

Abstract
The generation of memory B cells by vaccination plays a critical role in maintaining antigen-specific antibodies and producing antibody responses upon re-exposure to a pathogen. B-cell populations contributing to antibody production and protection by vaccination remain poorly defined. We used influenza virus-like particle (VLP) vaccine in a transgenic mouse model that would identify germinal centre-derived memory B cells with the expression of yellow fluorescent protein (YFP+ cells). Immunization with influenza VLP vaccine did not induce significant increases in YFP+ cells although vaccine antigen-specific antibodies in sera were found to confer protection against a lethal dose of influenza A virus (A/PR8). In addition, CD43+ B220− populations with low YFP+ cells mainly contributed to the production of vaccine antigen-specific IgG isotype-switched antibodies whereas CD43− B220+ populations with high YFP+ cells were able to produce vaccine antigen-specific IgM antibodies. Challenge infection of immunized transgenic mice with live influenza A virus resulted in significant increases in YFP+ cells in the B220− populations of spleen and bone marrow cells. These results suggest that CD43+ B220− B cells generated by vaccination are important for producing influenza vaccine antigen-specific antibodies and conferring protection.
Keywords: influenza virus, memory B cells, plasma cells, protection, vaccine
Introduction
Antibodies offer the first line of defence against infection with pathogens. Many licensed vaccines that provide long-term protection also induce prolonged antibody responses.1,2 Continuous production of antibodies is required to sustain a level of antigen-specific antibodies since the half-life of antibodies is < 3 weeks.3–5 Long-lived plasma cells, a terminally differentiated form of B cells, are mainly responsible for maintaining the level of serum antibodies.6,7 Plasma cells do not express typical B-cell markers.8 A conventional model postulates that the maintenance of serum antibody requires the proliferation and differentiation of memory B cells into antibody-secreting plasma cells.9
Memory B cells have been traditionally considered a product of germinal centre (GC) reaction upon primary antigen exposure.10,11 Selection of B cells with a high-affinity receptor depends on the cognate interaction with helper CD4 T cells and intraclonal competition for antigen retained as immune complexes on follicular dendritic cells.12 Memory B cells were traditionally defined as isotype-switched receptor-expressing B cells that underwent affinity maturation in the GC.13 Recent studies demonstrated that T-cell-dependent immune responses can generate both IgM+ and isotype-switched memory B-cell subsets.14,15 In addition, some memory B cells without somatic mutations can be generated in a GC-independent manner.16 There are also differences in the generation, affinity maturation, function and longevity of heterogeneous memory B-cell populations with different immunoglobulin isotypes.17
A previous study reported a novel transgenic mouse model that could identify GC-derived memory B cells by expression of β-galactosidase (β-gal+) and cre recombinase in a GC-specific fashion.18 The β-gal+ B cells from mice that were immunized with hapten as a model antigen were found to be effective in generating antibody-forming cells.18 It was also demonstrated that a substantial portion of the secondary antibody-forming cell responses was composed of β-gal− B cells.18 However, memory B-cell populations after immunization with relevant vaccine antigens have not been investigated.
To understand production of GC-experienced vaccine antigen-specific memory B cells and protective immunity, we used a modified transgenic mouse model that expresses yellow fluorescent protein (YFP) as a tractable marker instead of β-galactosidase. Influenza virus-like particle (VLP) vaccines have been well demonstrated to induce protective antibody responses and to confer long-term protection against influenza A viruses.19–24 Immunization of YFP transgenic mice with influenza VLP induced protective levels of antibodies but did not induce significant increases in YFP+ B-cell populations. Analysis of fractionated cells showed that the CD43+ B220− populations with low YFP+ cells produced vaccine antigen-specific IgG isotype-switched antibodies in vitro. Results in this study suggest that the generation of CD43+ B220− populations significantly contributes to inducing protective antibody responses after vaccination.
Materials and methods
Cells and virus
Spodoptera frugiperda SF9 insect cells (American Type Culture Collection, Manassas, VA; CRL-1711) for the production of recombinant baculoviruses and VLPs were maintained in SF900-II serum-free medium (Invitrogen, Carlsbad, CA) at 27°. Influenza H1N1 virus A/PR/8/34 (A/PR8) was propagated in the allantoic cavity of 10-day-old embryonated hen's eggs. Egg allantoic fluids were harvested at 3 days post-infection, kept at 4° overnight. The harvested fluids were centrifuged to remove cell debris and frozen at −80° until used. Mice were infected with serial dilutions of A/PR8 virus and the 50% of lethal dose (LD50) was then determined. Inactivation of the sucrose-gradient-purified virus was performed by mixing the virus with formalin at a final concentration of 1:4000 (volume/volume) as described previously.25
Preparation of influenza VLPs
The preparation of influenza VLPs has been described previously.19 SF9 insect cells were co-infected with recombinant baculoviruses expressing A/PR8 haemagglutinin and M1 proteins, and culture supernatants containing released VLPs were harvested after 3 days of infection. After removing cell debris, VLPs in culture supernatants were concentrated by an ultrafiltration system based on a QuixStand hollow fibre device (GE Healthcare, Piscataway, NJ) and then purified by sucrose gradient ultracentrifugation. Influenza VLPs containing A/PR8 haemagglutinin were characterized by Western blot analysis as previously described.19
Immunization and challenge
ROSA transgenic mice were generated and maintained as described previously,18 and kindly provided by Dr Joshy Jacob (Emory University). BALB/c mice were purchased from Harlan Laboratories (Indianapolis, IN) and C57BL/6 mice were from the Jackson Laboratory (Bar Harbor, ME). ROSA transgenic mice were intramuscularly immunized with influenza A/PR8 VLPs (5 μg/mouse) at weeks 0 and 12. Mice were killed 9 days after prime or boost immunization. For protection experiments, immunized ROSA transgenic mice were intranasally challenged with A/PR8 virus (5 × LD50). The protective efficacy of whole immune sera was assessed by modified passive transfer as previously described.19,26,27 Briefly, sera from unimmunized naive, prime or boost immunized ROSA transgenic mice were heat inactivated at 56° for 30 min (final fourfold diluted) and mixed with a lethal dose of influenza A/PR8 virus (15 × LD50). After incubation of the mixture at room temperature for 30 min, 7- to 8-week-old naive female BALB/c mice were intranasally infected with a mixture of A/PR8 virus and sera. Infected mice were observed daily for 2 weeks to monitor body weight changes and survival rates. Mice were killed when 25% of body weight loss was observed, in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines. All animal studies were approved and conducted under the guidelines of the Emory and Georgia State University's IACUC (approval nos 179–2008 and A11026, respectively).
