

Abstract
CD13/Aminopeptidase N is a transmembrane metalloproteinase that is expressed in many tissues where it regulates various cellular functions. In inflammation, CD13 is expressed on myeloid cells, is up-regulated on endothelial cells at sites of inflammation and mediates monocyte/endothelial adhesion by homotypic interactions. In animal models the lack of CD13 alters the profiles of infiltrating inflammatory cells at sites of ischaemic injury. Here, we found that CD13 expression is enriched specifically on the pro-inflammatory subset of monocytes, suggesting that CD13 may regulate trafficking and function of specific subsets of immune cells. To further dissect the mechanisms regulating CD13-dependent trafficking we used the murine model of thioglycollate-induced sterile peritonitis. Peritoneal monocytes, macrophages and dendritic cells were significantly decreased in inflammatory exudates from global CD13KO animals when compared with wild-type controls. Furthermore, adoptive transfer of wild-type and CD13KO primary myeloid cells, or wild-type myeloid cells pre-treated with CD13-blocking antibodies into thioglycollate-challenged wild-type recipients demonstrated fewer CD13KO or treated cells in the lavage, suggesting that CD13 expression confers a competitive advantage in trafficking. Similarly, both wild-type and CD13KO cells were reduced in infiltrates in CD13KO recipients, confirming that both monocytic and endothelial CD13 contribute to trafficking. Finally, murine monocyte cell lines expressing mouse/human chimeric CD13 molecules demonstrated that the C-terminal domain of the protein mediates CD13 adhesion. Therefore, this work verifies that the altered inflammatory trafficking in CD13KO mice is the result of aberrant myeloid cell subset trafficking and further defines the molecular mechanisms underlying this regulation.
Keywords: adhesion molecules, cell trafficking, inflammation, transgenic/knockout mice
Introduction
Inflammation is a controlled response of the host to infection or injury that involves various molecular, cellular and physiological changes and is coordinated primarily by specific cell adhesion molecules and chemoattractants. Monocytes circulate in the blood, bone marrow and spleen during homeostasis where they patrol for inflammatory adhesion molecules up-regulated in response to infection or tissue damage on the endothelium lining the blood vessels.1 Upon detection of inflammation these patrolling cells migrate from the blood into the tissues and produce cytokines to attract monocytes and a constellation of other inflammatory cells, thereby establishing the particular inflammatory environment of the tissue. In the mouse and in humans two phenotypically and functionally distinct subsets of monocytes have been identified either as inflammatory/classical (CD11b+ Gr1hi) or reparative/non-classical (CD11b+ Gr1lo) monocytes that traffic to the site of inflammation in temporally distinct waves depending on the tissue and type of injury.2,3 Although this model of inflammatory trafficking is well established, the mechanisms and key players that control preferential monocyte migration and their role in disease progression and healing remain to be elucidated.
CD13 is a transmembrane ectopeptidase that is ubiquitously expressed in specific cell types of a variety of tissues and, pertinent to this study, CD13 expression is induced on the endothelium lining blood vessels in response to tissue injury or infection. Activation of monocytic CD13 with cross-linking antibodies induces CD13 phosphorylation by Src, leading to CD13-dependent homotypic monocyte/endothelial adhesion, so implicating CD13 as an inflammatory adhesion molecule.4,5 At baseline, global CD13 knockout (CD13KO) mice are phenotypically unremarkable6 but demonstrate clearly altered pathologies when challenged in different models of ischaemic injury.7,8 For example, we have seen that the numbers and profiles of infiltrating myeloid populations are skewed in CD13KO mice in response to permanent coronary occlusion, contributing to adverse remodelling and diminished cardiac repair.7 Furthermore, the lack of CD13 leads to compromised reperfusion and muscle regeneration after hindlimb ischaemia due in part to a CD13-dependent reduction in specific myeloid cell subsets, in particular Gr1hi inflammatory monocytes and dendritic cells.8 In the current study, using adoptive transfer, antibody blocking and gain of function models we demonstrate that both endothelial and monocytic CD13 expression is essential for inflammatory cell infiltration in response to thioglycollate (TG) -induced peritonitis, confirming that altered trafficking contributes to the phenotypes found in our in vivo studies.7,8 In addition, species- and domain-specific chimeras verified that the CD13 C-terminus determines monocyte/endothelial adhesion and myeloid cell trafficking. Therefore, this investigation confirms the requirement for CD13 expression for adhesion and trafficking of myeloid cell subsets and further clarifies the molecular mechanisms underlying CD13-mediated monocyte/endothelial adhesion during the process of cellular migration in vivo.
Materials and methods
Animals
The global FVB CD13 CD13KO mouse was generated at the Gene Targeting and Transgenic Facility as published.6 All animals were housed under specific pathogen-free conditions with a 12 hr light/dark cycle and controlled temperature at the University of Connecticut Health Center animal facilities and all procedures were performed in accordance with Institutional and Office of Laboratory Animal Welfare guidelines.
Cell lines
Mouse monocytic cell line, WEHI 78/24, human cervical cancer cell lines, C33a and human leukaemic monocytic cell line, U937 were used in this study.
TG-induced sterile peritonitis
Wild-type CD13 (CD13WT) and CD13KO FVB/N, age-matched male mice 6–8 weeks of age were injected with 1 ml of 3% TG intraperitoneally. Peritoneal lavage cells were obtained after 48 hr for flow cytometric or immunofluorescence analysis.
