

Abstract
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation and synovial hyperplasia leading to progressive joint destruction. Fibroblast-like synoviocytes (FLS) are central components of the aggressive, tumour-like synovial structure termed pannus, which invades the joint space and cartilage. A distinct natural killer (NK) cell subset expressing the inhibitory CD94/NKG2A receptor is present in RA synovial fluid. Little is known about possible cellular interactions between RA-FLS and NK cells. We used cultured RA-FLS and the human NK cell line Nishi, of which the latter expresses an NK receptor repertoire similar to that of NK cells in RA synovial fluid, as an in vitro model system of RA-FLS/NK cell cross-talk. We show that RA-FLS express numerous ligands for both activating and inhibitory NK cell receptors, and stimulate degranulation of Nishi cells. We found that NKG2D, DNAM-1, NKp46 and NKp44 are the key activating receptors involved in Nishi cell degranulation towards RA-FLS. Moreover, blockade of the interaction between CD94/NKG2A and its ligand HLA-E expressed on RA-FLS further enhanced Nishi cell degranulation in co-culture with RA-FLS. Using cultured RA-FLS and the human NK cell line Nishi as an in vitro model system of RA-FLS/NK cell cross-talk, our results suggest that cell-mediated cytotoxicity of RA-FLS may be one mechanism by which NK cells influence local joint inflammation in RA.
Keywords: CD94/NKG2A, fibroblast-like synoviocytes, natural killer cytotoxicity, NKG2D
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by synovial inflammation and progressive destruction of cartilage and bone.1 The healthy synovial lining that encapsulates the joint cavity is only a few cell layers thick and is composed of two main cellular subsets termed fibroblast-like synoviocytes (FLS) and macrophage-like synoviocytes. In RA the synovial membrane becomes hyperplastic, including a tumour-like proliferation of activated FLS (RA-FLS) together with infiltrating leucocytes such as macrophages, T cells, natural killer (NK) cells and B cells. Together, these cells form a synovial pannus, which aggressively spreads into the joint cavity, invading the cartilage and bone, which subsequently results in structural tissue damage.2
The RA-FLS play an essential role in driving cartilage erosion.3 As recently reviewed by Niedermeier et al.,4 these cells can directly infiltrate cartilage and release mediators responsible for proteolytic matrix degradation, including matrix metalloproteinases. Moreover, RA-FLS contribute to RA pathogenesis by production of pro-inflammatory cytokines, including interleukin-6 (IL-6), IL-15 and granulocyte–macrophage colony-stimulating factor, as well as secretion of chemokines, including stromal cell-derived factor 1 (CXCL12), IP10 (CXCL10), monocyte chemotactic protein 1 (MCP1), IL-8 (CXCL8), and RANTES (CCL5).5–9
Natural killer cells are bone marrow-derived lymphocytes that are important in immune defence. Upon activation, they rapidly produce a range of cytokines and mediate cytotoxic responses against infected, stressed or malignantly transformed cells.10–13 It is becoming increasingly appreciated that NK cells may play a role in the modulation of a multitude of immune responses through their capacity to promote differentiation of monocytes into osteoclasts and dendritic cells, and to promote polarization of T-cell and B-cell responses.14–17 Furthermore, recent studies in mice have shown that NK cells eliminate subsets of activated T cells via cell-mediated cytotoxic responses.18,19 Natural killer cell-mediated killing of activated T cells is dampened by the interaction with its antigen (in mouse this is Qa-1, the mouse equivalent of human HLA-E) expressed on activated T cells, and inhibitory CD94/NKG2A receptors expressed on NK cells and subsets of CD8+ T cells.20 Consistent with in vitro data, disrupting the interaction between CD94/NKG2A and Qa-1 in mouse models of RA and multiple sclerosis (e.g. in collagen-induced arthritis and experimental-allergic encephalomyelitis models), using antibodies against CD94/NKG2A or Qa-1 ameliorated disease,18,19,21 and in one study in experimental-allergic encephalomyelitis it led to a reduction in the number of follicular helper T cells and T helper 17 cells.19 Therapeutic enhancement of NK cell-mediated cytotoxic responses therefore appears to be beneficial in certain experimental models of human chronic inflammatory diseases.
Human NK cells may be divided into two main subsets based on surface expression of CD56 and CD16 (FcγRIII). CD56dimCD16+ NK cells make up approximately 90% of peripheral blood (PB) NK cells, whereas the remaining 10% are CD56bright CD16dim/− NK cells.22–24 The CD56bright subset is, however, the dominant subset in secondary lymphoid organs such as tonsils and lymph nodes.25,26 Moreover, NK cells with a CD56bright phenotype are over-represented at inflammatory sites in numerous diseases,27,28 including synovial fluid (SF) of RA patients.29,30
Previous studies suggest that RA-FLS promote the migration, activation and survival of PB NK cells in vitro.31 Based on the finding that the vast majority of RA SF NK cells display an activated phenotype in vivo, together with the observation that RA-FLS express MHC class I-chain related protein A (MIC-A) and MIC-B, ligands for the activating NK cell receptor NKG2D,32,33 we investigated the cross-talk that takes place between NK cells and RA-FLS in vitro. Using the human NK cell line Nishi, we investigated whether RA-FLS can stimulate NK cell degranulation that reflects NK cell-mediated cytotoxicity in vitro, and if so, which receptor–ligand interactions were involved.
Materials and methods
Ethics statement
Collection of human tissue, SF and blood samples was approved by the local ethics committee at each site (listed below for each sample) and informed consent was obtained from all participating patients.
Clinical RA synovial samples
Synovial tissue biopsies for the isolation of RA-FLS were provided by Dr Henning Bliddal and Dr Martin Andersen from the Rheumatology Department, Frederiksberg Hospital, Copenhagen, Denmark, and obtained from the small joints of RA patients undergoing arthroscopic synovectomy (eight patients, approved by The Ethics Committee for Copenhagen and Frederiksberg Municipalities, Denmark). Samples of SF for in vitro-derived RA-FLS and enrichment of autologous NK cells were collected from patients at the Rheumatology Clinic, Rigshospitalet Hospital (Copenhagen, Denmark) by Dr Bo Baslund and Dr Lars Juul (two patients, approved by The Ethics Committee for Copenhagen, Denmark). Paired SF and PB samples for cell surface staining of NK cells were from the Rheumatology unit of Karolinska University Hospital (eight patients, approved by the Ethics Review Board in Stockholm). Synovial fibroblasts from non-inflamed tissue of healthy donors without RA were purchased from Asterand (two donors, under full compliance with all UK Human Tissue Authority Codes of Practice and directives).
Isolation and culture of cells
The RA-FLS were derived from human synovial tissue as previously described.34 Isolated RA-FLS and healthy FLS were cultured in RPMI-1640 GlutaMAX™ medium (Gibco, Paisley, UK) supplemented with 10% heat-inactivated fetal calf serum (Gibco), 2% heat-inactivated human serum (Sigma-Aldrich, St Louis, MO), 100 IU/ml penicillin, 100 μg/ml streptomycin and 2 nm basic fibroblast growth factor (Invitrogen, Carlsbad, CA).35 For two experiments (Fig. 5e), RA-FLS were isolated from RA SF by adding SF directly to the above cell culture medium, after which RA-FLS adhered to the tissue culture flask. The RA-FLS were passaged at 90% confluency (approximately every 4–7 days) and passaged no more than five times for use in experiments. Nishi is a human NK cell line derived from the peripheral blood mononuclear cells of a boy with chronic active Epstein–Barr virus infection complicated with NK cell leukaemia.36 Nishi NK cells were grown in 24-well plates in IMDM GlutaMAX™ medium (Gibco) supplemented with 10% heat-inactivated fetal calf serum (Gibco), 2% heat-inactivated human serum (Sigma-Aldrich), 100 IU/ml penicillin, 100 μg/ml streptomycin and 10 ng/ml recombinant human IL-15 (PeproTech, Rocky Hill, NJ). NK cells were enriched from RA SF by centrifuging SF, resuspending cells in the above-described medium and enriching for NK cells by negative selection magnetic separation (Miltenyi Biotec, Bergisch Gladbach, Germany). Enriched SF NK cells were cell surface stained and analysed by flow cytometry; or expanded in the same culture medium for up to 3 weeks before co-culture with autologous SF-derived RA-FLS. Mononuclear cells were prepared from paired RA PB and RA SF samples by Ficoll separation (Pharmacia, Stockholm, Sweden), frozen in 10% DMSO (Sigma-Aldrich) and 90% fetal calf serum (Life Technologies, Carlsbad, CA), and stored in liquid nitrogen for further analysis by flow cytometry.