Serum antibody responses
Blood samples collected by retro-orbital plexus puncture with heparinized microcapillaries (Drummond Scientific Company, Broomall, PA) were harvested after anaesthetizing mice with isoflurane (Baxter, Deerfield, IL) inhalation 9 days after immunization and 5 days after challenge and stored at −20° until analysis. Influenza virus-specific IgG, IgG1, IgG2a, IgG2b and IgG2c (Southern Biotechnology, Birmingham, AL) were determined in sera by standard ELISA methods as described previously.21,28 Briefly, horseradish peroxidase-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgG2b and IgG2c were used as secondary antibodies with O-phenylenediamine (Zymed, San Francisco, CA) in citrate–phosphate buffer, pH. 5·0 containing 0·03% H2O2 (Fisher Scientific, Fair Lawn, NJ) as a substrate and the optical density at 450 nm was read by ELISA reader (BioTek ELx800; BioTek Instruments Inc., Winooski, VT). Antibody concentrations were determined by comparing the readings for experimental samples with the standard curves for purified mouse IgG, IgG1, IgG2a, IgG2b and IgG2c antibodies.
CD43 cell fractionation and adoptive transfer
The B220 positive and negative cell fractions were isolated from the splenocytes of unimmunized naive, prime and boost immunized mice using the MidiMACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Briefly, splenocytes were incubated first with CD4, CD8 and CD11c microbeads (Miltenyi Biotec) and then applied onto the column in a magnetic field. Flow-through cells that did not bind CD4, CD8, CD11c microbeads were then incubated with CD43 microbeads (Miltenyi Biotec). Cells bound (considered as CD43+ B220− B cells) or unbound (considered as CD43− B220+ B cells) to the magnetic column were collected and nucleated cells were counted using haemocytometers after Trypan blue dead-cell exclusion.
The CD43+ or CD43− cell fractions were enriched from the spleen cells of naive C57BL/6 mice as described above using the MidiMACS system. Enriched CD43+ (2·2 × 107/mouse) or CD43− (3·3 × 107/mouse) cells were intravenously injected via tail vein into recipient naive MHCII-deficient mice (I-Aβ-/-; Jackson Laboratory), which were then immunized with 10 μg of A/PR8 VLPs intramuscularly 4 hr after adoptive cell transfer. Blood samples were collected 2 weeks after immunization and IgG antigen-specific antibody levels were determined by ELISA.
In vitro cultures, antibody production and flow cytometry
Isolated CD43+ and CD43− fractionated cells were cultured in vitro with A/PR8 VLP stimulator (1 μg/well) for 1, 3 or 5 days. Culture supernatants were collected after 3 or 5 days and used for analysis of in vitro antibody (IgG, IgG isotypes, IgM) production using ELISA. For flow cytometry analysis, live lymphocytes were gated according to their sizes and granularity defined in the forward light scatter (FSC) and side light scatter (SSC) plots. Dead cells were excluded by staining with near-infrared fluorescence reactive dye (Invitrogen, Eugene, OR). Endogenously expressed YFP+ cells were detected in the same channel of FITC and the percentages of YFP+ cells were gated and determined by FACS analysis from each B220+ (RA3-6B2; BD Pharmingen, Franklin Lakes, NJ) or B220− stained B cells. Cell acquisition was performed with a multi-laser multi-parameter analysis cytometer (LSR-II; BD Biosciences, Mountain View, CA) and analysed using the FlowJo program (Version 7.6.4; Tree Star Inc., Ashland, OR).
Splenocytes and bone marrow cells were collected from vaccinated ROSA transgenic mice before and after A/PR8 influenza H1N1 virus challenge infection. Spleen and bone marrow cells were stained with fluorescence-conjugated B220 antibody (RA3-6B2, BD Pharmingen) and YFP+ cells were calculated after being gated in B220+ or B220− B cells by FACS.
ELISpot assay
Spleens and bone marrow cells were obtained from the mice killed after boost immunization with influenza A/PR8 VLPs (3 μg/mouse) and single cell suspensions were prepared by mechanical disruption. IgG-secreting and IgM-secreting cell spots were determined on Multi-screen 96-well plates (Millipore, Billerica, MA) coated with immunoglobulin-specific capture antibodies as described previously.19 Briefly, 5 × 105 spleen or bone marrow cells (per well) were cultured in medium (blank control) or A/PR8 VLP (400 ng/well) pre-coated plate. After 36 hr of incubation, the number of IgG-secreting or IgM-secreting cell spots was counted using a Bioreader 5000-Eβ (Biosys, Miami, FL) and data were analysed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA).
Statistics
Statistical analysis was performed using a two-tailed Student's t-test and one-way analysis of variance when comparing two or more different groups, respectively. Data were analysed using Prism software (GraphPad software). Values of P < 0·05 were considered to be statistically significant. Unless otherwise stated, all results were presented as means ± SE.
Results
Virus antigen-specific antibody responses in transgenic mice marking memory B cells
A previous study demonstrated a novel transgenic mouse model in which a tractable marker in the ROSA locus in GC B cells was expressed when B cells were exposed to antigens.18 In this study, we used transgenic ROSA mice that express a directly tractable fluorescent marker protein (YFP) so that vaccine-exposed GC B cells (YFP+ B cells) could be traced and quantified. Our goal was to investigate the YFP+ B-cell populations in spleen and bone marrow after influenza vaccination. Recombinant influenza VLP vaccines have been described in our previous studies.19–22 The transgenic mice were intramuscularly immunized with 5 μg of influenza VLP vaccine with haemagglutinin derived from influenza A/PR/8/34 strain (PR8 VLP). After prime-immunization of ROSA transgenic mice with PR8 VLP, influenza virus (A/PR/8/34) specific IgG antibodies were induced at substantial levels compared with naive controls (Fig. 1a). We compared IgG and IgM antibody levels after prime and boost immunizations. There was a good response of IgM antibodies after priming (Fig. 1b) whereas significantly higher levels of IgG antibodies were induced after boosting. There was a significant increase in IgG antibodies after boosting but not in IgM antibodies (Fig. 1b). When IgG isotype antibodies were analysed, IgG2b and IgG2c (an equivalent to IgG2a in BALB/c mice) were predominantly detected (Table 1, Fig. 1c), indicating the induction of T helper type 1-like IgG isotypes. A moderate level of the IgG2a isotype was also detected, probably as the result of cross-reactivity (Table 1, Fig. 1c). The IgG1 isotype was not induced at a significant level after priming (Table 1, Fig. 1c). Total IgG and isotype antibodies specific for viral antigen were increased approximately 10-fold after boost immunization (Fig. 1). These results indicate that influenza VLP vaccination induces not only IgG2b and IgG2c antibodies but also the IgG1 subclass after boost immunization, representing T helper type 1 and type 2 immune responses with preference of the T helper type 1 immune responses.
Figure 1.