Flow cytometry
Cells were isolated from peritoneal lavage, spleen, bone marrow and peripheral blood 48 hr after TG administration and stained with a cocktail of antibodies at 4° for 30 min: anti-CD45.1-phycoerythrin (PE), anti-F480-FITC, anti Gr-1-APCe780, anti-CD3/CD19/NK1.1/Ly6G-AF700, anti-CD11c-PECy7 and anti-CD11b-Pacific blue and UV live dead dye (to eliminate dead cells). Flow cytometry was performed on LSRII (Becton Dickinson, Franklyn Lakes, NJ) and the data were analysed with FlowJo software (Tree Star, Ashland, OR). Live/dead cells were negatively gated for T cells, B cells, natural killer (NK) cells and Ly6G+ cells to eliminate lymphocytes, NK cells and neutrophils, respectively. Next the CD45+ cells were analysed for macrophages (F480+ CD11b+), dendritic cells (CD11c+ CD11b+), inflammatory monocytes (Gr-1hi CD11b+) and reparative monocytes (Gr-1lo CD11b+).
Chimeric CD13 expression constructs
The murine/human (M/H) CD13 chimeric molecule was created using the hCD13 TOPO vector and digested stepwise with NotI and then EcoRI to eliminate the N-terminal half. The NotI site is not conserved between species, so a mouse CD13 forward primer was designed to introduce a NotI site. Digestion was performed by creating forward primers and reverse primers for both the C-terminal and N-terminal half of the mouse cDNA. H/M CD13 chimera used an EcoRI and PmeI site to eliminate the C-terminal half of human CD13. All human, mouse, M/H, H/M constructs/chimera cDNAs were cloned into the pcDNA/V5/GW/D-TOPO (Invitrogen, San Diego, CA) according to the manufacturer's instructions.
Retroviral vector construction and infection
The V5 tagged CD13 was excised and cloned into the retroviral expression vector pBM-IRES-Puro. High titre virus preparations were obtained using a Phoenix amphotropic packaging cell line (Orbigen, San Diego, CA) as previously described.5 For infection of C33a, 50% confluent dish was treated with 10 ml virus stock in the presence of 5 μg/ml polybrene. C33a were subject to virus medium for 24 hr and allowed to recover for 48 hr before selection. For infection of WEHI 78/24 cells, 1 × 105 cells were resuspended in 5 ml virus stock in 15 ml conical tube and centrifuged at 800 g for 30 min at 32° in the presence of 5 μg/ml polybrene. After retroviral infection, cells were cultured for an additional 18 hr in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, antibiotics and l-glutamine. CD13-V5 over-expressing cells were enriched by puromycin selection (1 μg/ml for 36 hr).
Quantitative PCR
Gr-1hi, Gr-1int and Gr-1lo monocyte cell populations were sorted by FACS and analysed for CD13 expression. RNA was isolated using Trizol according to the manufacturer's instructions (Invitrogen Corporation, Carlsbad, CA). The PCR primers for CD13 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were as follows:
CD13 F– 5'-TCACAGTGATAACGGGAAAGCCCA-3'
CD13 R– 5'-ATAAGCTCCGTCTCAGCCAATGGT-3'
GAPDH F– 5'-ACCACAGTCCATGCCATCAC-3'
GAPDH R– 5'-TCCACCACCCTGTTGCTGTA-3'
Western blot analysis
WEHI 78/24 and C33a cell lysates were separated by SDS–PAGE and probed for CD13. GAPDH and β-actin were used as loading controls.
Flow cytometry analysis
C33a and WEHI 78/24 cells were stained with human CD13 monoclonal antibody (mAb) 452 followed by goat anti-rat Alexa488 and then analysed for human CD13 by flow cytometry.
Quantitative adhesion assay
C33a epithelial cell constructs were seeded (0·5 × 106/ml) in 96-well plates and grown to confluency for 24 hr. Calcein-labelled (30 min labelling at 37°) WEHI 78/24 cell constructs and treated with CD13 cross-linking mAb (452) or IgG control (30 min at 37°) were allowed to interact/adhere with C33a cells for 30 min at 37°. Unbound WEHI 78/24 cells were washed extensively. The adherent cells were lysed and fluorescence was quantified by a fluorescence plate reader (Bio-Rad, Model 680; Bio-Rad, Hercules, CA). Antibody cross-linking was also performed on the epithelial C33a layer to the various CD13 constructs used as specified in the figure legend. Each condition was assayed in triplicate.
Isolation of splenic myeloid cells
Total splenocytes from CD13WT and CD13KO mice were run through a Ficoll-Paque gradient (GE Healthcare, Piscataway, NJ) and cells collected from the interface (lymphocytes and myeloid cells) were incubated with anti-CD3/CD19/NK1.1-PE antibodies followed by treatment with PE-conjugated-microbeads and passed through a MACS column to exclude the T/B/NK1.1 cells according to the manufacturer's protocol (Miltenyi Biotec, Auburn, CA).
Adoptive transfer of splenic myeloid cells and mCD13 WEHI 78/24
CD13WT and CD13KO splenic myeloid cells or WEHI 78/24 transfectants were differentially labelled with the fluorescent dyes PKH67 (green) and PKH26 (red) as indicated according to the manufacturer's instructions (Sigma-Aldrich, St Louis, MO). The mCD13 WEHI 78/24 cells were either untreated or treated with 1 μg of SL-13 mAb. Equal numbers of differentially labelled cells were adoptively transferred via tail vein followed by TG injection. After 48 hr, peritoneal lavage cells were subjected to cytospin for immunofluorescence analysis and flow cytometry.
