyrophilic cytoplasmic granules in putative NK cells. Blood. 1984;63:305–9. [PubMed] [Google Scholar]
- 104.Itala M, Vainio O, Remes K. Functional abnormalities in granulocytes predict susceptibility to bacterial infections in chronic lymphocytic leukaemia. Eur J Haematol. 1996;57:46–53. doi: 10.1111/j.1600-0609.1996.tb00489.x. [DOI] [PubMed] [Google Scholar]
- 105.Schlesinger M, Broman I, Lugassy G. The complement system is defective in chronic lymphatic leukemia patients and in their healthy relatives. Leukemia. 1996;10:1509–13. [PubMed] [Google Scholar]
- 106.Caligaris-Cappio F. Chronic lymphocytic leukemia “Cinderella” is becoming a star. Mol Med. 2009;15:67–9. doi: 10.2119/molmed.2008.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tripodo C, Porcasi R, Guarnotta C, Ingrao S, Campisi V, Florena AM, Franco V. C1q production by bone marrow stromal cells. Scand J Immunol. 2007;65:308–9. doi: 10.1111/j.1365-3083.2006.01871.x. [DOI] [PubMed] [Google Scholar]
- 108.Espinoza CR, Feeney AJ. The extent of histone acetylation correlates with the differential rearrangement frequency of individual VH genes in pro-B cells. J Immunol. 2005;175:6668–75. doi: 10.4049/jimmunol.175.10.6668. [DOI] [PubMed] [Google Scholar]
- 109.Oppezzo P, Dumas G, Lalanne AI, Peyelle-Brogard B, Magnac C, Pritsch O, Dighiero G, Vuillier F. Different isoforms of BASAP regulate expression of AID in normal and chronic lymphocytic leukemia B cells. Blood. 2005;105:2495–503. doi: 10.1182/blood-2004-09-3644. [DOI] [PubMed] [Google Scholar]
- 110.Rothenberg EV, Telfer JC, Anderson MK. Transcriptional regulation of lymphocyte lineage commitment. BioEssays. 1999;21:726–42. doi: 10.1002/(SICI)1521-1878(199909)21:9<726::AID-BIES4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 111.Kardava L, Yang Q, St Leger A, Foon KA, Lentzch S, Vallejo AN, Milcarek C, Borghesi L. The B lineage transcription factor E2A regultes apoptosis in chronic lymphocytic leukemia (CLL) cells. Int Immunol. 2001;23:375–84. doi: 10.1093/intimm/dxr027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Quong MW, Martensson A, Langerak AW, Rivera RR, Nemazee D, Murre C. Receptor editing and marginal zone B cell development are regulated by the helix-loop-helix Protein, E2A. J Exp Med. 2004;199:1101–12. doi: 10.1084/jem.20031180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lin YC, Jhunjhunwala S, Benner C, et al. A global network of transcription factors, involving E2A, EBF1 and Foxp1 that orchestrates the B cell fate. Nat Immunol. 2010;11:635–43. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klenle D, Benner A, Läufle C, et al. Gene expression factors as predictors of genetic risk and survival in chronic lymphocytic leukemia. Haematologica. 2010;95:102–9. doi: 10.3324/haematol.2009.010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lloberas J, Soler C, Celada A. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunol Today. 1999;20:184. doi: 10.1016/s0167-5699(99)01442-5. [DOI] [PubMed] [Google Scholar]
- 116.Bassuk AG, Leiden JM. The role of ETS transcription factors in the development and function of the mammalian immune system. Adv Immunol. 1997;64:65. doi: 10.1016/s0065-2776(08)60887-1. [DOI] [PubMed] [Google Scholar]
- 117.Mankai A, Bordron A, Renaudineau Y, Martins-Carvalho C, Takahashi S, Ghedira I, Berthou C, Youinou P. Purine-rich box-1 mediated reduced expression of CD20 alters rituximab induced lysis of chronic lymphocytic leukemia B cells. Cancer Res. 2008;68:7512–9. doi: 10.1158/0008-5472.CAN-07-6446. [DOI] [PubMed] [Google Scholar]
- 118.Iwasaki H, Somoza C, Shigematsu H, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Polli M, Dakic A, Light A, Wu L, Tarlingon DM, Nutt SL. The development of functional B lymphocytes in conditional PU.1 knock-out mice. Blood. 2005;106:2083. doi: 10.1182/blood-2005-01-0283. [DOI] [PubMed] [Google Scholar]
- 120.Medina KL, Ponguabala JM, Reddy KL, et al. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–17. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 121.Yuki H, Ueno S, Tatetsu H, et al. Pu.1 is a potent tumor suppressor in classical Hodgkin lymphoma cells. Blood. 2013;121:962–70. doi: 10.1182/blood-2012-05-431429. [DOI] [PubMed] [Google Scholar]
- 122.Travis A, Amsterdam A, Belangeer C, Grosschedl R. LEF-1, a gene encoding a lymphoid-specific protein with an HMG domain, regulates T cell receptor α enhance function. Genes Dev. 1991;5:880–94. doi: 10.1101/gad.5.5.880. [DOI] [PubMed] [Google Scholar]