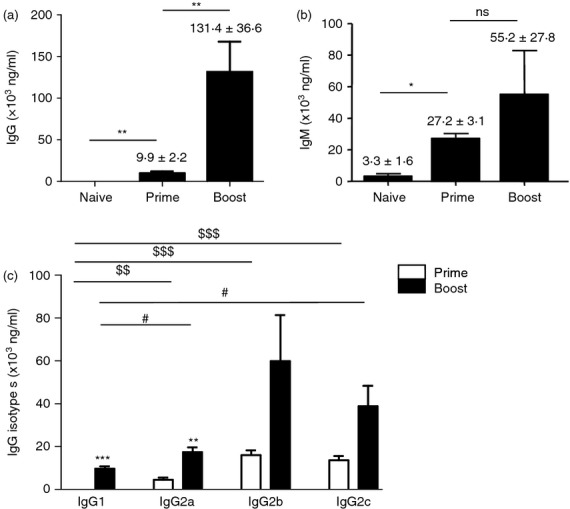
Immunization of ROSA yellow fluorescent protein transgenic (YFP Tg) mice induces vaccine-specific IgG antibodies (a) Total IgG and (b) IgM antibodies specific for inactivated influenza (A/PR8) virus antigen day 9 post prime and boost immunization (n = 5). Naive or immune sera were used to determine relative antibody concentrations by a standard ELISA. Averages ± standard errors are marked on each column. **P < 0·01, *P < 0·05, and ns; not significant between groups. (c) Isotype switched IgG antibodies specific to inactivated influenza (A/PR8) virus antigen after prime and boost immunization. A representative result is shown out of three independent experiments. Total quantitative concentrations of isotype antibodies are presented in Table 1 (ng/ml). **P < 0·01 and ***P < 0·001 compared with prime, respectively. $$P < 0·01 and $$$P < 0·001 compared with prime IgG1, respectively. #P < 0·05 compared with boost IgG1.
Table 1.
Comparison of immunoglobulin isotypes from prime and boost ROSA transgenic mice
| Prime1 | Boost2 | Fold increase | |
|---|---|---|---|
| IgG1 | ND | 9722·53 ± 1100·31 | – |
| IgG2a | 4543·92 ± 966·63 | 17496·62 ± 2108·06** | 3·9 |
| IgG2b | 16059·70 ± 2222·36 | 59960·20 ± 21453·82 | 3·7 |
| IgG2c | 13655·18 ± 1989·32 | 38888·51 ± 9446·98 | 2·8 |
All data represent mean ± SE (ng/ml).
ND, not detectable.
9 days post immunization.
9 days after 2nd immunization (3 month after the first immunization).
P < 0·01 compared to prime.
To determine vaccine antigen-specific antibody-producing cell responses, an in vitro culture was performed with spleen and bone marrow cells for 1 day (Fig. 2). Although vaccine antigen-specific antibodies were induced after priming, antibody-secreting cell responses specific for the vaccine antigen after in vitro cultures were low at day 9 post prime immunization (Fig. 2). Importantly, significant levels of antibodies secreted into culture supernatants were observed after 1-day in vitro cultures of spleen and bone marrow cells from boosted mice (Fig. 2). After boost immunization, vaccine-specific antibody-forming cell spots were approximately 150 (IgG + IgM) per million total spleen cells and 130 (IgG + IgM) per million bone marrow cells, representing 0·013–0·015% of total cell populations (data not shown). These results indicate that priming with influenza A/PR8 VLP vaccine was effective in generating memory B cells, which were expanded and differentiated into antibody-secreting plasma cells after boost vaccination. Therefore, influenza VLPs provide an appropriate vaccine antigen to study YFP+ marking of B cells using this transgenic mouse model after vaccination, and their roles in providing protection.
Figure 2.
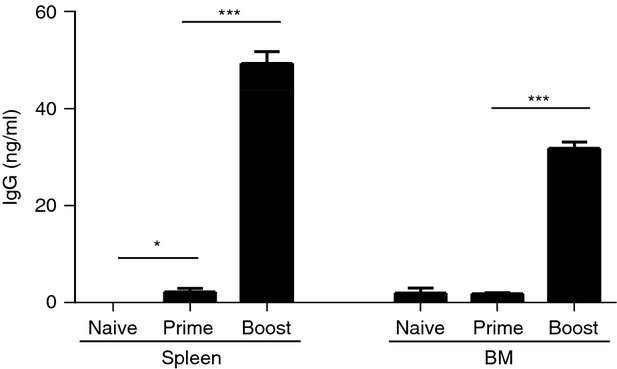
In vitro antibody production from spleen and bone marrow cells from ROSA yellow fluorescent protein transgenic (YFP Tg) mice. Splenocytes or bone marrow cells (5 × 105 cells/well in 96-well plates) at day 9 post prime or boost immunization (n = 3) were subjected to ex vivo cultures for 24 hr in influenza virus-like particle (VLP) -coated 96-well culture plates. A/PR8 VLP-specific IgG antibodies were measured by ELISA and total concentrations of IgG antibodies for each naive, prime, and boost immunized group from spleen or bone marrow were calculated accordingly. A representative is shown out of two independent experiments. *P < 0·05; ***P < 0·0001.
YFP positively marked cells in CD43+ B220− cells and in CD43− B220+ cells during in vitro culture
Both β-gal+ and YFP+ ROSA transgenic mouse models express transgenes (YFP, β-gal) under the same MHC class II promoter. Monitoring YFP expression would be more sensitive than β-gal because YFP fluorescence is directly measured without substrate reactions. β-Gal+ transgenic mice were known to have some backgrounds of transgene expression up to 20% or more before immunization.18 We analysed YFP+ B220+ and YFP+ B220− populations in spleens and bone marrow after prime and boost immunizations (Fig. 3a,b). Although there was a moderate increasing pattern in splenic YFP+ B220+ populations and bone marrow YFP+ B220− populations after immunization, no statistical significance was observed in YFP+ B220+ and YFP+ B220− populations between primed and boosted mice compared with naive mice (Fig. 3).
Figure 3.
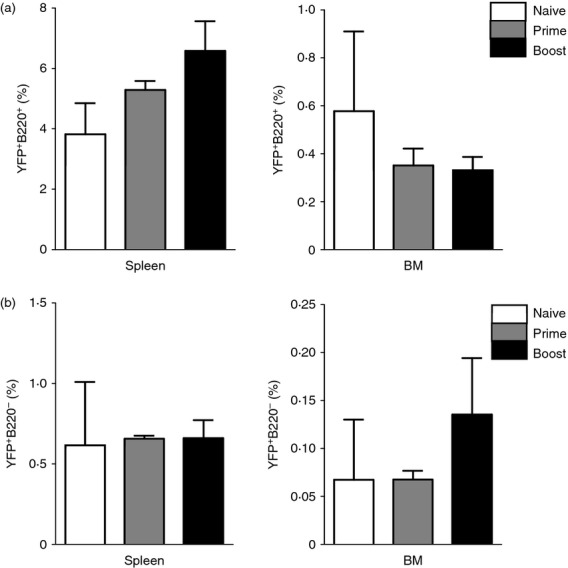
Endogenous yellow fluorescent protein-positive (YFP+) populations in spleens and bone marrow before and after immunization. Splenocytes and bone marrow cells were harvested 9 days after prime and boost immunization. The cells were stained with surface marker antibodies and analysed by flow cytometry. YFP+ B220+ (a) and YFP+ B220− (b) cell population in naive, primed, or boosted ROSA transgenic mice. A representative is shown out of two independent experiments.