In vitro membrane dye labelling
Fluorescence of differently labelled cells with PKH67 (green) and PKH26 (red) dye was quantified. Thirty non-overlapping fields at 20× were individually counted for green and red dye. Images were photographed with an Optronics camera attached to Ziess Axioskop 2 plus microscope using the Zeiss Achroplan 40× objective and photographed with an Axiocam MRC camera (0·63× magnification) attached to a Zeiss Axioplan 2 microscope using a 10×, 20×, 40× and 63× objective.
Statistical analysis
Results are presented as mean ± SEM. Statistical analysis was performed using an unpaired, two-tailed t-test. Differences were considered significant at P < 0·05.
Results
Inflammatory cell profiles are skewed in CD13KO animals in response to TG-induced peritonitis
We have previously shown that CD13 acts as a homotypic adhesion molecule regulating monocyte/endothelial adhesion in vitro and that mice lacking CD13 show altered inflammatory cell profiles in injury models, suggesting that it may participate in in vivo inflammatory processes via its adhesive properties. To directly address this possibility, we initially evaluated inflammatory monocyte profiles in the bone marrow, spleen, peripheral blood and peritoneal exudates of CD13WT and CD13KO mice 48 hr after TG injection by flow cytometry (Fig. 1a,b). At this time point, monocyte trafficking predominates with little contribution from neutrophils.9 Although profiles of CD11b+ Gr1hi and CD11b+ Gr1lo cells from the bone marrow and spleen were indistinguishable, a clear difference was observed in the cells infiltrating the peritoneum. The CD11b+ Gr1hi pro-inflammatory monocyte population was diminished by nearly 70% in the CD13KO animals compared with wild type, while there was no significant difference between the CD11b+ Gr1lo reparative monocyte subsets (Fig. 1b,c). Importantly, analysis of CD13WT and CD13KO bone marrow, spleen, peripheral blood and lavage showed equivalent levels of immune cells under resting conditions (data not shown). Interestingly, the inflammatory monocyte population was elevated in the peripheral blood of CD13KO animals, perhaps indicating an inability of this subset to enter the peritoneum (Fig. 1d). In agreement with this notion, while the percentages of reparative monocytes were equivalent in either genotype, the percentages of F4/80+ macrophages and CD11c+ dendritic cells were also significantly decreased in the peritoneal exudates of the null animals (Fig. 1c) with a concomitant increase of these subpopulations in the blood (Fig. 1d and data not shown).
Figure 1.

Inflammatory cell profiles are altered in thioglycollate-elicited peritoneal exudate of CD13 knockout (CD13KO) mice. (a) Pseudo-colour plots of thioglycollate-elicited peritoneal exudates show sequential gating to obtain a CD45+ haematopoietic cell population. T/B lymphocytes, Ly6G+ neutrophils and natural killer cells were gated out of the live cell population and the remaining CD45+ cells were analysed. (b) Bone marrow, spleen, peripheral blood and 48 hr-post-thioglycollate injected peritoneal lavage of CD13 wild-type (CD13WT) and CD13KO animals were analysed by flow cytometry. Pseudo-colour plots of infiltrating inflammatory monocytes-Gr-1hi CD11b+ and reparative monocytes- Gr-1lo CD11b+, in the live CD45+ cell population were analysed in the different organs of CD13WT and CD13KO mice. (c) Quantification of percentage of infiltrating inflammatory monocytes-Gr-1hi CD11b+, reparative monocytes- Gr-1lo CD11b+, macrophages – F4/80hi CD11b+, and dendritic cells – CD11chi CD11b+ in the live CD45+ cell population in the peritoneal lavage and (d) peripheral blood of CD13WT and CD13KO mice. Data represent average of three independent experiments. Error bars represent mean ± SEM for CD13WT (n = 8) and CD13KO (n = 8) mice; *P < 0·05, **P < 0.01.
Mouse inflammatory monocyte subsets in the inflamed peritoneum are highly CD13 positive
FACS-sorting and quantitative RT-PCR of peritoneal exudate cells isolated from CD13WT mice after 48 hr of TG administration revealed that CD11b+ Gr1hi monocytes expressed markedly higher levels of CD13 mRNA (Fig. 2a,b) compared with the reparative CD11b+ Gr1lo monocytes. This is consistent with our observations that the lack of CD13 primarily affected infiltration of CD13-positive immune cell populations including inflammatory monocytes, macrophages and dendritic cells and supports the contribution of CD13 to preferential trafficking of subsets of inflammatory cells in vivo.7,10
Figure 2.