- 123.Raya T, O'Riordan M, Okamura R, Devaney E, Willert K, Nusse R, Grosschedl R. Wnt signaling regultes B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. 2000;13:15–24. doi: 10.1016/s1074-7613(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 124.Gutierrez A, Jr, Tscumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B cell lymphocytosis. Blood. 2010;116:2975–83. doi: 10.1182/blood-2010-02-269878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gutierrez A, Jr, Arendt BK, Tschumper RC, Kay NE, Zent CS, Jelinek DF. Differentiation of chronic lymphocytic leukemia B cells into immunoglobulin secreting cells decreases LEF-1 expression. Plos One. 2011;6:e26056. doi: 10.1371/journal.pone.0026056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jansen K, Rother MB, Brusletto BS, Olstad OK, Dalsbotten Aass HC, Van Zelm MC, Kierulf P, Gautvik KM. Increased ID2 levels in adult precursor B cells as compared with children is associated with impaired Ig locus contraction and decreased bone marrow output. J Immunol. 2013;191:1210–9. doi: 10.4049/jimmunol.1203462. [DOI] [PubMed] [Google Scholar]
- 127.Husson H, Carideo EG, Neubeg D, et al. Gene expression profiling lymphoma and normal germinal center B cells using cDNA arrays. Blood. 2002;99:282–9. doi: 10.1182/blood.v99.1.282. [DOI] [PubMed] [Google Scholar]
- 128.Mayers G, Ng YS, Bannock JM, et al. Activation-induced cytidine deaminase (AID) is required for B cell tolerance in humans. Proc Natl Acad Sci USA. 2011;108:11554–9. doi: 10.1073/pnas.1102600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Durandy A, Cantaert T, Kracker S, Meffre E. Potential roles of activation-induced cytidine deaminase in promotion or prevention of autoimmunity in humans. Autoimmunity. 2013;46:148–56. doi: 10.3109/08916934.2012.750299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oppezzo P, Vuillier F, Vasconcelos Y, Dumas G, Magnac C, Payelle-Brogard B, Pritsch O, Dighiero G. Chronic lymphocytic leukemia B cells expressing AID display dissociation between class switch recombination and somatic hypermutation. Blood. 2003;101:4029–32. doi: 10.1182/blood-2002-10-3175. [DOI] [PubMed] [Google Scholar]
- 131.Patten PE, Chu CC, Albesiano E, et al. IGHV-unmuteted and IGHV mutated chronic lymphocytic leukemia cells produce activation-induced deaminase protein with a full range of biologic functions. Blood. 2012;120:4802–11. doi: 10.1182/blood-2012-08-449744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hu H, Wang B, Borde M, Nardone J, Maika S, Allred L, Tucker PW, Rao A. FOXP1 is an essential transcriptional regulator of B cell development. Nat Immunol. 2006;7:819–26. doi: 10.1038/ni1358. [DOI] [PubMed] [Google Scholar]
- 133.Chen Z, Xiao Y, Zhang J, et al. Transcription factors E2A, FOXO1 and FOXP1 regulate recombination activating gene expression in cancer cells. PLoS One. 2011;6:e20475. doi: 10.1371/journal.pone.0020475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sargardoy A, Martinez-Ferrandis JL, Roa S, et al. Downregulation of FOXP1 is required during germinal center B cell function. Blood. 2013;121:4311–20. doi: 10.1182/blood-2012-10-462846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Karnowski A, Cao C, Matthias G, Carotta S, Corcoran LM, Martensson IL, Skok JA, Matthias P. Silencing and nuclear repositioning of the λ5 gene locus at the pre-B cell stage requires Aiolos and OFB-1. PLoS One. 2008;3:e3568. doi: 10.1371/journal.pone.0003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sun J, Matthias G, Mihatsch MJ, Georgopoulos K, Matthias P. Lack of the transcriptional coactivator OBF-1 prevents the development of systemic lupus erythematosus like phenotypes in Aiolos mutant mice. J Immunol. 2003;170:1699–706. doi: 10.4049/jimmunol.170.4.1699. [DOI] [PubMed] [Google Scholar]
- 137.Billot K, Soeur J, Chereau F, et al. Deregulation of Aiolos expression in chronic lymphcytic leukemia is associated with epigenetic modifications. Blood. 2011;117:1917–27. doi: 10.1182/blood-2010-09-307140. [DOI] [PubMed] [Google Scholar]
- 138.Nagy M, Chapuis B, Matthes T. Expression of transcription factors Pu.1 Spi-B, Blimp-1, BSAP and OcT2 in normal human plasma cells and in multiple myeloma cells. Br J Haematol. 2002;116:429–35. doi: 10.1046/j.1365-2141.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 139.Saito T, Chiba S, Ichikawa M, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–85. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 140.Thomas MD, Kremer CS, Ravichandran KS, Rajewsky K, Bender TP. C-Myb is critical for B cell development and maintenance of follicular B cells. Immunity. 2005;23:275–86. doi: 10.1016/j.immuni.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 141.Golay J, Luppi M, Songia S, et al. Expression of A-myb, but not c-myb and B-myb, is restricted to Burkitt's lymphoma, sIg+ B acute lymphoblastic leukemia and a subset of chronic lymphocytic leukemias. Blood. 1996;87:1900–11. [PubMed] [Google Scholar]
