

See Borgkvist et al. (doi:10.1093/brain/awu150) for a scientific commentary on this article.
D2 autoreceptors and L-type calcium channels are both implicated in Parkinson’s disease, but how they interact is unclear. Dragicevic et al. reveal that L-type calcium channels can modulate D2-autoreceptor responses via the neuronal calcium sensor NCS-1. This dopamine-dependent signalling network is altered in Parkinson’s disease and could represent a therapeutic target.
Keywords: D2-autoreceptor, isradipine, Parkinsons disease, l-DOPA, cocaine
Abstract
Dopamine midbrain neurons within the substantia nigra are particularly prone to degeneration in Parkinson’s disease. Their selective loss causes the major motor symptoms of Parkinson’s disease, but the causes for the high vulnerability of SN DA neurons, compared to neighbouring, more resistant ventral tegmental area dopamine neurons, are still unclear. Consequently, there is still no cure available for Parkinson’s disease. Current therapies compensate the progressive loss of dopamine by administering its precursor l-DOPA and/or dopamine D2-receptor agonists. D2-autoreceptors and Cav1.3-containing L-type Ca2+ channels both contribute to Parkinson’s disease pathology. L-type Ca2+ channel blockers protect SN DA neurons from degeneration in Parkinson’s disease and its mouse models, and they are in clinical trials for neuroprotective Parkinson’s disease therapy. However, their physiological functions in SN DA neurons remain unclear. D2-autoreceptors tune firing rates and dopamine release of SN DA neurons in a negative feedback loop through activation of G-protein coupled potassium channels (GIRK2, or KCNJ6). Mature SN DA neurons display prominent, non-desensitizing somatodendritic D2-autoreceptor responses that show pronounced desensitization in PARK-gene Parkinson’s disease mouse models. We analysed surviving human SN DA neurons from patients with Parkinson’s disease and from controls, and detected elevated messenger RNA levels of D2-autoreceptors and GIRK2 in Parkinson’s disease. By electrophysiological analysis of postnatal juvenile and adult mouse SN DA neurons in in vitro brain-slices, we observed that D2-autoreceptor desensitization is reduced with postnatal maturation. Furthermore, a transient high-dopamine state in vivo, caused by one injection of either l-DOPA or cocaine, induced adult-like, non-desensitizing D2-autoreceptor responses, selectively in juvenile SN DA neurons, but not ventral tegmental area dopamine neurons. With pharmacological and genetic tools, we identified that the expression of this sensitized D2-autoreceptor phenotype required Cav1.3 L-type Ca2+ channel activity, internal Ca2+, and the interaction of the neuronal calcium sensor NCS-1 with D2-autoreceptors. Thus, we identified a first physiological function of Cav1.3 L-type Ca2+ channels in SN DA neurons for homeostatic modulation of their D2-autoreceptor responses. L-type Ca2+ channel activity however, was not important for pacemaker activity of mouse SN DA neurons. Furthermore, we detected elevated substantia nigra dopamine messenger RNA levels of NCS-1 (but not Cav1.2 or Cav1.3) after cocaine in mice, as well as in remaining human SN DA neurons in Parkinson’s disease. Thus, our findings provide a novel homeostatic functional link in SN DA neurons between Cav1.3- L-type-Ca2+ channels and D2-autoreceptor activity, controlled by NCS-1, and indicate that this adaptive signalling network (Cav1.3/NCS-1/D2/GIRK2) is also active in human SN DA neurons, and contributes to Parkinson’s disease pathology. As it is accessible to pharmacological modulation, it provides a novel promising target for tuning substantia nigra dopamine neuron activity, and their vulnerability to degeneration.
Introduction
Substantia nigra dopaminergic neurons project to the dorsal striatum and are important for voluntary movement. In contrast with neighbouring dopamine midbrain neurons in the ventral tegmental area, SN DA neurons are particularly prone to degeneration in Parkinson’s disease (Bjorklund and Dunnett, 2007; Schultz, 2010). The cause for this selective vulnerability of SN DA neurons is still unclear, and the typical motor symptoms of Parkinson’s disease occur when ∼70% of SN DA neurons are already lost (Collier et al., 2011; Shulman et al., 2011; Sulzer and Surmeier, 2012). Consequently, there is still no cure for Parkinson’s disease available, and current Parkinson’s disease therapies try to compensate the progressive loss of striatal dopamine by administering its precursor l-DOPA (still the gold standard in Parkinson’s disease therapy) and/or dopamine D2 receptor agonists (Collier et al., 2011; Gazewood et al., 2013; Olanow and Schapira, 2013). Understanding the molecular mechanisms of substantia nigra dopamine function and signalling in health and disease is the prerequisite for the development of novel neuroprotective Parkinson’s disease therapies. Dopamine D2-autoreceptors and Cav1.3 L-type-Ca2+ channels, calcium load and metabolic stress in SN DA neurons have already been identified as trigger factors, contributing to the progressive loss of SN DA neurons, striatal dopamine, and thus to Parkinson’s disease pathology (Liss and Roeper, 2010; Surmeier et al., 2012; Ford, 2014), but their interplay is still unclear.