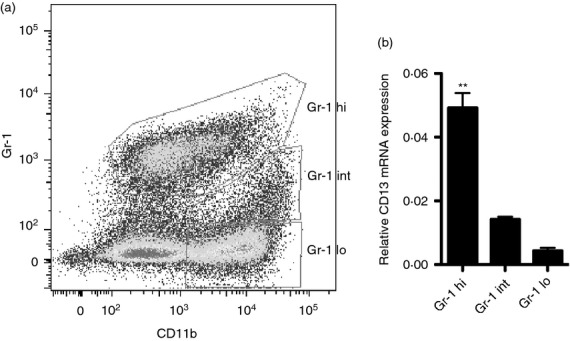
(a) CD13 is highly expressed in the inflammatory monocytes isolated from the inflamed peritoneum. Thioglycollate (TG) -administered peritoneal lavage was isolated from four CD13 wild-type (CD13WT) mice and pooled for FACS sorting. Pseudo-colour plot of infiltrating inflammatory monocytes – Gr-1hi CD11b+, intermediate population – Gr-1int CD11b+ and reparative monocytes – Gr-1lo CD11b+ in the live CD45+ (T/B/Ly6G/natural killer)− cell population. (b) Relative CD13 mRNA levels in Gr1hi CD11b+, Gr1int CD11b+ and Gr1lo CD11b+ monocyte subsets; **P < 0.01.
Peritoneal infiltration of CD13KO myeloid cells is reduced following adoptive transfer
While our results in intact animals are consistent with impaired peritoneal infiltration, it is formally possible that cells lacking CD13 are differentially released from the bone marrow or other haematopoietic reservoirs.2 To directly address whether CD13 controls inflammatory cell infiltration in vivo, we adoptively transferred mixtures of equal numbers of differentially labelled CD13WT (green) and CD13KO (red) isolated primary splenic myeloid cells (T/B/NK-depleted) or total bone marrow cells directly into the circulation of wild-type animals immediately before TG injection (Fig. 3a). Equivalent labelling with the membrane dyes was confirmed by microscopic analysis of cytospun cells before injection (Fig. 3a). Flow cytometric analysis of inflammatory exudates after 48 hr showed that fewer total CD13KO splenic myeloid cells (Fig. 3b,c), or bone marrow cells (data not shown) entered the peritoneum. Further analysis of the peritoneal lavage revealed a specific reduction in the numbers of Gr-1hi inflammatory monocytes, F4/80+ macrophages and CD11c+ dendritic cells of CD13KO labelled cells compared with the CD13WT cells (Fig. 3d), although ratios were equivalent before injection.6 These results indicate that monocytic CD13 was necessary for optimal trafficking despite the fact that the wild-type endothelium expresses CD13. Identical results in experiments where the dyes were interchanged argued against dye-specific effects on trafficking (data not shown). Therefore, CD13 regulates inflammatory cell egress from the circulation into the peritoneum.
Figure 3.

CD13 regulates trafficking of inflammatory cells to the peritoneum. Equal numbers of differentially labelled CD13 wild-type (CD13WT) and CD13 knockout (CD13KO) splenic myeloid cells were adoptively transferred in thioglycollate-challenged CD13WT mice. (a) Microscopic analysis of cytospun green-labelled CD13WT and red-labelled CD13KO splenic myeloid cells before injection and in the thioglycollate -and cell-injected peritoneal lavage of CD13WT mice. Objectives; 63 × and 20 ×. Blue = DAPI nuclear stain. (b, c) Flow cytometric analysis of total green-labelled CD13WT and red-labelled CD13KO cells in the peritoneal exudates of CD13WT animals. The contribution of background autofluorescence for both red and green cells was subtracted from total cells. Data represent number of labelled cells ± SEM, n = 4 mice/group; **P < 0·01. (d). Percent of labelled green-CD13WT and red-CD13KO cells in Gr-1hi CD11b+ inflammatory monocytes, Gr-1lo CD11b+ reparative monocytes F4/80+ CD11b+ macrophages and CD11c+ CD11b+ dendritic cells. Data represent average of three independent experiments. Error bars represent mean ± SEM for CD13WT (n = 5) mice; **P < 0·01.
CD13 expression in monocytes is a prerequisite for optimal cell trafficking in vivo
To determine whether enforced CD13 expression confers a trafficking advantage to monocytes and confirm that infiltration of myeloid cells into the peritoneal cavity is indeed CD13 specific, we adopted a gain-of-function approach. Adoptive transfer of pools of CD13-negative murine monocytic WEHI78/24 cells infected with murine CD13 cDNA expression vectors (mCD13; red) or control cells infected with empty vector (EV; green) into the circulation of wild-type animals resulted in significantly more peritoneal-resident mCD13-expressing cells than vector control cells, suggesting that CD13 expression positively regulates monocyte trafficking (Fig. 4a). Similarly, pre-treatment of mCD13-expressing WEHI cells with an anti-CD13 blocking mAb (SL13; green) resulted in comparable reductions in the infiltration of treated cells, confirming that the effect is indeed CD13 dependent (Fig. 4b). Importantly, adoptively transferred cells were not cleared by the immune system as equal numbers of both populations were present in the peripheral blood of unchallenged animals (Fig. 4c).
Figure 4.