Figure 5.

NKG2D, DNAX accessory molecule 1 (DNAM-1), NKp44 and NKp46 are involved in Nishi natural killer (NK) cell degranulation towards rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS). (a) RA-FLS were seeded at 1 × 104 cells/well in 48-well plates, grown to confluency (approximately 4·8 × 104 cells/well) for 72 hr, and Nishi were added at the indicated ratios overnight. Adherent cells were fixed with 4% paraformaldehyde and stained with eosin. An ImmunoSpot Image Analyzer was used to take images and quantify % of well area covered. Figure and graph are duplicates of one donor, and representative of n = 2 donors. (b) RA-FLS were seeded at 2 × 104 cells/well in 96-well plates, the following day 6 × 103 cells/well Nishi were added to the approximately 4·8 × 104 RA-FLS/well giving an effector : target ratio of 1 : 4. RA-FLS and Nishi were co-cultured overnight. Wells were analysed as described. Representative of n = 3. (c, d) RA-FLS were seeded at 3 × 104 cells/well in 96-well plates, the following day 9 × 104 Nishi/well were added. Nishi and RA-FLS were co-cultured as described and blocked with monoclonal antibodies (mAbs) or isotype-matched control immunoglobulin as indicated. Nishi were identified as viable, single CD3− CD16− CD56+ cells. Data points are a combination of several experiments and a total of n = 8 donors. (e) RA synovial fluid mononuclear cells (SFMC) were enriched for NK cells by antibody-coated bead separation and cultured in interleukin-15 (IL-15) for up to 3 weeks. RA-FLS were seeded at 1·5 × 104 cells/well in 96-well plates, the following day 4·5 × 104 autologous enriched CD3− CD56+ RA SF NK cells were added per well. NK cells and FLS were co-cultured as described. Results from two donors are shown. NK cells were identified as viable, single CD3− CD56+ cells.
Flow cytometry
The following antibodies were used for surface staining of either RA-FLS (detached using 2 mm EDTA) or NK cells at 10 μg/ml or 1 : 100 dilution, together with Near-IR LIVE/DEAD® Fixable Dead Cell Stain (Invitrogen): FITC anti-CD3 (HIT3A, BD Biosciences, Franklin Lakes, NJ), FITC-conjugated anti-CD158a/b (EB6, BD Biosciences), FITC-conjugated anti-CD158b1/b2 (DX27, BD Biosciences), FITC-conjugated anti-CD158e1 (DX9, BD Biosciences), FITC-conjugated anti-DNAX accessory molecule-1 (DNAM-1; DX11, BD Biosciences), phycoerythrin (PE) -conjugated anti-NKG2D (1D11, BD Biosciences), PE-conjugated anti-CD85j (HP-F1, BD Biosciences), PE-conjugated anti-CD244 (2B4, BD Biosciences), PE-Cy7-conjugated anti-CD56 (B159, BD Biosciences), anti-HLA-E (3D12, eBioscience, San Diego, CA), anti-MHC class I (DX17, BD Biosciences), PE-conjugated anti-CD48 (BJ40, BioLegend, San Diego, CA), anti-MIC-A (159227, R&D Systems, Minneapolis, MN), anti-MIC-B (236511, R&D Systems), anti-UL-16 binding protein 1 (ULBP-1; 170818, R&D Systems), anti-ULBP-2/5/6 (165903, R&D Systems), anti-ULBP-3 (166510, R&D Systems), anti-CD112 (R2.525, BD Biosciences), PE-conjugated anti-CD155 (SKII.4, BioLegend), allophycocyanin (APC) -conjugated anti-CD54 (HA58, BD Biosciences), anti-KIR2D (NKVFS1, Miltenyi Biotec), PE-conjugated anti-CD55 (A10, BD Biosciences), APC-conjugated anti-CD68 (Y1/82A, BD Biosciences), anti-HLA-G (87G, BioLegend) and PE-conjugated anti-NKG2C (134591, R&D Systems). Uncojugated antibodies were detected by subsequent incubation with APC-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA). Mouse IgG1, mouse IgG2a and mouse IgG2b were used as isotype controls (R&D Systems). The specificities of the anti-ULBP-1, -2 and -3 monoclonal antibodies (mAbs; R&D Systems) were tested on BA/F3 or K562 cells transfected with ULBP-1, -2, -3, -4 or -6. We found that the anti-ULBP-1 and anti-ULBP-3 mAbs specifically recognize only ULBP-1 and ULBP-3, respectively, whereas anti-ULBP-2 binds ULBP-2, ULBP-5 and ULBP-6 (data not shown). Flow cytometry was performed on a BD LSR II or BD FACSCanto II (BD Biosciences). Data were analysed using FlowJo software (TreeStar Inc., Ashland, OR).
NK degranulation assays
Adherent RA-FLS (passaged up to five times) were trypsinized (Gibco) and seeded at 3 × 104 cells/well in flat-bottom 96-well plates. The next day, NK cells were added at 9 × 104 cells/well. Cells were co-cultured for 5 hr at 37° with FITC-conjugated anti-CD107a mAb (H4A3, BD Biosciences), FITC-conjugated anti-CD107b mAb (H4B4, BD Biosciences) and BD GolgiStop™ containing monensin (BD Biosciences). The following mouse anti-human mAbs were added at 10 μg/ml where indicated: anti-NKG2D (149810, R&D Systems), anti-NKG2A (humanized mAb Z270 (NNC0141-0100), isotype human IgG4, Novo Nordisk A/S, Måløv, Denmark), anti-LIR-1 (GHI/75, BioLegend), anti-2B4 (C1.7, BioLegend), anti-DNAM-1 (DX11, BioLegend), anti-NKp30 (P30-15, BioLegend), anti-NKp44 (P44-8, BioLegend), anti-NKp46 (9E2, BioLegend) and anti-lymphocyte function-associated antigen 1 (LFA-1/CD11a; HI111, BioLegend). Mouse IgG1 (R&D Systems), mouse IgG2b (R&D Systems) and human IgG4 (Sigma-Aldrich) were used as isotype controls. It was verified that none of the antibodies resulted in CD107a/b expression when added to NK cells alone (data not shown). Following the 5-hr CD107 assay, non-adherent cells were stained with Peridinin chlorophyll protein-conjugated anti-CD3 (SK7, BD Biosciences), Pacific Blue-conjugated anti-CD16 (3G8, BD Biosciences), PE-conjugated anti-NKG2A (Z199, Beckman Coulter, Pasadena, CA), Alexa Fluor 700 anti-CD56 (B159, BD Biosciences) and Near-IR LIVE/DEAD® Fixable Dead Cell Stain (Invitrogen). NK cells were gated as viable, single, CD3− CD56+ CD16+ cells. Results are expressed as % CD107a/b+ NK cells.