Dopamine release from striatal axon terminals (and from soma and dendrites) of SN DA neurons is controlled by their electrical activity (Beckstead et al., 2007; Rice et al., 2011; Ford, 2014). In synaptic isolation, SN DA neurons display a so-called ‘pacemaker activity’ that is intrinsically generated by a concerted interplay of a range of voltage-gated ion channels, however, with still unclear individual contributions (Khaliq and Bean, 2010; Liss and Roeper, 2010; Surmeier et al., 2012). Pacemaker activity of SN DA neurons is inhibited by dopamine itself, via therapeutically relevant dopamine D2-autoreceptors, through activation of inwardly rectifying potassium channels (GIRK2) (Luscher and Slesinger, 2010; Anzalone et al., 2012; Gantz et al., 2013). Mouse models with mutations in PARK genes, which lead to familial forms of Parkinson’s disease (Gasser et al., 2011), show altered substantia nigra dopamine D2-autoreceptor responses (Goldberg et al., 2005; Tong et al., 2009), further linking D2-autoreceptor function to Parkinson’s disease pathology.
The contribution of, in particular, the Cav1.3 L-type-Ca2+ channels to pacemaker activity is more controversial than that of D2-autoreceptors. More precisely, a suggested postnatal age-dependent switch from a HCN (hyperpolarization-activated cyclic nucleotide gated) channel driven pacemaker in juvenile postnatal mouse SN DA neurons to a metabolically more challenging and Cav1.3 L-type-Ca2+ channel driven pacemaker in adult SN DA neurons, which would render them more vulnerable to Parkinson’s disease triggers, is highly disputed (Chan et al., 2007; Puopolo et al., 2007; Guzman et al., 2009; Drion et al., 2011; Surmeier et al., 2012). No other calcium mediated physiological functions of L-type-Ca2+ channels in SN DA neurons are known. However, blood–brain barrier permeable L-type-Ca2+ channel blockers (e.g. isradipine) do protect highly vulnerable SN DA neurons from degeneration in Parkinson’s disease and its chronic rodent models (Ritz et al., 2010; Ilijic et al., 2011; Surmeier et al., 2012), presumably by reducing substantia nigra dopamine calcium load and metabolic mitochondrial stress during pacemaking (Guzman et al., 2010; Surmeier and Schumacker, 2013); and they are already in clinical trials for pharmacological neuroprotective Parkinson’s disease therapy (Parkinson Study Group, 2013; ClinicalTrials. gov Identifier: NCT00909545). In contrast to wild-type, SN DA neurons from adult PARK7 (DJ-1) knockout mice display not only increased L-type-Ca2+ channel-generated metabolic stress levels, but also rapidly desensitizing somatodendritic D2-autoreceptor responses (Goldberg et al., 2005; Guzman et al., 2010), suggesting a possible link between L-type-Ca2+ channels and D2-autoreceptor function. D2 receptor signalling in general is complex, depending on additional factors such as Ca2+ and the neuronal calcium sensor 1 (NCS-1) (Kabbani et al., 2002), and the signalling mechanisms of somatodendritic D2-autoreceptors remain poorly understood (Beaulieu and Gainetdinov, 2011; Ford, 2014).
Here, we detected dramatically higher messenger RNA levels of D2-autoreceptors and GIRK2 in SN DA neurons from post-mortem Parkinson’s disease brains compared to those of controls. To address a possible homeostatic plasticity of D2-autoreceptor responses, in age-dependent interplay with L-type-Ca2+ channels, we analysed isolated somatodendritic D2-autoreceptor responses as well as related L-type-Ca2+ channel function in SN DA neurons from juvenile (postnatal Day 13) and adult (postnatal Day 90) mice in vitro – under control conditions and 3–4 days after inducing a transient elevation of extracellular dopamine levels by a single in vivo injection of either l-DOPA or cocaine. We detected prominent desensitization of D2-autoreceptor responses in juvenile SN DA neurons that were absent in adult mice. Non-desensitizing, adult-like D2-autoreceptor responses could be induced in juvenile substantia nigra (but not ventral tegmental area) dopaminergic neurons by transient high-dopamine states in vivo. Cav1.3 L-type-Ca2+ channel function and Ca2+-dependent NCS-1/D2-autoreceptor interactions were crucial for this effect. Ncs1 messenger RNA levels were significantly higher in mouse juvenile SN DA neurons after in vivo cocaine, as well as in surviving human SN DA neurons in patients with Parkinson’s disease. Thereby, our findings provide a novel functional link in SN DA neurons between Cav1.3 L-type-Ca2+ channels and D2-autoreceptor activity, controlled by NCS-1 levels. They indicate that this signalling complex (Cav1.3/NCS-1/D2-autoreceptors) is tuning substantia nigra dopamine activity (and likely long-term vulnerability), in response to changes in extracellular dopamine levels in mice (cocaine, l-DOPA) as well as in humans (Parkinson’s disease). Thus, pharmacological modulation of this signalling network could emerge as a promising novel approach for modulating substantia nigra dopamine activity, as well as their vulnerability.