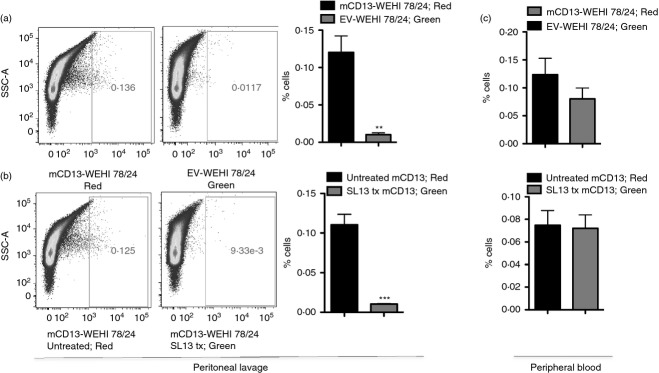
CD13 expression in monocytic cell line drives myeloid cell infiltration into the peritoneal cavity. Adoptive transfer of differentially labelled murine monocytic WEHI78/24 cells infected with either (a) mouse CD13 (mCD13-red) and empty vector control (EV-green) or (b) untreated mouse CD13 (mCD13-red) and SL-13 blocking antibody-treated mouse CD13 (SL13 tx mCD13-green) into thioglycollate-administered CD13 wild-type (CD13WT) mice. (a, b) Preferential trafficking of cells expressing CD13 to the peritoneum was analysed by flow cytometry. (c) Percentage of cells present in the peripheral blood of unchallenged CD13WT animals. Data represent average of three independent experiments. Error bars represent mean ± SEM for CD13WT (n = 5) per mice group; **P < 0·01, ***P < 0·001.
CD13 mAb binds in a species- and domain-specific manner
To investigate if the binding of CD13 mAb is species and domain specific, we used chimeras of mouse or human CD13 constructs. CD13 is a type II protein comprised of seven domains (Fig. 5a–f); domain I consists of a short cytosolic tail, followed by the membrane spanning domain II that is highly conserved among various species.11 Extracellular domains III–VII contain the catalytic site of the enzyme and exhibit a greater degree of species variability (Fig. 5f). We constructed two different chimeric expression constructs, one containing the murine CD13 N-terminus (including domains I–V and 50% of domain VI) fused to the human CD13 C-terminus (including 50% of the remaining domain VI and domain VII), designated as M/H (Fig. 5d–f). The reciprocal chimera is designated H/M and contains the human CD13 N-terminus and the C-terminus of murine CD13 (Fig. 5e,f). WEHI monocytic and C33A adherent epithelial cells were infected with chimeric or control constructs. Flow cytometric and immunoblot analysis of the various cell lines verified the expression of the infected molecules and indicated that exogenous human and mouse CD13 was highly expressed in both the adherent and monocytic pools (Fig. 5g,h). The high degree of species specificity of our CD13 reagents was apparent as the anti-hCD13 only recognizes lines expressing hCD13 while the mouse anti-CD13 mAb only recognizes mCD13 (Fig. 5g,h). Finally, analysis of cell lines containing chimeric molecules indicated that these were expressed at lower levels than their respective control lines and that both of these antibodies recognize the C-terminal region of CD13 (Fig. 5g,h).
Figure 5.

Species- and domain-specific constructs of CD13. Figure represents (a) the different domains of CD13, (b) full-length human CD13, (c) full length mouse CD13, (d, f) M/H CD13 chimera containing mouse CD13 at the N-terminal and human CD13 at the C-terminal, (e, f) H/M CD13 chimera containing human CD13 at the N-terminal and mouse CD13 at the C-terminal. (g) Representative histogram of C33a and WEHI 78/24 expressing human CD13 and M/H CD13 chimera with anti-human CD13 monoclonal antibody 452 by flow cytometry. (h) Representative immunoblot analysis of C33a and WEHI 78/24 expressing mouse and H/M CD13 constructs with anti-mouse SL13 antibody. Data represent average of two independent experiments.
The C-terminal domain of CD13 participates in cellular trafficking in response to inflammation
To identify the domain responsible for CD13-dependent cell trafficking, we intravenously injected differentially labelled cells from each of the CD13-expressing monocyte lines or empty vector control cells into CD13WT mice followed by TG injection. Flow cytometric analysis of the labelled peritoneal cells in the lavage indicated markedly higher numbers of cells expressing the H/M CD13 chimera (murine C-term) compared with either vector control or M/H CD13 chimera (Fig. 6a,b). However, the percentage of cells in the circulation in either chimeras or the control vector remained the same (Fig. 6a,b). These results directly demonstrate that the C-terminal domain of CD13 participates in inflammatory cell trafficking.
Figure 6.

The C-terminal domain of CD13 is responsible for cell infiltration into the inflamed peritoneum. A mixture of equal number of (a) WEHI 78/24 cells transfected with H/M CD13-red and empty vector-green or a mixture of (b) H/M CD13-red and M/H CD13-green were injected into CD13 wild-type (CD13WT) mice and cell trafficking to the injured peritoneum and peripheral blood was analysed by flow cytometry 48 hr after thioglycollate injection. Data represent average of three independent experiments. Error bars represent mean ± SEM for CD13WT (n = 5) mice; *P < 0·05, **P < 0·01.
The C-terminal domain of CD13 mediates homotypic monocyte/endothelial cell adhesion
We have shown that basal adhesion of primary CD13KO macrophages to endothelial cells is compromised and that CD13 activation on either monocytes or endothelial cells with cross-linking mAbs induces CD13-dependent adhesion in vitro.4–6 To verify that the effects on trafficking obtained with our chimeric monocytic cell lines in vivo are the result of CD13 homotypic adhesion, we performed an in vitro adhesion assay using our engineered lines and the activating anti-hCD13 mAb 452. We added the various CD13-expressing monocyte cell lines to hCD13-expressing adherent epithelial or vector control cells that had been treated with the activating anti-hCD13 mAb 452 or isotype control antibodies and assessed adhesion (Fig. 7). Our results indicated that, similar to our in vivo results, monocytes that express the hCD13 C-terminus (hCD13 and M/H-CD13) mediate homotypic adhesion to hCD13+ monolayer cells treated with the activating anti-hCD13 mAb 452 (Fig. 7a). Similarly, activation of the U937 human monocytic cell line with the 452 antibody induced homotypic adhesion to monolayers expressing either hCD13 or M/H CD13, but not to mCD13 or H/M-expressing monolayers (Fig. 7b). Likewise, activation of the WEHI M/H CD13 with the human CD13 mAb 452 induced strong adhesion in the monolayer cells expressing total hCD13 or the chimera containing the human C-terminus, although at decreased levels (Fig. 7c), likely due to the reduced expression levels of CD13 protein produced from this chimeric construct (Fig. 5g,h). Therefore, the C-terminal domain of CD13 on both monocytes and the monolayer cells mediates activated adhesion.