RA-FLS ‘clearance’ assays
The RA-FLS were trypsinized (Gibco), counted and seeded at 1 × 104 cells/well in 48-well plates, grown to confluency (approximately 4·8 × 104 cells/well) for 72 hr, and Nishi were added at the indicated effector : target ratios overnight. In another experiment, RA-FLS were seeded at 2 × 104 cells/well in 96-well plates, the following day, 6 × 103 cells/well Nishi were added, RA-FLS and Nishi were co-cultured overnight. Following overnight co-cultures of RA-FLS and Nishi, non-adherent cells were removed by washing with PBS, and the remaining adherent cells were fixed with 4% paraformaldehyde and stained with Eosin. The image analysis was performed using a Series CTL-ImmunoSpot S5 Micro Analyzer (Cellular Technology, Bonn, Germany). Images were analysed and quantified using ImmunoSpot software (CTL-Europe, Bonn, Germany).
Statistical analysis
Statistical significances were determined by an analysis of variance test, and a Tukey post-test (determined using GraphPad Prism software; GraphPad Prism, San Diego, CA). All values are given as average ± SEM. Statistical significance is defined as the following: *P < 0·05; **P < 0·01; ***P < 0·001.
Results
Synovial fibroblasts express multiple ligands for NK cell receptors
We first evaluated whether synovial fibroblasts derived during surgery of non-inflamed joints from healthy donors without RA are potential targets for NK cells. The synovial lining layer contains both FLS and macrophage-like synoviocytes.37 The complement-regulatory molecule CD55 has previously been shown to specifically mark FLS located in the synovial lining,38,39 whereas CD68 is a well-known macrophage marker that is present in macrophage-like synoviocytes. Flow cytometry analyses showed that cultured synoviocytes derived from non-inflamed healthy synovial tissue express CD55, but not CD68, consistent with the FLS phenotype (Fig. 1a). We found that FLS from non-inflamed synovium from two donors express numerous ligands for several activating NK cell receptors, including ULBP-1, ULBP-2/5/6 and ULBP-3, but not MIC-A or MIC-B, all known ligands for the activating NK cell receptor NKG2D (Fig. 1b). Among the ligands for the activating receptor DNAM-1, FLS from donors without RA expressed CD155 (poliovirus receptor) but not CD112 (poliovirus-related receptor 2). Efficient cytolytic function and cytokine production by NK cells are dependent on the adhesion molecule integrin β2 LFA-1 binding to CD54 (intercellular adhesion molecule 1) on target cells,40 and we detected high levels of CD54 on non-inflamed FLS; as well as CD48, a ligand for the activating 2B4 (CD244) receptor (Fig. 1c). Next we analysed whether non-inflamed FLS express MHC class I ligands, which can be recognized by inhibitory NK cell receptors, and we found that FLS expressed high levels of classical MHC class I molecules (i.e. HLA-A, -B and -C), but not HLA-E, the latter of which is the ligand for the inhibitory NK cell receptor CD94/NKG2A (Fig. 1d).
Figure 1.
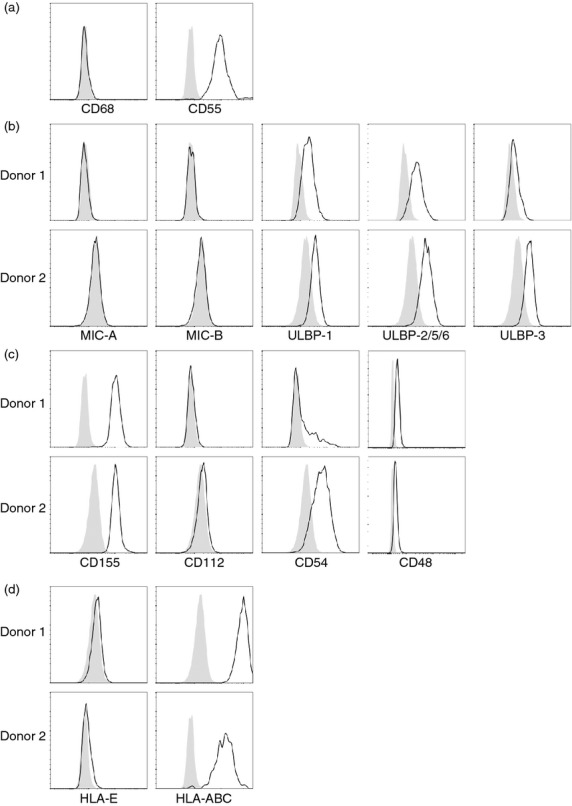
Synovial fibroblasts from non-inflamed synovium express numerous ligands for natural killer (NK) cell receptors. Synovial fibroblasts derived from tissue of non-inflamed joints were surface stained for expression of (a) fibroblast-marker CD55 and macrophage-marker CD68, ligands for activating NK cell receptors (b) NKG2D [MHC class I-chain related protein A,B (MIC-A,B) and UL-16 binding protein 1 (ULBP-1; ULBP-2/5/6,3)] and (c) DNAX accessory molecule 1 (DNAM-1; CD155 and CD112), lymphocyte function-associated antigen 1 (LFA-1; CD54) and 2B4/CD244 (CD48); and (d) ligands for inhibitory NK cell receptors. Filled histograms represent isotype-matched control immunoglobulin. Figure shows stainings from two donors.
To assess whether synovial fibroblasts from an inflammatory setting express similar ligands we raised several short-term adherent cell lines from the small joints of RA synovial tissue biopsies. We confirmed that tissue-derived RA-FLS cultured in vitro are CD68− CD55+ (shown in Fig. 2a for a representative donor) and similar to non-inflamed FLS, the RA-FLS also express the NKG2D ligands ULBP-1, ULBP-2/5/6 and ULBP-3 (Fig. 2b), as well as the DNAM-1 ligand CD155, the LFA-1-ligand CD54 and the 2B4 (CD244) ligand CD48 (Fig. 2c). We detected expression of the NKG2D ligand MIC-A in two of nine RA donors. Furthermore, we found that RA-FLS express both high levels of classical MHC class I molecules (HLA-A, -B, -C), and HLA-E, the ligand for CD94/NKG2A (Fig. 2d).
Figure 2.

Rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) express numerous ligands for natural killer (NK) cell receptors. RA-FLS derived from overgrowth of RA synovial tissue were surface stained for expression of (a) fibroblast-marker CD55 and macrophage-marker CD68 (histograms are representative of n = 3 to n = 5 donors), ligands for activating NK cell receptors (b) NKG2D [MHC class I-chain related protein A,B (MIC-A,B) and UL-16 binding protein 1 (ULBP-1; ULBP-2/5/6,3) and (c) DNAX accessory molecule 1 (DNAM-1; CD155 and CD112), lymphocyte function-associated antigen 1 (LFA-1; CD54) and 2B4/CD244 (CD48); and (d) ligands for inhibitory NK cell receptors. Filled histograms represent isotype-matched control immunoglobulin. Histograms in (b) to (d) are representative of the number of donors indicated in plots of median fluorescence intensity (MFI) beneath.
Overall, FLS from the non-inflamed synovium of healthy donors as well as RA-FLS express numerous ligands for both activating and inhibitory NK cell receptors, and are hence capable of forming a cytolytic synapse. Interestingly, it has recently been shown that both mouse and human NKG2D ligands are subject to regulation by proliferative signals, as serum starvation or inhibition of proliferation (via inhibition of cyclin-dependent kinases by Roscovitine) reduced expression of the NKG2D ligand RAE-1ε on primary mouse fibroblasts and tumour cell lines, as well as MICA/B and ULBP-2 on a human tumour cell line.41 In our hands, serum starvation affected the expression of the detected ligands by RA-FLS in a variable pattern (see Supporting information; Fig. S1a). However, we found that inhibition of proliferation by Roscovitine did reduce, but not abrogate, the expression of ULBP-1 and ULBP-2/5/6 by RA-FLS (Fig. S1b). This suggests that the expression of CD155, CD54, HLA-E and HLA-A, -B, -C on RA-FLS is not regulated by proliferative signals, whereas the expression of ULBP-1 and ULBP-2/5/6 is partially regulated in connection with proliferation.