It was reported that the marker positive (β-gal+) B-cell populations were segregated into distinct B220+ and B220− cells.18 To analyse B220+ and B220− subpopulations contributing to inducing vaccine-specific antibody responses, we fractionated spleen cells that were obtained from prime and boost influenza VLP-vaccinated YFP+ ROSA mice (Fig. 4). T-cell-depleted, B-cell-enriched whole splenocytes were then fractionated into CD43+ and CD43− cell sub-populations using the MACS fractionation system (Fig. 4). Flow cytometry profiles indicated 89% of purity for CD43+ and 97% of purity for CD43− cells. CD43+ populations contained a majority of B220− cells (93% CD43+ B220− cells) whereas CD43− populations contained B220+ conventional B cells (81% CD43− B220+ cells) as shown in Fig. 4.
Figure 4.
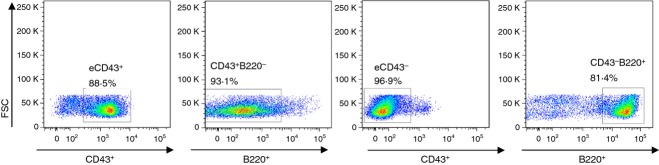
Flow cytometry profile of B220 cells in CD43+ and CD43− population. Enriched CD43+ (eCD43+ B220−) or CD43− (eCD43− B220+) fractionated cells were isolated from splenocytes by MACS and stained with fluorescence-conjugated B220 antibody. B220+ or B220− cell populations were determined after gating CD43+ or CD43− cells. The representative cell populations are shown out of three independent experiments.
These major B-cell fractions were each subjected to in vitro culture for 1, 3 or 5 days in the presence of PR8 VLP vaccines (Fig. 5). Both B220− (CD43+ fraction) and B220+ (CD43− fraction) subpopulations were analysed for the presence of YFP+ B cells during 1–5 days of in vitro culture (Fig. 5). In splenocytes from primed YFP+ ROSA mice, more YFP+ cells (2·4–3·3%) were found in the B220+ cell fraction than those in the B220− cell fraction (1·5–1·6%). Approximately twofold lower percentages of YFP+ cells were detected in the B220− cell fractions compared with those in B220+ cell fractions from spleen cells of boosted YFP+ ROSA mice at days 1, 3 and 5 in vitro culture periods (Fig. 5). No significant changes were observed in percentages of YFP+ cells during 5 days of culture although total cell numbers dropped substantially, probably as the result of apoptotic cell death.
Figure 5.

Flow cytometry yellow fluorescent protein-positive (YFP+) profiles of CD43− B220+ and CD43+ B220− fractionated populations during in vitro cultures. For the B-cell isolation, fresh splenocytes isolated at 2 weeks after prime and boost immunization of ROSA transgenic mice (n = 5) were separated by MACS. Splenocytes were incubated with a CD4, CD8 and CD11c microbeads cocktail and then unlabelled cells from the magnetic column were labelled with CD43 microbeads. After isolation of CD43 magnetic beads unbound (considered as B220+ B cells) or bound (considered as B220− B cells) cells, each fraction was in vitro cultured with A/PR8 virus-like particle (VLP) stimulator (1 μg/well) for 1, 3 or 5 days (D1, D3, and D5, respectively). Endogenously expressed YFP+ cells were determined by FACS analysis from each gated B220+ or B220− stained B cells.
Vaccine-specific antibody production dominantly from CD43+ subpopulations during in vitro culture
It is important to determine the vaccine-specific antibody-producing functionality of CD43+ B220− cells and CD43− B220+ cells during in vitro culture. Levels of IgG, IgM and IgG isotype antibodies secreted into culture supernatants were measured by ELISA (Fig. 6). After 3 days of culture, vaccine-specific IgG antibody was produced at a substantial level from CD43+ B220− cell fractions of 2-week boosted spleen cells but not from CD43− B220+ cell fractions (D3, Fig. 6a). On the other hand, a higher level of IgM antibody was observed in the CD43− (B220+ populations) at both 3 and 5 days compared with those in the CD43+ (B220− populations) fraction (Fig. 6b). Therefore, it is indicated that IgM antibody-producing CD43− B220+ cells are at an earlier stage of B-cell differentiation than the IgG antibody-producing CD43+ B220− cell populations.
Figure 6.
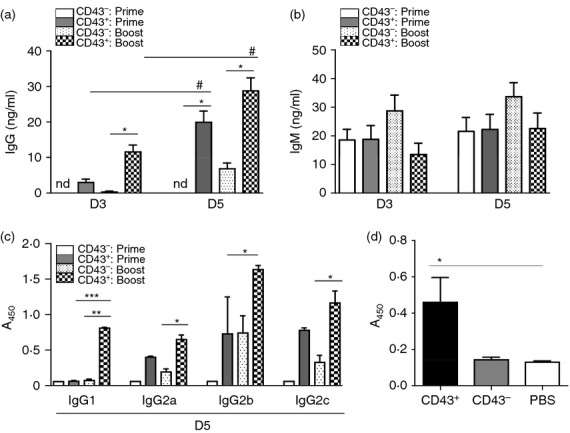
CD43+ B220− populations are major cell types contributing to in vitro antibody production. CD43+ or CD43− isolated cell fractions of splenocytes from ROSA yellow fluorescent protein transgenic mice (n = 3) by the MACS system were pooled and cultured for 3 (D3) or 5 (D5) days with A/PR8 virus-like particle (VLP) stimulator (1 μg/well). In vitro culture supernatants (100 μl) were used to measure the relative antibody concentrations for influenza virus (A/PR8) -specific IgG (a), IgM (b) and IgG isotype antibodies (c) determined by ELISA. CD43+: prime, CD43 positive splenocytes from 2 week prime immunized mice; CD43−: prime, CD43 negative splenocytes from 2 week prime immunized mice; CD43+: boost, CD43 positive splenocytes from 2 weeks boost immunized mice; CD43−: boost, CD43 negative splenocytes from 2 weeks boost immunized mice; nd, not detectable. This pattern of in vitro antibody production was consistent and the data shown were a representative out of three independent experiments. (d) IgG antibody responses in MHCII-deficient mice with adoptive transfer of CD43+ and CD43− fractionated populations from C57BL/6 mice followed by vaccination. Immunization of MHCII-deficient mice influenza VLP vaccine (A/PR8 VLP) was followed 4 hr after adoptive transfer. Serum antibody levels were determined at 2 weeks after immunization and presented as optical densities at 450 nm. (a–d) * or #, ** and ***; P < 0·05, 0·01 and 0·001, respectively. Horizontal statistical bars indicate comparing groups (*, **, *** comparison between CD43− versus CD43+ prime or boost; # comparison between D3 and D5).