Figure 7.

The C-terminal domain of CD13 on both monocyte and the epithelium is necessary for homotypic cell adhesion. (a) C33a cells expressing empty vector or human CD13 were cross-linked with control IgG or activating anti-hCD13 monoclonal antibody 452. Calcein-labelled WEHI 78/24 expressing empty vector, human CD13, mouse CD13, M/H CD13 or H/M CD13 constructs were added to the activated monolayer and homotypic cell adhesion was measured as fluorescence intensity at optical density at 485 and 530 nm. WEHI expressing H CD13 and M/H CD13 showed significant increase in adhesion with 452-activated C33a expressing HCD13 compared with WEHI expressing EV, M CD13 and H/M CD13. (b) Calcein-labelled human monocytic cell line U937, was cross-linked with IgG or 452 and extent of adhesion with the C33a monolayer expressing empty vector, human CD13, mouse CD13, M/H CD13 or H/M CD13 constructs was assessed as in (a). C33a expressing H CD13 and M/H CD13 showed significant increase in adhesion with 452-activated U937 cells. (c) Similarly, calcein-labelled WEHI 78/24 expressing empty vector, human CD13 or M/H construct were activated with IgG or 452, followed by interaction with the monolayer, the C33a cells expressing empty vector, human CD13, mouse CD13, M/H and H/M CD13 constructs. C33a expressing H CD13 and M/H CD13 showed significant increase in adhesion with 452 activated WEHI expressing HCD13 and M/H CD13 compared with C33a expressing EV, M CD13 and H/M CD13. Homotypic adhesion of monocyte and the monolayer were determined fluorometrically. Data represent ratio of relative fluorescence unit (RFU) measured upon activation with 452 to control IgG. Data represents average of three independent experiments. Error bars represent mean ± SEM; **P < 0·01.
CD13 expression on both monocytes and endothelium is critical for optimal cell trafficking in vivo
To further explore the requirement for CD13 expression on both monocytes and the endothelium in immune cell infiltration in response to injury, we intravenously injected differentially labelled wild-type and CD13KO splenic myeloid populations (T/B/NK-depleted) into either CD13WT or CD13KO recipients along with peritoneal TG administration. Microscopic (Fig. 8a) and flow cytometric (Fig. 8b) analysis of cells present in the peritoneal lavage after 48 hr exhibited significantly reduced numbers of CD13KO labelled cells in the CD13WT recipient, confirming a monocyte-specific role for CD13 (Fig. 8a,b). Further, there was a similar reduction in CD13WT cells in CD13KO recipients (compared with CD13WT recipient) as indicated by a significant decrease in infiltrating cells of both genotypes (Fig. 8a,b). Therefore, in agreement with our in vitro data, endothelial CD13 is also critical for monocyte trafficking from the circulation, despite positive CD13 expression on monocytes. Importantly, infiltrating CD13KO monocytes were further decreased in CD13KO recipients, consistent with our in vitro findings that CD13 on both monocytes and the endothelium plays a functional role in inflammatory cell trafficking to the site of injury.4
Figure 8.
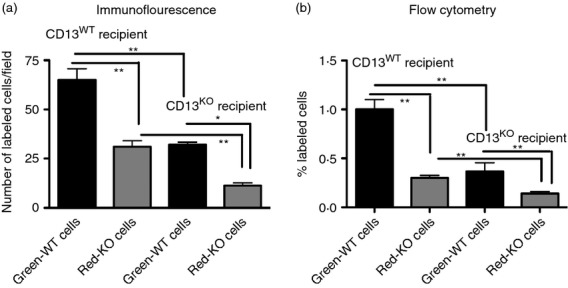
CD13 expression on both monocyte and the endothelium is essential for optimal cell trafficking in vivo. A mixture of equal number of CD13 wild-type (CD13WT) and CD13 knockout (CD13KO) splenic myeloid cells differentially labelled with green and red dye respectively were adoptively transferred to thioglycollate-administered CD13WT and CD13KO recipients. After 48 hr, peritoneal exudates from recipients of both genotypes were isolated, cytospun and analysed by microscopy (a) and flow cytometry (b). (a) Total number of green-WT or red-KO cells was calculated in the recipient mice (n = 5/genotype and 30 non-overlapping fields per animal were calculated). (b) Flow cytometric analysis of percentage of infiltrating green-WT and red-KO cells in the peritoneal lavage of CD13WT and CD13KO recipients (n = 3/mice per group) Data represent average of three independent experiments. Error bars represent mean ± SEM; *P < 0.05, **P < 0·01.