Characterization of SF NK cell receptor repertoire
It has been previously reported that RA SF NK cells primarily exhibit a CD56bright phenotype, and are phenotypically similar to the CD56bright NK cell subset in PB.29,30 We characterized the receptor repertoire of NK cells from paired peripheral blood mononuclear cells and SF mononuclear cells (SFMC) samples from RA patients, and confirmed that RA SF NK cells exhibit a CD56bright phenotype and express LIR-1 and KIR to a lower frequency and CD94/NKG2A to a higher frequency, compared with RA PB NK cells (Fig. 3). Furthermore, similarly to RA PB NK cells, RA SF NK cells are primarily DNAM-1+ NKG2D+, whereas the expression of 2B4 or NKG2C were more heterogeneous in the RA SF NK cell population compared with NK cells derived from PB (Fig. 3).
Figure 3.

Expression of natural killer (NK) cell receptors on paired rheumatoid arthritis synovial fluid mononuclear cell (RA SFMC) and peripheral blood mononuclear cell (PBMC) samples. Frozen samples of paired SFMC and PBMC from n = 4 to n = 8 RA donors were stained for expression of various NK cell receptors as indicated. Dot plots in (a) are representative examples of NK receptor expression and (b) shows % NK cells positive for a given receptor, relative to isotype-matched control immunoglobulin. NK cells were gated as viable, single, CD3− CD14− CD56+ cells.
We next wished to analyse the receptor–ligand interactions between NK cells and RA-FLS. Due to difficulties in isolating sufficient numbers of RA SF NK cells to perform such experiments, we evaluated whether the human NK cell line Nishi could serve as an appropriate effector cell model. We analysed the expression of NK cell receptors on Nishi cells and found that they are phenotypically nearly identical to RA SF NK cells, in that Nishi cells express the activating receptors NKG2D, DNAM-1, 2B4 and LFA-1 (Fig. 4a). Previous studies have shown that RA SF NK cells also express the natural cytotoxicity receptors (NCRs) NKp30 and NKp44, the latter of which is an activation marker.42 Both NKp30 and NKp44, as well as NKp46, were also expressed by Nishi cells (Fig. 4a). Similarly to RA SF NK cells, Nishi cells are also KIR2D−CD94/NKG2A+ (Fig. 4b). In contrast, although we could only detect a small subset of RA SF NK cells positive for LIR-1, all Nishi cells express this inhibitory receptor (Figs 3 and 4). Based on these phenotypic similarities, we reasoned that Nishi cells could mimic RA SF NK cells in an in vitro set-up aimed at investigating the receptor–ligand interactions between NK cells and RA-FLS.
Figure 4.
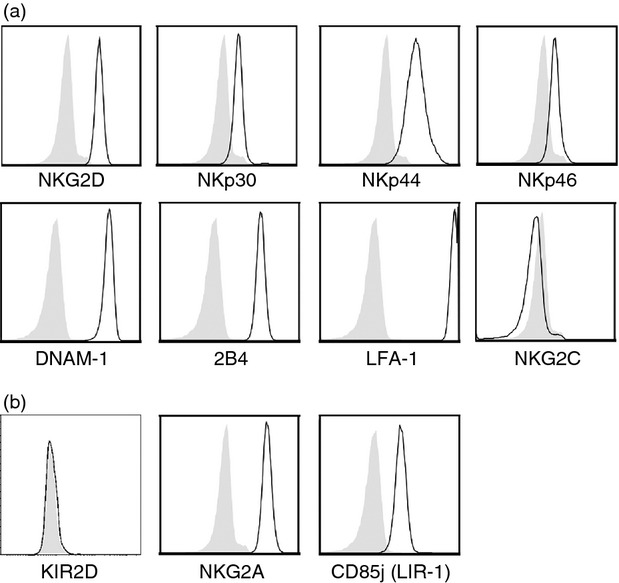
Expression of natural killer (NK) cell receptors by the Nishi NK cell line. Nishi NK cells were stained for surface expression of (a) activating and (b) inhibitory NK cell receptors (black line) or isotype-matched control immunoglobulin (filled histogram). Nishi NK cells were gated as viable, single CD56+ cells.
Nishi cells induce clearance of RA-FLS in an in vitro culture
To assess NK cell interactions with RA-FLS, the latter were grown to confluence. Subsequently Nishi were added at different effector : target (E : T) ratios for a 24-hr co-culture. As shown in Fig. 5(a), an almost complete disappearance of adherent RA-FLS was observed at an E : T of 1 : 1, whereas decreasing the E : T ratio led to a gradual increase in the number of adherent RA-FLS (Fig. 5a). These results suggest that Nishi cells are mediating ‘clearance’ of adherent RA-FLS in vitro, and although previous studies suggest that adherent cells detach following apoptosis induction in vitro,43 further experiments are required to confirm that this clearance of RA-FLS is indeed a result of Nishi NK cell-mediated cytotoxicity.
Assuming that this ‘clearance’ of RA-FLS is dependent on receptor–ligand interactions between RA-FLS and Nishi cells, we used an anti-human NKG2A mAb to block the interaction between the inhibitory receptor CD94/NKG2A and its ligand HLA-E, expressed on RA-FLS. We reasoned that blocking this inhibitory signal in an overnight Nishi/RA-FLS co-culture would lead to increased activation of Nishi cells, and therefore also to an increased clearance of RA-FLS, as CD94/NKG2A is the primary inhibitory NK cell receptor expressed by both RA SF NK cells and Nishi cells. As shown in Fig. 5(b), blocking the interaction between CD94/NKG2A and its ligand HLA-E with an anti-NKG2A mAb did indeed lead to increased clearance of RA-FLS.
Nishi cells degranulate in response to RA-FLS via multiple activating receptors
To confirm that Nishi/RA-FLS interactions result in activation of NK cell functions, i.e. cytotoxic responses against RA-FLS, we analysed cell surface expression of CD107a [lysosomal-associated membrane protein (LAMP-1)], and CD107b (LAMP-2), molecules, which become exposed on the NK cell surface following exocytosis of lytic granules,44 and which are known to correlate with specific target cell lysis. As shown in Fig. 5(c), Nishi cells cultured alone do not degranulate (1·4 ± 0·2% are CD107a/b+), but an average of 12·0 ± 1·4% Nishi cells degranulate in response to RA-FLS, supporting the theory that RA-FLS trigger Nishi NK cell activation. In an identical set-up using FLS derived from the non-inflamed synovium of two healthy donors, we found that non-RA FLS can also stimulate Nishi NK cell degranulation, as 15·1% and 6·2% of Nishi NK cells are CD107a/b+ following co-culture with healthy donors 1 and 2, respectively (see Supporting information, Fig. S2).
Successful triggering of NK cell functions results from an integration of both activating and inhibitory signals.45 To evaluate which receptor–ligand interactions are involved in the NK/RA-FLS cross-talk, a set of neutralizing mAbs targeting various activating and inhibitory receptors was added to the co-cultures described above. The antibodies were chosen to cover a panel of NK cell receptors shown to be expressed on RA SF NK cells, as well as on Nishi cells (Fig. 5d). The addition of these antibodies, or immunoglobulin-matched isotype controls, to Nishi cells in the absence of RA-FLS did not result in Nishi cell degranulation (data not shown). As shown in Fig. 5(d), the addition of anti-NKG2D reduced Nishi cell degranulation by an average of 46·4% ± 6·5%, relative to isotype control. Moreover, masking the NCRs NKp44 and NKp46 reduced degranulation by 21·8% ± 2·5% and 49·2% ± 9·0%, respectively. The activating receptor DNAM-1 also plays an important role in the interaction between Nishi cells and RA-FLS, as adding an anti-DNAM-1 mAb to the co-culture reduced degranulation by 57·3% ± 7·9%. Furthermore, we found that blocking the interaction between the inhibitory CD94/NKG2A receptor and its ligand HLA-E enhanced Nishi cell degranulation towards RA-FLS by 82·9% ± 20·0% relative to isotype control (Fig. 5d). Masking the NCR NKp30, the 2B4 receptor or the inhibitory LIR-1 receptor did not affect Nishi degranulation towards RA-FLS (data not shown). Overall, this suggests that NKG2D, DNAM-1, NKp44 and NKp46 are involved in Nishi degranulation towards RA-FLS, and that RA-FLS expression of the ligand for CD94/NKG2A, HLA-E, inhibits Nishi cell degranulation.