After 5 days of culture, IgG antibody secreted into culture supernatants was found at a just detectable level in CD43− B220+ boosted but not primed cell fractions, which was much lower than that detected in the population of CD43+ B220− cells (D5, Fig. 6a). IgG antibody was produced at a significantly higher level from CD43+ B220− cell fractions of 2-week primed and boosted spleen cells at 5 days of culture compared with those at 3 days of culture. Also, CD43+ populations of primed and boosted splenocytes showed higher in vitro antibody production than those of corresponding primed and boosted CD43– fractionated splenocytes (D5, Fig. 6a).
We further determined the pattern of antibody isotypes in culture supernatants of CD43 MACS fractionated spleen cells. IgG1 was detected in the CD43+ (B220−) population with low YFP+ cell percentages of boosted ROSA YFP mice (Fig. 6c). IgG2a, IgG2b and IgG2c isotype antibodies were also found at much higher levels in CD43+ (B220−) cell fractions with low YFP+ cell percentages from both primed and boosted ROSA YFP mice compared with those in CD43− (B220+) cell fractions with higher YFP+ cell percentages. It is also interesting to note that IgG2b and IgG2c antibodies were produced at detectable levels in the CD43− (B220+ population) cell fraction of boosted splenocytes, although their levels were lower compared with those in CD43+ cell fractions (Fig. 6c). These results indicate that YFP+ ROSA mice can generate vaccine antigen-specific memory B cells capable of differentiating to IgG and isotype antibody-secreting plasma cells after VLP vaccination.
In vivo antibody production from CD43+ fractionated cells
To better understand the in vivo contribution of CD43+ and CD43– B-cell subpopulations to inducing antibodies, we determined the capacity of in vivo antibody production by CD43+ (B220− with low YFP+ cells) and CD43− (B220+ with high YFP+ cells) cells. MHCII-deficient mice were found to be defective in inducing vaccine-specific antibody responses after immunization (unpublished data). CD43+ and CD43− subpopulations were adoptively transferred to naive MHCII-deficient mice that were followed by subsequent immunization with influenza VLP vaccines (Fig. 6d). As expected, the group of mice with adoptive transfer of the CD43+ subpopulation induced significantly higher levels of vaccine-specific IgG antibodies in response to influenza VLP immunization (CD43+, Fig. 6d). Similar to the mock control (PBS, Fig. 6d), significant levels of IgG antibodies were not detected in the MHCII-deficient mouse group with a CD43− subpopulation transferred adoptively after immunization with influenza VLP (CD43−, Fig. 6d). Both groups did not respond to boost immunization probably because of the limited lifespan of adoptively transferred cells (data not shown). These results provide in vivo evidence that CD43+ (B220− with low YFP+ cells) subpopulations mainly contribute to producing IgG antibody responses to vaccination.
Increased YFP+ cell populations in immunized ROSA mice after virus challenge
It is expected that virus challenge of vaccinated mice would stimulate the production of YFP+ cells because replicating virus is considered to be a stronger inducer of immune responses than non-replicating vaccine antigens. Spleen and bone marrow cells were harvested from boost-vaccinated YFP+ ROSA mice at day 5 post challenge infection with A/PR8 influenza virus. Percentages of YFP+ cells were found to be increased by two- to threefold in both spleen and bone marrow after virus challenge compared with those found before challenge (Fig. 7). Significant increases in YFP+ cell populations were observed in B220– cell populations but not in B220 positively gated B cells, although B220+ gated B-cell populations showed higher levels of YFP+ cells than B220− cell populations by over threefold (Fig. 7). Significant changes in YFP+ cells in both B220+ and B220− cell populations were not observed in the unvaccinated naive mice before and after influenza virus infection (data not shown). These results indicate that YFP+ marked cell populations can be increased in vaccinated mice after influenza virus challenge, in particular, in the CD43+ B220− populations.
Figure 7.
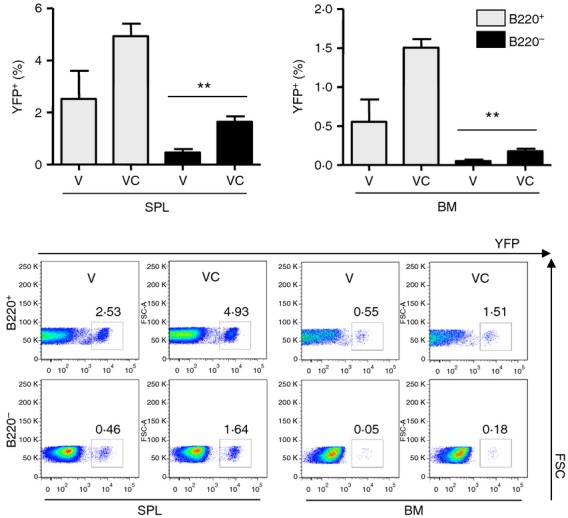
Yellow fluorescent protein-positive (YFP+) populations in the spleen and bone marrow cells from vaccinated ROSA transgenic mice after challenge infection. Splenocytes and bone marrow cells were collected from vaccinated ROSA transgenic mice before and after A/PR8 influenza H1N1 virus challenge infection. Spleen and bone marrow cells were stained with fluorescence-conjugated B220 antibody and YFP+ cells were calculated in B220+ or B220− B cells by FACS. The representative images for each group were shown. V: vaccinated mice at weeks 0 and 12 (n = 7). VC: vaccinated mice at week 0 and week 12, and challenged 5 days before killing (n = 3). Statistical significances were compared with vaccine-only groups of B220+ and B220− spleen and bone marrow cells. **, P < 0·01.