Discussion
CD13 was originally identified as a marker of myeloid cells where it was shown to be expressed to varying degrees on myeloid progenitors and their differentiated progeny.12 Our subsequent investigations have demonstrated that in the haematopoietic system this molecule is expressed at relatively higher levels on particular myeloid cell subsets such as CD8+ dendritic cells where it regulates receptor-mediated antigen uptake and subsequent cross-presentation to T cells.10 Similarly, the high CD13 expression on circulating monocytes led us to explore its potential role as an inflammatory adhesion molecule in models of heart and skeletal muscle ischaemic injury.7,8 In these studies we found that a global lack of CD13 led to distorted inflammatory cell profiles, so contributing to adverse healing and aberrant functional recovery. Interestingly, while the ratios of infiltrating cells were clearly skewed in both tissues, myeloid subsets were differentially affected, supporting the notion that myeloid cell trafficking patterns may be injury- and tissue-specific,3 and importantly, that in part, CD13 expression may dictate these patterns. In the current study we extend these observations using the TG-induced peritonitis model and directly demonstrate that CD13 is a physiological regulator of cell trafficking in vivo and identify the particular domain(s) of the protein that mediates monocyte adhesion.
CD13 is identical to aminopeptidase N, a cell surface peptidase that cleaves neutral amino acids from the N-terminus of small peptides and participates in hydrolysis of bioactive peptides in various tissues such as neuropeptides, peptides of the renin–angiotensin system and intestinal dietary peptides.13–15 However, studies from our laboratory and others have determined that in addition to its ability to hydrolyse small peptides, CD13 is a multifunctional protein that plays both enzyme-dependent and -independent roles (reviewed in ref. 16). CD13 serves as a species-specific receptor for a subtype of coronaviruses in a number of species, and this function is not affected by inhibitors of its enzymatic activity.17 More recently, we have identified CD13 as a homotypic adhesion molecule mediating monocytic/endothelial CD13 adhesion induced by activation upon cross-linking by mAbs or virus binding, which is also enzyme-independent.4 We have further shown that cross-linking induces Src-dependent phosphorylation of the CD13 cytoplasmic tail and CD13 is phosphorylated in inflammatory tissues, supporting the notion that CD13 activation and adhesion is physiological and participates in inflammatory cell mobilization.5 In the current study, flow cytometric analyses of infiltrating cells isolated from CD13WT and CD13KO TG-induced peritoneal exudates along with gain/loss of function adhesion studies with cells engineered to express mCD13 or treated with the blocking mAb SL13 strongly supports that the lack of CD13 directly affects monocyte–endothelial cell adhesion and subsequent trafficking.
As mentioned above, mammalian CD13 is a receptor for group 1 coronaviruses including the human coronavirus HCov229E, feline coronavirus, porcine transmissible gastroenteritis coronavirus, canine coronavirus, each of which exhibit highly species-specific binding to CD13 of their natural host species.18,19 An extensive study using cross-species chimeric expression constructs mapped the CD13 residues dictating the host range to small, species-specific amino acid differences in the CD13 C-terminus.20 We have found that CD13-dependent adhesion is also highly species-specific. We expressed chimeric expression constructs of the N- and C-terminal domains of human and mouse CD13 in adherent cells and monocytes to identify the region of the protein that mediates adhesion. In vitro monocyte/monolayer adhesion assays indicated that the C-terminal half of the protein mediates adhesion on both the monocytes and adherent cells. Furthermore, adoptive transfer of differentially labelled cells expressing the mixed chimeras determined that in vivo cell mobilization to the damaged tissue in peritonitis is also dependent on this large C-terminal region. Further study will determine if the specific regions that dictate virus host range also determine species-specific adhesion.
In acute inflammation, circulating monocytes migrate across the endothelium to gain access to the site of infection or injury. This interaction involves initial monocyte adhesion to activated endothelial cells and subsequent transendothelial migration by mechanisms mediated by adhesion molecules and activating chemokines. It has been well established that two distinct subsets of monocytes exist in mice and humans that are differentially channelled in steady-state versus inflammatory conditions. The ‘inflammatory’ monocytes (Gr-1high Ly6Chigh, mouse; CD14+ CD16−, human), home to sites of inflammation or infection and can differentiate into both activating or inhibitory macrophage and dendritic cell subsets, depending on signals from the specific microenvironment or the nature of the infection.3,21 The second group, the resident/reparative monocytes (Gr-1lo CD11b+) populate resting tissues and replenish tissue macrophages or dendritic cells. In rodents, the numbers of inflammatory monocytes specifically increase in the circulation in response to infection or injury and infiltrate more readily or often exclusively compared with the reparative subset.22–25 For example, studies have demonstrated that only inflammatory/classical monocytes migrate to sites of chemical injury of skeletal muscle26 or the site of intracutaneous injection of latex beads,27 while in the infarcted heart, both inflammatory and reparative monocytes traffic in distinct temporal patterns to perform their functions during inflammation and healing.2 Finally, the reparative subset has also been demonstrated to perform a patrolling function in the steady state where these cells monitor the healthy endothelium for sites of inflammation and rapidly extravasate to initiate the inflammatory response induced by irritants, aseptic wounding, or infection.1 Trafficking of the two subsets has been shown to be distinctly regulated by signals elicited by ligation of specific chemokine receptors CCR2 and CX3CR1 as well as adhesion molecules such as LFA-1,1 contributing to coordinated remodelling of injured tissue. However, given the complex nature of subset trafficking, additional regulatory molecules must be involved. In the current study, we have shown that CD13 is more highly expressed on the inflammatory monocyte subset and it is the migration of this population that is predominantly affected in CD13KO animals, suggesting that CD13 may constitute one of these auxiliary regulatory proteins. Moreover, the relative contributions of the distinct patterns of trafficking, differential differentiation of cells once at the site of inflammation, and sequential trafficking strategies in the different tissues are currently unknown and will require the identification of the markers and mechanisms by which these are regulated. The possibility that the selective expression of CD13 on myeloid cells as well as the inflamed endothelium may orchestrate some element of this process is intriguing and is the subject of active investigation in our laboratory.