IL-15-activated RA SF NK cells degranulate in response to autologous RA-FLS in in vitro co-cultures
Another approach taken to assess the interaction between RA-FLS and RA SF NK cell cross-talk was to derive in vitro RA-FLS and RA SF NK cells from the same SF sample to operate in an autologous set-up. We were able, from SF samples from two RA patients, to derive RA-FLS and enrich the SFMC for RA SF NK cells using IL-15-stimulated SF cultures for up to 3 weeks. We reasoned that IL-15 stimulation would mimic the inflammatory conditions in the RA joint as IL-15 has been shown to be in abundance in an RA joint.46,47 These IL-15-stimulated RA-SFMC were co-cultured with autologous RA-FLS in a CD107 assay, and NK cells were subsequently identified as CD3− CD56+ IL-15-activated SFMC. These IL-15-stimulated RA SF NK cells degranulated in response to autologous RA-FLS, and masking CD94/NKG2A with an anti-NKG2A mAb led to a significant increase in NK cell degranulation in both samples (Fig. 5e). This suggests that RA SF NK cells, which have been IL-15-activated in vitro, can also degranulate in response to RA-FLS, a process that is further enhanced upon blocking CD94/NKG2A.
Discussion
It is becoming increasingly appreciated that NK cells play an important role in regulating adaptive immune responses. In this study, we have set-up a model system to investigate NK cell/RA-FLS interactions in vitro. We have shown that the human NK cell line Nishi degranulates upon co-culture with RA-FLS in vitro, and found that DNAM-1 and NKG2D are key activating NK cell receptors involved in mediating this response, because blocking these receptors with mAbs drastically reduced Nishi NK cell degranulation. NKG2D and DNAM-1 are well-characterized receptors, which have previously been shown to play a role in NK cell-mediated lysis of macrophages,48 tumour cells,49 microglia,50 dendritic cells,51 activated CD4+ T cells52–56 and regulatory T cells.57 The NCRs NKp44 and NKp46 are also involved in NK cell recognition of RA-FLS, because blocking these receptors also reduced Nishi cell degranulation.
We detected ligands for NKG2D and DNAM-1 on RA-FLS from all donors. Of the NKG2D ligands, we detected surface expression of ULBP-1 and ULBP-2/5/6, but not MIC-A/B, despite previous studies showing expression of MIC-A/B in RA synoviocytes by immunohistochemistry.32,33 This discrepancy could be due to a low level of MIC-A/B expression, which can be detected by immunohistochemistry but not by flow cytometry. Our findings suggest that further studies using ULBP- and MIC-specific mAbs should be performed to conclude which members of the NKG2D-ligand family are present at inflammatory sites in RA. Endogenous ligands for the NCRs NKp44 and NKp46 are still obscure, although viral haemagglutinins have been proposed as possible ligands,58,59 but attempts to identify the ligand(s) on RA-FLS have so far been unsuccessful. Ligands for activating NK cell receptors are typically induced upon cellular stress, viral infection or tumorigenesis.60 Our data suggest that the expression of the NKG2D ligands ULBP-1 and ULBP-2/5/6 on RA-FLS can be regulated by proliferative signals, which supports the theory that RA-FLS may indeed express these ligands in vivo, as RA-FLS are highly proliferative in RA synovium.61 A recent report using mouse fibroblasts41 showed that the expression of these ligands is also affected by serum starvation. In our hands there was only a variable effect of serum starvation on ligand expression. Interestingly, we could detect expression of the same ligands for activating NK receptors on FLS derived from non-inflamed synovium of two healthy donors, suggesting that the expression of these ligands on RA-FLS may not be disease-specific, as they may also be up-regulated as a cellular stress response following isolation, culture and passaging of FLS in vitro. Indeed, our finding that Nishi NK cells also degranulate in response to FLS derived from non-inflamed synovium, despite a limitation of only having FLS from two healthy donors in this study, emphasizes that further experiments are required to analyse freshly isolated FLS from both RA and healthy donors, which have not been cultured in vitro, for expression of these ligands, as well as their ability to trigger NK cell cytotoxicity. It remains to be seen whether the pathogenic characteristics of RA-FLS are a result of the chronic inflammatory environment, or whether these cells are intrinsically abnormal. A previous study, using a SCID mouse model of RA, showed that RA-FLS are able to degrade normal articular cartilage in the absence of immune cells and a pro-inflammatory environment,3 suggesting that RA-FLS are of an autonomous, activated phenotype.
We could not find a correlation between expression level of HLA-E on RA-FLS, and the effect of blocking with an anti-NKG2A mAb. This is not surprising, however, as HLA-E is only expressed at the cell surface in conjunction with bound peptides, typically leader peptides derived from signal sequences of classical MHC class I molecules.62 The nature of the bound peptide affects the stability and cell surface expression of the HLA-E/peptide complex,63 which subsequently determines the extent to which it is recognized by NKG2A, or an anti-HLA-E antibody. The expression level of HLA-E detected by flow cytometry is therefore not necessarily indicative of the strength of its engagement with NKG2A.
Rheumatoid arthritis SF NK cells are known to exhibit a CD56bright NKG2A+KIR− phenotype30 and produce high levels of cytokines following IL-2 stimulation.29 Although PB CD56bright NK cells are typically cited as being less cytotoxic compared with CD56dim NK cells due to lower levels of perforin, granzymes and cytolytic granules,64 numerous studies have shown that, following cytokine activation, CD56bright NK cells are equally, if not more, cytotoxic compared with CD56dim NK cells.23,56,65 Indeed, we found not only that RA SF NK cells have a CD56bright NKG2A+KIR− phenotype, but also that they are LIR-1− DNAM-1+ NKG2D+. Natural killer cells with a CD56bright phenotype are found at inflammatory sites in a number of diseases,27,30,66–69 and an expansion in CD56bright NK cells has been associated with clinical remission in autoimmune diseases such as multiple sclerosis,70–72 implicating an immune-regulatory role for CD56bright NK cells in chronic inflammation. In addition to their CD56bright NKG2A+ KIR− phenotype,30 RA SF NK cells have also been shown to be CD69+ NKp44+,29 suggesting an activated phenotype. Under steady-state conditions, NK cells in healthy donors are resting, and hence CD69− NKp44−, highlighting an important difference between RA SF NK cells and healthy resting NK cells. This suggests that despite our finding that Nishi NK cells also degranulated in response to healthy FLS in vitro, this is unlikely to happen in vivo, for two reasons: first, normal non-inflamed synovium is relatively acellular and contains very few lymphocytes73 and it is therefore unlikely that there would be any NK cells present; and second, NK cells under resting conditions will not elicit cytotoxicity towards healthy FLS, even if the latter express ligands for activating NK cell receptors, as NK cells require cytokines for efficient activation. Further studies are required to assess whether primary RA SF NK cells, as well as primary resting NK cells from healthy donors, can kill freshly isolated FLS from both healthy donors and RA patients ex vivo. The accumulation of CD56bright NK cells in RA SF may be a result of preferential recruitment from blood, local differentiation, or selective survival. CD56bright NK cells have been shown to have a higher resistance to apoptosis and to survive better in oxidant-rich surroundings,74,75 such as in an inflamed RA joint.