Immune sera of ROSA mice vaccinated with PR8 VLP confer protection against influenza virus infection
Virus-specific antibody responses play a critical role in providing protection against influenza virus infection. We determined whether levels of serum antibodies induced by prime or prime-boost immunization of ROSA mice would be protective. A modified assay for passive transfer of immune sera mixed with a lethal dose of influenza virus was used as previously described.29–31 Naive mice were infected with a mixture of a lethal dose of influenza A/PR8 virus and naive sera of unimmunized mice, or prime or boost immune sera from PR8 VLP vaccine-immunized ROSA mice. Mice that received prime or boost immune sera did not show body weight loss (Fig. 8a) and were fully protected against lethal infection (Fig. 8b). It is worth noting that immune sera from primed mice conferred relatively good protection. This is consistent with results from our previous study demonstrating that a single immunization of mice with influenza VLP vaccines could provide protection against lethal challenge.22 The group of mice that was infected with a mixture of influenza virus and naive sera showed severe body weight loss and had to be killed by day 6–8 after infection. Therefore, these results indicate that antibodies most probably contributed by CD43+ B220− populations induced after prime immunization of ROSA mice can confer protection against influenza virus infection.
Figure 8.
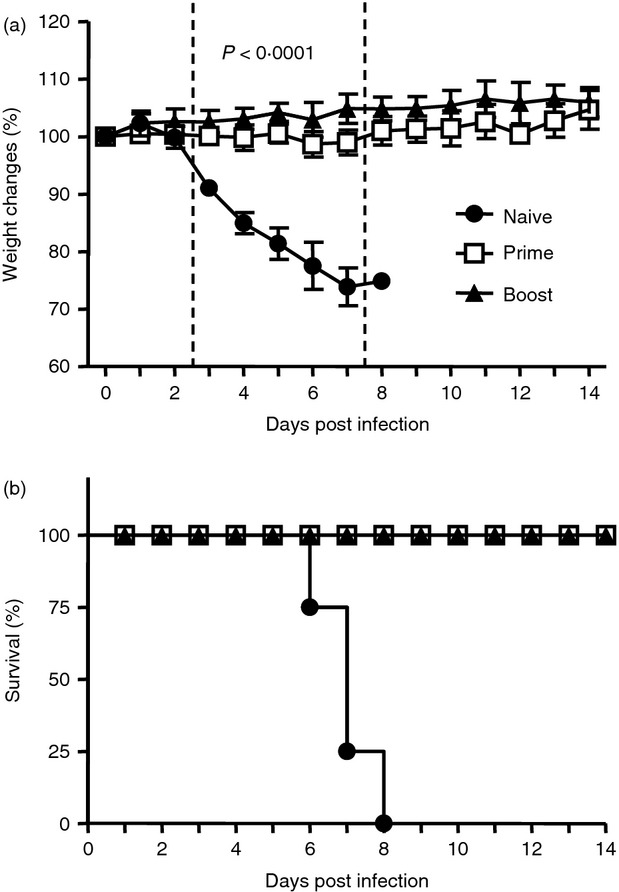
Protective efficacy of immune sera. Immune sera were collected from unimmunized naive, prime or boost immunized ROSA transgenic mice with influenza A/PR8 virus-like particle (VLP) intramuscularly. Heat-inactivated immune sera were incubated with a lethal dose of influenza A/PR8 virus (15 × LD50) at room temperature for 30 min. Groups of naive BALB/c mice (n = 4) were intranasally infected with sera (final fourfold diluted) and virus mixture. Body weight changes (a) and survival rate (b) were monitored for 2 weeks. Naive, sera from unvaccinated naive mice; prime, sera from prime vaccinated mice; boost, sera from boost vaccinated mice. Mice were killed when they lost 25% of initial body weight. Statistical significance (P < 0·0001) indicates both prime and boost immune sera groups compared to naive group.
Discussion
Germinal centre reactions are known to be important for inducing isotype-switched antibodies, and generating long-lived memory B cells and antibody-secreting plasma cells.10,11,13 Most of our understanding about the generation of GC B cells was derived from studies using high doses (50–100 μg) of model antigens in the presence of experimental adjuvants,11,18,32 or infectious viral pathogens,6,33–35 which may not represent the process of inducing protective immunity after vaccination. Hence, it is highly significant to study GC B cells and the induction of antibodies after immunization with relevant vaccine antigens, and their correlation with protective immunity after challenge infection.
A double transgenic mouse model was previously described, in which a tractable marker (β-gal) was permanently expressed in GC B cells using a cre-lox recombination strategy.18 VLPs as a non-replicating subunit vaccine were demonstrated to confer protection against influenza virus, suggesting that they can serve as a protective vaccine antigen in a mouse model for challenge and protection. In the present study, using the transgenic mouse model in which YFP is expressed in GC B cells as a tractable marker without the additional step of substrate incubation, we investigated the production of vaccine antigen-specific antibody-secreting B cells in vivo and in vitro. We found that YFP+ B cells were found in both spleen and bone marrow tissues. Although influenza VLP vaccination of YFP transgenic mice induced protective antibody responses, levels of YFP+ cells in vaccinated mice were not significantly increased as compared with those in unvaccinated control mice (Fig. 3). It was an important finding that B220− phenotypic cells with low YFP+ percentages were a major population contributing to vaccine-specific antibody production in vitro and showed significant increases in YFP+ cells after challenge infection of vaccinated mice.
Approximately 1–5 μg of recombinant influenza VLP vaccines have been shown to induce protection in BALB/c mice.22,36,37 Prime immunization of YFP+ mice induced substantial levels of vaccine antigen-specific antibodies with T helper type 1 IgG2b and IgG2c isotype antibodies being dominant (Fig. 1b), and boost immunization further increased those vaccine-specific antibodies. Levels of antibodies induced by prime or prime-boost immunization were found to be sufficient for conferring protection. Therefore, the use of a 5-μg VLP vaccine dose should be appropriate to investigate the level of GC B cells and antibody-forming cell responses that would be protective after vaccination. We found that there were no significant increases in percentages of YFP+ cells in spleens or bone marrow after prime or boost immunization of YFP+ ROSA mice compared with those of unimmunized mice (Fig. 3). In a previous study using this ROSA transgenic mouse model, 50 μg of alum-adjuvanted hapten conjugate as a model antigen was used to induce markedly increased levels of β-gal+ cells.18 Hence, it is assumed that a protective level of immunity by vaccination with 5 μg VLP might not need to induce significantly higher levels of YFP+ memory B cells compared with those of unimmunized YFP+ ROSA naive control mice. In support of this argument, the number of vaccine-specific antibody-secreting cells was very small, approximately 30 IgG antibody-secreting cells per million spleen cells (0·003%), a level that is approximately 10-fold less than those observed after infection of mice with a live lymphocytic choriomeningitis virus.33,34 Another possibility is that a protective level of vaccine-specific memory B cells may be too low to be reflected by significant differences in YFP+ memory B cells by flow cytometry analysis.