- 142.Xiao C, Calado DP, Galler G, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–59. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 143.Wang M, Tan LP, Dijkstra MK, et al. miRNA analysis in B cell chronic lymphocytic leukemia: proliferation centers characterized by low miR-150 and high BIC/miR-155 expression. J Pathol. 2008;215:13–20. doi: 10.1002/path.2333. [DOI] [PubMed] [Google Scholar]
- 144.Tang LP, Wang M, Robertus JL, et al. miRNA profiling of B cell subsets: specific miRNA profile for germinal center B cells with variation between centroblast and centrocytes. Lab Invest. 2009;89:708–76. doi: 10.1038/labinvest.2009.26. [DOI] [PubMed] [Google Scholar]
- 145.Ochai K, Maienschein-Cline M, Simonetti G, et al. Transcriptional regulation of germinal center B cells and plasma cell fates by dynamical control of IRF4. Immunity. 2013;38:918–29. doi: 10.1016/j.immuni.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Havelange V, Pekarsky Y, Nakamura T, Palamarchuk A, Alder H, Rassenti L, Kipps T, Croce CM. IRF4 mutations in chronic lymphocytic leukemia. Blood. 2011;118:2827–9. doi: 10.1182/blood-2011-04-350579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chung-Che C, Lorek J, Sabath DE, Ying L, Chitambar CR, Logan B, Kampalath B, Cleveland RP. Expression of MUM1/IRF4 correlates with clinical outcome in patients with B cell chronic lymphocytic leukemia. Blood. 2002;100:4671–5. doi: 10.1182/blood-2002-01-0104. [DOI] [PubMed] [Google Scholar]
- 148.Alinikula J, Nera KP, Junttilla S, Lassila O. Alternate pathways for BCL6-mediated regulation of B cells to plasma cell differentiation. Eur J Immunol. 2011;41:2404–13. doi: 10.1002/eji.201141553. [DOI] [PubMed] [Google Scholar]
- 149.Zhou Z, Li A, Wang Z, Pei F, Xia Q, Liu G, Ren Y, Hu Z. Blimp-1 siRNA inhibits B cell differentiation and prevents the development of lupus in mice. Hum Immunol. 2013;74:297–301. doi: 10.1016/j.humimm.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 150.Martinez A, Pittaluga S, Rudelius M, et al. Expression of the interferon regulatory factor 8/ICSBP-1 in human reactive lymphoid tissues and B cell lymphomas: a novel germinal center marker. Am J Surg Pathol. 2008;32:1190–200. doi: 10.1097/PAS.0b013e318166f46a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 promotes tolerance by restricting survival of anergic anti-DNA B cells, yet is also required for their activation. J Immunol. 2013;190:1447–56. doi: 10.4049/jimmunol.1202115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Miles K, Heaney J, Sibinska Z, Salter D, Savill J, Gray D, Gray M. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci USA. 2012;109:887–92. doi: 10.1073/pnas.1109173109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tromp JM, Tonino SH, Elias JA, et al. Dichotomy in NF-κB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene. 2010;29:5071–82. doi: 10.1038/onc.2010.248. [DOI] [PubMed] [Google Scholar]
- 154.Bertrand FE, Eckfeldt CE, Lysholm AS, LeBien TW. Notch-1 and Notch-2 exhibit unique patterns of expression in human B-lineage cells. Leukemia. 2000;14:2095–102. doi: 10.1038/sj.leu.2401942. [DOI] [PubMed] [Google Scholar]
- 155.Thomas M, Calamito M, Srivastava B, Maillard I, Pear WS, Allman D. Notch activity synergizes with B-cell-receptor and CD40 signaling to enhance B-cell activation. Blood. 2007;109:3342–50. doi: 10.1182/blood-2006-09-046698. [DOI] [PubMed] [Google Scholar]
- 156.Chavez JC, Sahakian E, Pinilla-Ibarz J. Ibrutinib: an evidence-based review of its potential in the treatment of advanced chronic lymphocytic leukemia. Core Evid. 2013;8:37–45. doi: 10.2147/CE.S34068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kharfan-Dabaja MA, Wierda WG, Cooper LJ. Immunotherapy for chronic lymphocytic leukemia in the era of BTK inhibitors. Leukemia. 2014;28:507–17. doi: 10.1038/leu.2013.311. [DOI] [PubMed] [Google Scholar]
- 158.Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B cell receptor signalling activity. Leukemia. 2014;28:649–47. doi: 10.1038/leu.2013.358. [DOI] [PubMed] [Google Scholar]