The net outcome of the cellular interactions between NK cells and RA-FLS in an inflamed synovium remains unclear. A previous study has shown that cell–cell contact between RA-FLS and NK cells stimulates NK cell survival, migration, and interferon-γ production, as well as the production of pro-inflammatory chemokines and matrix metalloproteinases.31 RA-FLS constitutively express IL-15,76,77 a cytokine that enhances NK cell cytotoxicity and proliferation.78 These studies imply that RA-FLS/NK contact-dependent and -independent interactions may influence joint inflammation at numerous levels. Despite a low number of donors available in this study, our results suggest that RA-FLS can stimulate degranulation of Nishi NK cells as well as autologous IL-15-stimulated RA SF NK cells in vitro. Further studies are required to confirm that NK cell degranulation leads to RA-FLS apoptosis, and not simply cell detachment in vitro. One can speculate that NK cells may also be able to kill RA-FLS in an inflamed RA joint in vivo, and that exploitation of the cytotoxic potential of NK cells by blocking CD94/NKG2A with an anti-NKG2A mAb may potentially yield an opportunity for therapeutic treatment of chronic inflammation. Indeed, interrupting the interaction between Qa-1 (the equivalent of HLA-E in rodents) and CD94/NKG2A, either with a genetic mutation or with a blocking anti-NKG2A F(ab')2 fragment, in mouse models of multiple sclerosis (experimental-allergic encephalomyelitis) and RA (collagen-induced arthritis), resulted in reduced activated CD4+ T-cell numbers and disease amelioration in a perforin-dependent fashion,18,19,21 supporting the hypothesis that promoting NK cell-mediated elimination of pro-inflammatory immune cells may also be a potential treatment strategy in humans. A prerequisite, however, for NK cell-mediated killing is expression of relevant ligands for activating receptors, and further studies by immunohistochemistry are required to confirm that RA-FLS express the relevant ligands for activating and inhibitory NK cell receptors in situ. Furthermore, as this study was based on a model system of Nishi NK cell degranulation towards cultured RA-FLS in vitro, further functional studies are required to confirm that primary RA SF NK cells can kill freshly isolated RA-FLS ex vivo.
The cellular interactions between NK cells, RA-FLS, and other immune cells in the synovium of a chronically inflamed joint, and how these interactions subsequently influence disease progression, remain unclear. This study is the first to suggest that NK cells may play a role in the elimination of RA-FLS, a process that is enhanced upon blocking the ability of HLA-E to engage the inhibitory CD94/NKG2A NK cell receptor. One can speculate that NK cell-mediated cytotoxicity of RA-FLS may therefore be one mechanism through which NK cells are involved in the regulation of local inflammation in RA.
Acknowledgments
We wish to thank Dr Lars Juul at Rigshospitalet for RA SF samples; Jesper Kastrup and Christina Stage for assistance in acquiring RA SFMC samples; and Jette Møller Frøsig for excellent technical assistance. EMB and HB were supported by the OAK Foundation. AF, YS, VM and LB were supported by the Swedish Research Council and the King Gustaf V 80 Year Foundation. NN and MA were supported by Novo Nordisk A/S and the Danish Agency for Science, Technology and Innovation.
Disclosures
EDG, PS and KS have assigned patents related to NKG2A and NNC0141-0100 (WO2007042573, 20070726; WO2007147898, 20071227; WO2008009545, 20080124; WO2009092805, 20090730).
Henning Bliddal has received a fee for participation as speaker at a symposium organized by Pfizer and has received research grants from Abbvie, Pfizer, Roche, Novo Nordisk A/S.
Véronique Pascal works for Novo Nordisk A/S and owns Novo Nordisk shares. No other conflicts of interest.
Natasja Nielsen received funding from Novo Nordisk A/S for her PhD, is currently employed by Novo Nordisk A/S and owns Novo Nordisk shares. No other conflicts of interest.
Elisabeth Douglas Galsgaard is employed by Novo Nordisk A/S and holds shares in Novo Nordisk A/S.
Pieter Spee at the time of the study was employed by Novo Nordisk A/S where the studies were performed and owns shares in Novo Nordisk A/S.
During the time of drafting the manuscript AERF has been employed as associated researcher at the Rheumatology Unit at Karolinska Institutet, Sweden, in parallel with a position at Novartis. The research done at Karolinska Institutet was independent and has not been in conflict with the work done at Novartis.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Effect of serum-free culture conditions and inhibition of proliferation on expression of ligands on rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS). (a) RA-FLS were cultured under normal conditions (RPMI + 10% fetal calf serum + 2% human serum + 2 nm basic fibroblast growth factor) or in FibroGRO serum-free media for 5 days, and were surface stained for expression of ligands as indicated. Plots indicate median fluorescence intensity (MFI) for n = 4 donors. (b) RA-FLS were cultured under normal conditions or with the addition of 25 μm Roscovitine to inhibit proliferation for 5 days, and subsequently surface stained for expression of ligands as indicated. Plots indicate MFI for six donors.
Figure S2. Nishi natural killer (NK) cells degranulate in response to fibroblast-like synoviocytes (FLS) derived from non-inflamed synovium of two healthy donors. FLS were seeded at 3 × 104 cells/well in 96-well plates, the following day 9 × 104 Nishi/well were added. Nishi and FLS were co-cultured as described. CD107a/b expression was detected on Nishi cells by flow cytometry. Nishi were gated as viable, single CD3− CD16− CD56+ cells. Nishi cultured alone (without FLS) did not express CD107a/b (data not shown).
References
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Zvaifler NJ, Boyle D, Firestein GS. Early synovitis–synoviocytes and mononuclear cells. Semin Arthritis Rheum. 1994;23:11–6. doi: 10.1016/0049-0172(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 3.Muller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–15. [PMC free article] [PubMed] [Google Scholar]
- 4.Niedermeier M, Pap T, Korb A. Therapeutic opportunities in fibroblasts in inflammatory arthritis. Best Pract Res Clin Rheumatol. 2010;24:527–40. doi: 10.1016/j.berh.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Pierer M, Rethage J, Seibl R, et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol. 2004;172:1256–65. doi: 10.4049/jimmunol.172.2.1256. [DOI] [PubMed] [Google Scholar]
- 6.Bradfield PF, Amft N, Vernon-Wilson E, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheum. 2003;48:2472–82. doi: 10.1002/art.11219. [DOI] [PubMed] [Google Scholar]
- 7.Nanki T, Nagasaka K, Hayashida K, Saita Y, Miyasaka N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Immunol. 2001;167:5381–5. doi: 10.4049/jimmunol.167.9.5381. [DOI] [PubMed] [Google Scholar]
- 8.Rathanaswami P, Hachicha M, Sadick M, Schall TJ, McColl SR. Expression of the cytokine RANTES in human rheumatoid synovial fibroblasts. Differential regulation of RANTES and interleukin-8 genes by inflammatory cytokines. J Biol Chem. 1993;268:5834–9. [PubMed] [Google Scholar]
- 9.Carrion M, Perez-Garcia S, Jimeno R, et al. Inflammatory mediators alter interleukin-17 receptor, interleukin-12 and -23 expression in human osteoarthritic and rheumatoid arthritis synovial fibroblasts: immunomodulation by vasoactive intestinal peptide. NeuroImmunoModulation. 2013;20:274–84. doi: 10.1159/000350892. [DOI] [PubMed] [Google Scholar]
- 10.Gillgrass A, Ashkar A. Stimulating natural killer cells to protect against cancer: recent developments. Expert Rev Clin Immunol. 2011;7:367–82. doi: 10.1586/eci.10.102. [DOI] [PubMed] [Google Scholar]