The transgenic mouse contains the SmaI-truncated MHC class II I-Eαd promoter, which drives the gene expression of YFP+ phenotype in B lineage cells restricted to the GC stage of B-cell development.18,38,39 Despite this expected restriction in the expression of a tractable marker gene under the MHC class II I-Eαd promoter, only a fraction of approximately 20% of β-gal+ cells was reported to have GC-specific surface markers.18 In other words, some GC B cells could be presented without a YFP+ phenotype and YFP+ cells could be found outside the GC regions. We analysed PNA+ CD95+ GC phenotypes in the YFP+ B220+ cells and CD138+ phenotype in the YFP+ B220− populations. Significant changes in these marker phenotypes were not detected after priming and boost immunizations (data not shown). Despite the lack of detectable changes in YFP+ cell populations after vaccination, we observed significant increases in vaccine-specific class-switched antibody responses in both serum samples as well as in in vitro cultures of spleen and bone marrow cells. In favour of this idea, both β-gal+ and β-gal− populations with a germinal centre marker (peanut agglutinin) were shown to have mutations in the complementarity-determining region.18
CD43-negative fractions have been used to determine the roles of conventional B cells by adoptive transfer in the presence of cognate CD4+ T cells.40 CD43 is known to be expressed on plasmablast-like B220− populations and non-conventional splenic memory B-1-like cells, but not on conventional peripheral B (B220+ B-2) cells.32,41,42 In our studies, higher percentages of YFP+ cells were found in the B220+ population than the B220− population in splenocytes after prime boost vaccination with influenza VLP vaccine, which is consistent with a previous study using the transgenic mice immunized with a hapten-conjugated model antigen.18 In the present study, we found that the CD43-positive fraction containing B220− cells with low YFP+ percentages mainly contributed to producing vaccine-specific IgG antibody responses, as shown by in vitro cultures of fractionated splenocytes. Importantly, this in vitro antibody production by CD43+ subpopulations was further supported by in vivo adoptive transfer experiments into MHCII-deficient mice, which also have CD4 T-cell deficiency (Fig. 6d). The in vitro production of antibodies in the CD43+ B220− populations was also found to be independent of the help from CD4 T cells. Therefore, this study suggests that these CD43+ B220− cells might be late-stage memory B cells that do not need helper T cells for differentiation into antibody-secreting plasma cells. CD43+ B220− cells are likely to be plasmablasts that differentiate into antibody-secreting plasma cells. Even the prime only B220− populations showed a higher capacity for producing antibodies in vitro than prime-boosted B220+ populations (Fig. 6a,c). In contrast, B220+ populations with high percentages of YFP+ cells in the CD43– fraction appeared to be effective in producing IgM antibodies in vitro, indicating an early stage of B-cell activation upon antigen exposure. Prime vaccination induced significant levels of IgM antibodies, even more than IgG antibodies (Fig. 1a,b). Protective capacity of prime immune sera suggests that both IgG and IgM antibodies might have contributed to the observed protection, in line with a previous study that a single vaccination would be beneficial for conferring protection.22 Which class of antibody, IgM or IgG, played a bigger role in conferring protection remains to be determined. It was shown that IgM antibodies partially contributed to controlling lung virus titres.43 We still expect that class-switched IgG antibodies would have strong virus-neutralizing activity and confer more effective protection. Although lung virus clearance was not measured, it is expected that boost immune sera would be more effective in controlling lung viral loads, which is considered a more sensitive assay.
Live virus infection of influenza VLP-vaccinated mice could provide effective stimulation of B220− populations with YFP+ cells. In this regard, it is worth noting that significant increases in YFP+ cells were observed only in the B220− populations of spleens and bone marrow after challenge infection. It is likely that some B220− populations with YFP+ cells are vaccine antigen-specific memory B cells contributing to the differentiation into antibody-forming cells in vivo. In support of this notion, it was previously reported that adoptive transfer of B220– memory B cells produced greater amounts of antibodies than their B220+ counterparts.32 Despite the observation of increased YFP+ cells in the B220− populations, the levels of antibodies in in vitro cultures of spleen and bone marrow cells were not significantly higher after vaccination and challenge compared with those from vaccination only before challenge (data not shown). One possibility is that the time of analysis at 5 days after challenge would not be sufficient to reflect the post-challenge boost of antibody production.
It is generally considered that differentiation of GC-derived memory B cells to antibody-forming cells contributes to the rapid increase in antigen-specific antibodies, which is typical of secondary immune responses. However, a previous study on ROSA transgenic mice reported significant amounts of unmarked (β-gal−) antibody-forming cells during secondary antigen exposure.18 Interestingly, B220+ populations showed a capacity to produce IgM antibodies in vitro, suggesting that B220+ populations (with YFP+) are an early stage of memory B cells before IgG isotype switching after antigen exposure. Hence, a substantial proportion of naive B cells and/or non-GC B cells is being produced during prime immunization as well as boost vaccination. This can be a possible reason why YFP+ cell populations were not observed at significantly increased levels during prime or boost immunization with VLP vaccines.
In summary, significant increases in YFP+ cell populations were not required for inducing protective levels of antibodies after vaccination. Hence, it is important to be cautious in studying antigen-specific memory B cells that are only restricted to GC-experienced B-cell populations. Live virus infection of immunized mice significantly stimulated the generation of YFP+ cells in the B220− population but not in the B220+ population. This study therefore provides evidence that CD43+ B220− populations are the major cell types contributing to the production of IgG isotype-switched antibodies, whereas CD43− B220+ cells are likely to be an early stage of B-cell activation mainly contributing to IgM antibody production.
Acknowledgments
This work was in part supported by NIH/NIAID grants AI105170 (S.M.K.), AI093772 (S.M.K.), and AI087782 (S.M.K.). The authors thank T. Kang for careful reading of the manuscript.
Disclosures
The authors declare no competing conflicts of interest.