Acknowledgments
This work was supported by Public Health Service grant HL-70694 from the National Heart, Lung and Blood Institute and the State of Connecticut Stem Cell Research Program grant #09-SCA-UCHC-009.
Author Contributions
MG, LHS, CG, KV designed experiments. MG, CG, MMR, KV, JS, FP, LC performed experiments. MG, LHS, CG, KV interpreted data. MG, LHS, CG wrote manu-script.
Disclosures
The authors declare that they have no competing interests.
References
- 1.Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 2.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingersoll MA, Platt AM, Potteaux S, Randolph GJ. Monocyte trafficking in acute and chronic inflammation. Trends Immunol. 2011;32:470–7. doi: 10.1016/j.it.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mina-Osorio P, Winnicka B, O'Conor C, et al. CD13 is a novel mediator of monocytic/endothelial cell adhesion. J Leukoc Biol. 2008;84:448–59. doi: 10.1189/jlb.1107802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramani J, Ghosh M, Rahman MM, Caromile LA, Gerber C, Rezaul M, Han D, Shapiro LH. Tyrosine phosphorylation of CD13 regulates inflammatory cell–cell adhesion and monocyte trafficking. J Immunol. 2013;191:3905–12. doi: 10.4049/jimmunol.1301348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winnicka B, O'Conor C, Schacke W, et al. CD13 is dispensable for normal hematopoiesis and myeloid cell functions in the mouse. J Leukoc Biol. 2010;88:347–59. doi: 10.1189/jlb.0210065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira FE, Cronin C, Ghosh M, et al. CD13 is essential for inflammatory trafficking and infarct healing following permanent coronary artery occlusion in mice. Cardiovasc Res. 2013;100:74–83. doi: 10.1093/cvr/cvt155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahman MM, Ghosh M, Subramani J, Fong GH, Carlson M, Shapiro LH. CD13 regulates anchorage and differentiation of the skeletal muscle satellite stem cell population in ischemic injury. Stem Cells. 2013 doi: 10.1002/stem.1610. doi: 10.1002/stem.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang J, Zarbock A, Gomez I, et al. Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood. 2011;118:786–94. doi: 10.1182/blood-2010-11-321406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh M, McAuliffe B, Subramani J, Basu S, Shapiro LH. CD13 regulates dendritic cell cross-presentation and T cell responses by inhibiting receptor-mediated antigen uptake. J Immunol. 2012;188:5489–99. doi: 10.4049/jimmunol.1103490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong AH, Zhou D, Rini JM. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J Biol Chem. 2012;287:36804–13. doi: 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shipp MA, Look AT. Hematopoietic differentiation antigens that are membrane-associated enzymes: cutting is the key! Blood. 1993;82:1052–70. [PubMed] [Google Scholar]
- 13.Look AT, Ashmun RA, Shapiro LH, Peiper SC. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J Clin Invest. 1989;83:1299–307. doi: 10.1172/JCI114015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noren K, Sjostrom H, Danielsen EM, Cowell GM, Skovbjerg H. Molecular and Cellular Basis of Digestion. Amsterdam: Elsevier; 1986. [Google Scholar]
- 15.Matsas R, Stephenson SL, Hryszko J, Kenny AJ, Turner AJ. The metabolism of neuropeptides. Phase separation of synaptic membrane preparations with Triton X-114 reveals the presence of aminopeptidase N. Biochem J. 1985;231:445–9. doi: 10.1042/bj2310445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mina-Osorio P. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med. 2008;14:361–71. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357:420–2. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tresnan DB, Levis R, Holmes KV. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J Virol. 1996;70:8669–74. doi: 10.1128/jvi.70.12.8669-8674.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tresnan DB, Holmes KV. Feline aminopeptidase N is a receptor for all group I coronaviruses. Adv Exp Med Biol. 1998;440:69–75. doi: 10.1007/978-1-4615-5331-1_9. [DOI] [PubMed] [Google Scholar]
- 20.Tusell SM, Schittone SA, Holmes KV. Mutational analysis of aminopeptidase N, a receptor for several group 1 coronaviruses, identifies key determinants of viral host range. J Virol. 2007;81:1261–73. doi: 10.1128/JVI.01510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–60. doi: 10.1038/nri2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 23.Sunderkotter C, Nikolic T, Dillon MJ, van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–7. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 24.Yrlid U, Jenkins CD, MacPherson GG. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J Immunol. 2006;176:4155–62. doi: 10.4049/jimmunol.176.7.4155. [DOI] [PubMed] [Google Scholar]
- 25.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu C, Edwards EW, Tacke F, et al. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med. 2004;200:1231–41. doi: 10.1084/jem.20032152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–69. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]