- 11.Sun JC, Beilke JN, Bezman NA, Lanier LL. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med. 2011;208:357–68. doi: 10.1084/jem.20100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhat R, Rommelaere J. NK-cell-dependent killing of colon carcinoma cells is mediated by natural cytotoxicity receptors (NCRs) and stimulated by parvovirus infection of target cells. BMC Cancer. 2013;13:367. doi: 10.1186/1471-2407-13-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dayanc BE, Bansal S, Gure AO, Gollnick SO, Repasky EA. Enhanced sensitivity of colon tumour cells to natural killer cell cytotoxicity after mild thermal stress is regulated through HSF1-mediated expression of MICA. Int J Hyperthermia. 2013;29:480–90. doi: 10.3109/02656736.2013.821526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soderstrom K, Stein E, Colmenero P, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci U S A. 2010;107:13028–33. doi: 10.1073/pnas.1000546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez NC, Lozier A, Flament C, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. 1999;5:405–11. doi: 10.1038/7403. [DOI] [PubMed] [Google Scholar]
- 16.Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. 2002;195:327–33. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morandi B, Bougras G, Muller WA, Ferlazzo G, Munz C. NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-γ secretion. Eur J Immunol. 2006;36:2394–400. doi: 10.1002/eji.200636290. [DOI] [PubMed] [Google Scholar]
- 18.Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity. 2007;26:593–604. doi: 10.1016/j.immuni.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leavenworth JW, Wang X, Wenander CS, Spee P, Cantor H. Mobilization of natural killer cells inhibits development of collagen-induced arthritis. Proc Natl Acad Sci U S A. 2011;108:14584–9. doi: 10.1073/pnas.1112188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu L, Kim HJ, Werneck MB, Cantor H. Regulation of CD8+ regulatory T cells: interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc Natl Acad Sci U S A. 2008;105:19420–5. doi: 10.1073/pnas.0810383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leavenworth JW, Schellack C, Kim HJ, Lu L, Spee P, Cantor H. Analysis of the cellular mechanism underlying inhibition of EAE after treatment with anti-NKG2A F(ab')2. Proc Natl Acad Sci U S A. 2010;107:2562–7. doi: 10.1073/pnas.0914732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanier LL, Le AM, Civin CI, Loken MR, Phillips JH. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J Immunol. 1986;136:4480–6. [PubMed] [Google Scholar]
- 23.Nagler A, Lanier LL, Cwirla S, Phillips JH. Comparative studies of human FcRIII-positive and negative natural killer cells. J Immunol. 1989;143:3183–91. [PubMed] [Google Scholar]
- 24.Gogali F, Paterakis G, Rassidakis GZ, Liakou CI, Liapi C. CD3–CD16–CD56bright immunoregulatory NK cells are increased in the tumor microenvironment and inversely correlate with advanced stages in patients with papillary thyroid cancer. Thyroid. 2013;23:1561–8. doi: 10.1089/thy.2012.0560. [DOI] [PubMed] [Google Scholar]
- 25.Fehniger TA, Cooper MA, Nuovo GJ, et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood. 2003;101:3052–7. doi: 10.1182/blood-2002-09-2876. [DOI] [PubMed] [Google Scholar]
- 26.Ferlazzo G, Munz C. NK cell compartments and their activation by dendritic cells. J Immunol. 2004;172:1333–9. doi: 10.4049/jimmunol.172.3.1333. [DOI] [PubMed] [Google Scholar]
- 27.Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A. CD56brightCD16– NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol. 2006;36:118–28. doi: 10.1002/eji.200535243. [DOI] [PubMed] [Google Scholar]
- 28.Carrega P, Morandi B, Costa R, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56bright CD16– cells and display an impaired capability to kill tumor cells. Cancer. 2008;112:863–75. doi: 10.1002/cncr.23239. [DOI] [PubMed] [Google Scholar]
- 29.de Matos CT, Berg L, Michaelsson J, Fellander-Tsai L, Karre K, Soderstrom K. Activating and inhibitory receptors on synovial fluid natural killer cells of arthritis patients: role of CD94/NKG2A in control of cytokine secretion. Immunology. 2007;122:291–301. doi: 10.1111/j.1365-2567.2007.02638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright, CD94bright, CD158– phenotype. Rheumatology (Oxford) 2003;42:870–8. doi: 10.1093/rheumatology/keg240. [DOI] [PubMed] [Google Scholar]
- 31.Chan A, Filer A, Parsonage G, et al. Mediation of the proinflammatory cytokine response in rheumatoid arthritis and spondylarthritis by interactions between fibroblast-like synoviocytes and natural killer cells. Arthritis Rheum. 2008;58:707–17. doi: 10.1002/art.23264. [DOI] [PubMed] [Google Scholar]
- 32.Schrambach S, Ardizzone M, Leymarie V, Sibilia J, Bahram S. In vivo expression pattern of MICA and MICB and its relevance to auto-immunity and cancer. PLoS ONE. 2007;2:e518. doi: 10.1371/journal.pone.0000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2003;100:9452–7. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nozaki T, Takahashi K, Ishii O, et al. Development of an ex vivo cellular model of rheumatoid arthritis: critical role of CD14-positive monocyte/macrophages in the development of pannus tissue. Arthritis Rheum. 2007;56:2875–85. doi: 10.1002/art.22849. [DOI] [PubMed] [Google Scholar]
- 35.Qu Z, Huang XN, Ahmadi P, et al. Expression of basic fibroblast growth factor in synovial tissue from patients with rheumatoid arthritis and degenerative joint disease. Lab Invest. 1995;73:339–46. [PubMed] [Google Scholar]
- 36.Cerboni C, Mousavi-Jazi M, Wakiguchi H, Carbone E, Karre K, Soderstrom K. Synergistic effect of IFN-γ and human cytomegalovirus protein UL40 in the HLA-E-dependent protection from NK cell-mediated cytotoxicity. Eur J Immunol. 2001;31:2926–35. doi: 10.1002/1521-4141(2001010)31:10<2926::aid-immu2926>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Iwanaga T, Shikichi M, Kitamura H, Yanase H, Nozawa-Inoue K. Morphology and functional roles of synoviocytes in the joint. Arch Histol Cytol. 2000;63:17–31. doi: 10.1679/aohc.63.17. [DOI] [PubMed] [Google Scholar]
- 38.Palmer DG, Selvendran Y, Allen C, Revell PA, Hogg N. Features of synovial membrane identified with monoclonal antibodies. Clin Exp Immunol. 1985;59:529–38. [PMC free article] [PubMed] [Google Scholar]
- 39.Stevens CR, Mapp PI, Revell PA. A monoclonal antibody (Mab 67) marks type B synoviocytes. Rheumatol Int. 1990;10:103–6. doi: 10.1007/BF02274823. [DOI] [PubMed] [Google Scholar]
- 40.Helander TS, Timonen T. Adhesion in NK cell function. Curr Top Microbiol Immunol. 1998;230:89–99. doi: 10.1007/978-3-642-46859-9_7. [DOI] [PubMed] [Google Scholar]
- 41.Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med. 2012;209:2409–22. doi: 10.1084/jem.20120565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantoni C, Bottino C, Vitale M, et al. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J Exp Med. 1999;189:787–96. doi: 10.1084/jem.189.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kulkarni GV, McCulloch CA. Serum deprivation induces apoptotic cell death in a subset of BALB/c 3T3 fibroblasts. J Cell Sci. 1994;107(Pt 5):1169–79. doi: 10.1242/jcs.107.5.1169. [DOI] [PubMed] [Google Scholar]
- 44.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 46.McInnes IB, al-Mughales J, Field M, et al. The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nat Med. 1996;2:175–82. doi: 10.1038/nm0296-175. [DOI] [PubMed] [Google Scholar]
- 47.McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat Med. 1997;3:189–95. doi: 10.1038/nm0297-189. [DOI] [PubMed] [Google Scholar]