References
- 1.Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. 2010;17:1055–65. doi: 10.1128/CVI.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 3.Fahey JL, Sell S. The immunoglobulins of mice. V. The metabolic (catabolic) properties of five immunoglobulin classes. J Exp Med. 1965;122:41–58. doi: 10.1084/jem.122.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talbot PJ, Buchmeier MJ. Catabolism of homologous murine monoclonal hybridoma IgG antibodies in mice. Immunology. 1987;60:485–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. 1988;18:313–6. doi: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 6.Slifka MK, Ahmed R. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr Opin Immunol. 1998;10:252–8. doi: 10.1016/s0952-7915(98)80162-3. [DOI] [PubMed] [Google Scholar]
- 7.Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J Immunol. 2003;171:4969–73. doi: 10.4049/jimmunol.171.10.4969. [DOI] [PubMed] [Google Scholar]
- 8.Abney ER, Cooper MD, Kearney JF, Lawton AR, Parkhouse RM. Sequential expression of immunoglobulin on developing mouse B lymphocytes: a systematic survey that suggests a model for the generation of immunoglobulin isotype diversity. J Immunol. 1978;120:2041–9. [PubMed] [Google Scholar]
- 9.Gray D, Siepmann K, van Essen D, Poudrier J, Wykes M, Jainandunsing S, Bergthorsdottir S, Dullforce P. B-T lymphocyte interactions in the generation and survival of memory cells. Immunol Rev. 1996;150:45–61. doi: 10.1111/j.1600-065x.1996.tb00695.x. [DOI] [PubMed] [Google Scholar]
- 10.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–8. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 11.Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J Exp Med. 1992;176:679–87. doi: 10.1084/jem.176.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McHeyzer-Williams MG. B cells as effectors. Curr Opin Immunol. 2003;15:354–61. doi: 10.1016/s0952-7915(03)00046-3. [DOI] [PubMed] [Google Scholar]
- 13.McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu Rev Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 14.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. 2012;209:597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–7. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New markers for murine memory B cells that define mutated and unmutated subsets. J Exp Med. 2007;204:2103–14. doi: 10.1084/jem.20062571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor JJ, Jenkins MK, Pape KA. Heterogeneity in the differentiation and function of memory B cells. Trends Immunol. 2012;33:590–7. doi: 10.1016/j.it.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chappell CP, Jacob J. Identification of memory B cells using a novel transgenic mouse model. J Immunol. 2006;176:4706–15. doi: 10.4049/jimmunol.176.8.4706. [DOI] [PubMed] [Google Scholar]
- 19.Quan FS, Huang C, Compans RW, Kang SM. Virus-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J Virol. 2007;81:3514–24. doi: 10.1128/JVI.02052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang SM, Song JM, Quan FS, Compans RW. Influenza vaccines based on virus-like particles. Virus Res. 2009;143:140–6. doi: 10.1016/j.virusres.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang SM, Yoo DG, Lipatov AS, et al. Induction of long-term protective immune responses by influenza H5N1 virus-like particles. PLoS ONE. 2009;4:e4667. doi: 10.1371/journal.pone.0004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quan FS, Yoo DG, Song JM, Clements JD, Compans RW, Kang SM. Kinetics of immune responses to influenza virus-like particles and dose-dependence of protection with a single vaccination. J Virol. 2009;83:4489–97. doi: 10.1128/JVI.02035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bright RA, Carter DM, Daniluk S, et al. Influenza virus-like particles elicit broader immune responses than whole virion inactivated influenza virus or recombinant hemagglutinin. Vaccine. 2007;25:3871–8. doi: 10.1016/j.vaccine.2007.01.106. [DOI] [PubMed] [Google Scholar]
- 24.Pushko P, Tumpey TM, Bu F, Knell J, Robinson R, Smith G. Influenza virus-like particles comprised of the HA, NA, and M1 proteins of H9N2 influenza virus induce protective immune responses in BALB/c mice. Vaccine. 2005;23:5751–9. doi: 10.1016/j.vaccine.2005.07.098. [DOI] [PubMed] [Google Scholar]
- 25.Sha Z, Compans RW. Induction of CD4+ T-cell-independent immunoglobulin responses by inactivated influenza virus. J Virol. 2000;74:4999–5005. doi: 10.1128/jvi.74.11.4999-5005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MC, Lee JS, Kwon YM, et al. Multiple heterologous M2 extracellular domains presented on virus-like particles confer broader and stronger M2 immunity than live influenza A virus infection. Antiviral Res. 2013;99:328–35. doi: 10.1016/j.antiviral.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song JM, Van Rooijen N, Bozja J, Compans RW, Kang SM. Vaccination inducing broad and improved cross protection against multiple subtypes of influenza A virus. Proc Natl Acad Sci USA. 2011;108:757–61. doi: 10.1073/pnas.1012199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song JM, Hossain J, Yoo DG, et al. Protective immunity against H5N1 influenza virus by a single dose vaccination with virus-like particles. Virology. 2010;405:165–75. doi: 10.1016/j.virol.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MC, Song JM, OE, Kwon YM, Lee YJ, Compans RW, Kang SM. Virus-like particles containing multiple M2 extracellular domains confer improved cross-protection against various subtypes of influenza virus. Mol Ther. 2013;21:485–92. doi: 10.1038/mt.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song JM, Wang BZ, Park KM, et al. Influenza virus-like particles containing M2 induce broadly cross protective immunity. PLoS ONE. 2011;6:e14538. doi: 10.1371/journal.pone.0014538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quan FS, Compans RW, Nguyen HH, Kang SM. Induction of heterosubtypic immunity to influenza virus by intranasal immunization. J Virol. 2008;82:1350–9. doi: 10.1128/JVI.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McHeyzer-Williams LJ, Cool M, McHeyzer-Williams MG. Antigen-specific B cell memory: expression and replenishment of a novel b220– memory B cell compartment. J Exp Med. 2000;191:1149–66. doi: 10.1084/jem.191.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol. 1995;69:1895–902. doi: 10.1128/jvi.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slifka MK, Ahmed R. Limiting dilution analysis of virus-specific memory B cells by an ELISPOT assay. J Immunol Methods. 1996;199:37–46. doi: 10.1016/s0022-1759(96)00146-9. [DOI] [PubMed] [Google Scholar]
- 35.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–72. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 36.Quan FS, Kim YC, Compans RW, Prausnitz MR, Kang SM. Dose sparing enabled by skin immunization with influenza virus-like particle vaccine using microneedles. J Control Release. 2010;147:326–32. doi: 10.1016/j.jconrel.2010.07.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quan FS, Kim YC, Vunnava A, Yoo DG, Song JM, Prausnitz MR, Compans RW, Kang SM. Intradermal vaccination with influenza virus-like particles by using microneedles induces protection superior to that with intramuscular immunization. J Virol. 2010;84:7760–9. doi: 10.1128/JVI.01849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Ewijk W, Ron Y, Monaco J, et al. Compartmentalization of MHC class II gene expression in transgenic mice. Cell. 1988;53:357–70. doi: 10.1016/0092-8674(88)90156-0. [DOI] [PubMed] [Google Scholar]
- 39.Burkly LC, Lo D, Cowing C, Palmiter RD, Brinster RL, Flavell RA. Selective expression of class II E α d gene in transgenic mice. J Immunol. 1989;142:2081–8. [PubMed] [Google Scholar]
- 40.Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. 2007;178:2415–20. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- 41.Wells SM, Kantor AB, Stall AM. CD43 (S7) expression identifies peripheral B cell subsets. J Immunol. 1994;153:5503–15. [PubMed] [Google Scholar]
- 42.Tarlinton D. B-cell memory: are subsets necessary? Nat Rev Immunol. 2006;6:785–90. doi: 10.1038/nri1938. [DOI] [PubMed] [Google Scholar]
- 43.Kopf M, Brombacher F, Bachmann MF. Role of IgM antibodies versus B cells in influenza virus-specific immunity. Eur J Immunol. 2002;32:2229–36. doi: 10.1002/1521-4141(200208)32:8<2229::AID-IMMU2229>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]