- 48.Schulz U, Kreutz M, Multhoff G, et al. Interleukin-10 promotes NK cell killing of autologous macrophages by stimulating expression of NKG2D ligands. Scand J Immunol. 2010;72:319–31. doi: 10.1111/j.1365-3083.2010.02435.x. [DOI] [PubMed] [Google Scholar]
- 49.El-Sherbiny YM, Meade JL, Holmes TD, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007;67:8444–9. doi: 10.1158/0008-5472.CAN-06-4230. [DOI] [PubMed] [Google Scholar]
- 50.Lunemann A, Lunemann JD, Roberts S, et al. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J Immunol. 2008;181:6170–7. doi: 10.4049/jimmunol.181.9.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magri G, Muntasell A, Romo N, et al. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood. 2011;117:848–56. doi: 10.1182/blood-2010-08-301374. [DOI] [PubMed] [Google Scholar]
- 52.Andresen L, Hansen KA, Jensen H, et al. Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J Immunol. 2009;183:897–906. doi: 10.4049/jimmunol.0803014. [DOI] [PubMed] [Google Scholar]
- 53.Vales-Gomez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008;68:1546–54. doi: 10.1158/0008-5472.CAN-07-2973. [DOI] [PubMed] [Google Scholar]
- 54.Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood. 2007;110:606–15. doi: 10.1182/blood-2006-10-052720. [DOI] [PubMed] [Google Scholar]
- 55.Molinero LL, Domaica CI, Fuertes MB, Girart MV, Rossi LE, Zwirner NW. Intracellular expression of MICA in activated CD4 T lymphocytes and protection from NK cell-mediated MICA-dependent cytotoxicity. Hum Immunol. 2006;67:170–82. doi: 10.1016/j.humimm.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 56.Nielsen N, Odum N, Urso B, Lanier LL, Spee P. Cytotoxicity of CD56bright NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS ONE. 2012;7:e31959. doi: 10.1371/journal.pone.0031959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy S, Barnes PF, Garg A, Wu S, Cosman D, Vankayalapati R. NK cells lyse T regulatory cells that expand in response to an intracellular pathogen. J Immunol. 2008;180:1729–36. doi: 10.4049/jimmunol.180.3.1729. [DOI] [PubMed] [Google Scholar]
- 58.Mandelboim O, Lieberman N, Lev M, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055–60. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 59.Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debre P, Vieillard V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood. 2013;122:2935–42. doi: 10.1182/blood-2013-03-489054. [DOI] [PubMed] [Google Scholar]
- 60.Bottino C, Castriconi R, Moretta L, Moretta A. Cellular ligands of activating NK receptors. Trends Immunol. 2005;26:221–6. doi: 10.1016/j.it.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Kurowska M, Rudnicka W, Kontny E, et al. Fibroblast-like synoviocytes from rheumatoid arthritis patients express functional IL-15 receptor complex: endogenous IL-15 in autocrine fashion enhances cell proliferation and expression of Bcl-x(L) and Bcl-2. J Immunol. 2002;169:1760–7. doi: 10.4049/jimmunol.169.4.1760. [DOI] [PubMed] [Google Scholar]
- 62.Pietra G, Romagnani C, Manzini C, Moretta L, Mingari MC. The emerging role of HLA-E-restricted CD8+ T lymphocytes in the adaptive immune response to pathogens and tumors. J Biomed Biotechnol. 2010;2010:907092. doi: 10.1155/2010/907092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brooks AG, Borrego F, Posch PE, et al. Specific recognition of HLA-E, but not classical, HLA class I molecules by soluble CD94/NKG2A and NK cells. J Immunol. 1999;162:305–13. [PubMed] [Google Scholar]
- 64.Jacobs R, Hintzen G, Kemper A, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. 2001;31:3121–7. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 65.Ellis TM, Fisher RI. Functional heterogeneity of Leu 19bright+ and Leu 19dim+ lymphokine-activated killer cells. J Immunol. 1989;142:2949–54. [PubMed] [Google Scholar]
- 66.Tak PP, Kummer JA, Hack CE, et al. Granzyme-positive cytotoxic cells are specifically increased in early rheumatoid synovial tissue. Arthritis Rheum. 1994;37:1735–43. doi: 10.1002/art.1780371205. [DOI] [PubMed] [Google Scholar]
- 67.Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol. 2004;173:6418–26. doi: 10.4049/jimmunol.173.10.6418. [DOI] [PubMed] [Google Scholar]
- 68.Katchar K, Soderstrom K, Wahlstrom J, Eklund A, Grunewald J. Characterisation of natural killer cells and CD56+ T-cells in sarcoidosis patients. Eur Respir J. 2005;26:77–85. doi: 10.1183/09031936.05.00030805. [DOI] [PubMed] [Google Scholar]
- 69.Schierloh P, Yokobori N, Aleman M, et al. Increased susceptibility to apoptosis of CD56dimCD16+ NK cells induces the enrichment of IFN-γ-producing CD56bright cells in tuberculous pleurisy. J Immunol. 2005;175:6852–60. doi: 10.4049/jimmunol.175.10.6852. [DOI] [PubMed] [Google Scholar]
- 70.Saraste M, Irjala H, Airas L. Expansion of CD56Bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-β. Neurol Sci. 2007;28:121–6. doi: 10.1007/s10072-007-0803-3. [DOI] [PubMed] [Google Scholar]
- 71.Vandenbark AA, Huan J, Agotsch M, et al. Interferon-β1a treatment increases CD56bright natural killer cells and CD4+ CD25+ Foxp3 expression in subjects with multiple sclerosis. J Neuroimmunol. 2009;215:125–8. doi: 10.1016/j.jneuroim.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 72.Wiendl H, Gross CC. Modulation of IL-2Rα with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol. 2013;9:394–404. doi: 10.1038/nrneurol.2013.95. [DOI] [PubMed] [Google Scholar]
- 73.Smith MD. The normal synovium. Open Rheumatol J. 2011;5:100–6. doi: 10.2174/1874312901105010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harlin H, Hanson M, Johansson CC, et al. J Immunol. 2007;179:4513–9. doi: 10.4049/jimmunol.179.7.4513. [DOI] [PubMed] [Google Scholar]
- 75.Thoren FB, Romero AI, Hermodsson S, Hellstrand K. The CD16–/CD56bright subset of NK cells is resistant to oxidant-induced cell death. J Immunol. 2007;179:781–5. doi: 10.4049/jimmunol.179.2.781. [DOI] [PubMed] [Google Scholar]
- 76.Harada S, Yamamura M, Okamoto H, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1508–16. doi: 10.1002/1529-0131(199907)42:7<1508::AID-ANR26>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 77.Miranda-Carus ME, Balsa A, Benito-Miguel M, de Perez AC, Martin-Mola E. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate. J Immunol. 2004;173:1463–76. doi: 10.4049/jimmunol.173.2.1463. [DOI] [PubMed] [Google Scholar]
- 78.Zwirner NW, Domaica CI. Cytokine regulation of natural killer cell effector functions. BioFactors. 2010;36:274–88. doi: 10.1002/biof.107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of serum-free culture conditions and inhibition of proliferation on expression of ligands on rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS). (a) RA-FLS were cultured under normal conditions (RPMI + 10% fetal calf serum + 2% human serum + 2 nm basic fibroblast growth factor) or in FibroGRO serum-free media for 5 days, and were surface stained for expression of ligands as indicated. Plots indicate median fluorescence intensity (MFI) for n = 4 donors. (b) RA-FLS were cultured under normal conditions or with the addition of 25 μm Roscovitine to inhibit proliferation for 5 days, and subsequently surface stained for expression of ligands as indicated. Plots indicate MFI for six donors.
Figure S2. Nishi natural killer (NK) cells degranulate in response to fibroblast-like synoviocytes (FLS) derived from non-inflamed synovium of two healthy donors. FLS were seeded at 3 × 104 cells/well in 96-well plates, the following day 9 × 104 Nishi/well were added. Nishi and FLS were co-cultured as described. CD107a/b expression was detected on Nishi cells by flow cytometry. Nishi were gated as viable, single CD3− CD16− CD56+ cells. Nishi cultured alone (without FLS) did not express CD107a/b (data not shown).