Materials and methods
Animals
Juvenile (postnatal Days 12–14) and adult (approximately postnatal Day 90) male C57BL/6, Cav1.2 DHP−/− mice and Cav1.3 knockout mice were bred and genotyped as described previously (Platzer et al., 2000; Sinnegger-Brauns et al., 2009). NCS-1−/− mice were kindly provided by Prof. Olaf Pongs, Hamburg. Mice were injected: subcutaneously with a mix of 100 mg/kg l-DOPA methyl-ester hydrochloride (Sigma) and 200 mg/kg of the peripheral decarboxylase-inhibitor benserazide hydrochloride (Sigma) or intraperitoneally with 15 mg/kg cocaine hydrochloride (Sigma) in saline (0.9% NaCl, Braun), or with saline (subcutaneously or intraperitoneally) as control. In contrast to cocaine, the l-DOPA administration for mice is less well-established. In divergence to a 4:1 ratio used in humans, we used l-DOPA and benserazide in 1:2 ratio, as in most studies for rodents (due to presumably faster metabolism). l-DOPA injections have been administered subcutaneously as this results in more consistent responses; response failures after intraperitoneal, but not subcutaneous, injections have been described (Lundblad et al., 2004, 2005; Lindgren et al., 2007; Prinz et al., 2013). Because l-DOPA and cocaine injections both led to the same effects on mouse substantia nigra dopamine D2-autoreceptor response, we continued our study with more robust and, for rodents, well-established cocaine paradigm (Saal et al., 2003). All animal procedures were approved by the German Regierungspräsidium Tübingen (Aktenzeichen: 35/9185.81-3; TV-No. 921 and 1043) or Darmstadt (V54-19c20/15-F40/28).
Ultraviolet laser microdissection and reverse transcription quantitative polymerase chain reaction analysis
UV laser microdissection of dopamine neurons from mouse and human brains using an LMD6000 system (Leica Microsystems), or cytoplasm harvest were performed essentially as described (Grundemann et al., 2011). Post-mortem human midbrains were obtained by the German Brain Bank (BrainNet). All available information on the respective Parkinson’s disease and control patient’s disease state, comorbidity, age, and tissue quality is summarized in Supplementary Table 1. We were not able to obtain information about the therapy of our analysed patients with Parkinson’s disease. However, it is likely that they were treated with l-DOPA (and/or D2-agonist).
All experiments on human material were approved by the ethic commission of Ulm University (247/10-UBB/bal).
Quantification of messenger RNA levels from UV laser microdissected samples was carried out by real time quantitative PCR after reverse transcription with a GeneAmp 7900HT (Applied Biosystems). For real-time quantitative PCR assays, TaqMan® probes were labelled with FAM as reporter and a non-fluorescent quencher. Quantitative PCR assay details, and assay-specific standard curve parameters are given in Supplementary Table 2. Standard curves for quantitative PCR quantification were generated using serial dilutions of complementary DNA (3, 0.3 and 0.03 ng), derived from human substantia nigra tissue RNA (Ambion, Applied Biosystems), or mouse midbrain messenger RNA (RNeasy® MINI kit, Qiagen), as template in duplicates in at least n = 3 independent quantitative PCR runs. The complementary DNA amount per cell in relation to the standard was calculated according to:
Where: S = serial dilution factor of the standard curve (i.e. 10), Nocells = number of harvested neurons per sample (i.e. 10 for mouse and 15 for human SN DA neurons), cDNA fraction = fraction of the UV laser microdissection complementary DNA reaction sample used as template in the individual quantitative PCR reactions (i.e. 5/17 for mouse, 1/11 for human substantia nigra dopaminergic neurons). The unit-magnitude corresponds to the respective standard used, which defines the unit at the y-intercept (i.e. pg equivalents of standard complementary DNA, derived from substantia nigra tissue messenger RNA). As primers for D1 and D5 dopamine receptors were not intron-spanning, a DNase digestion step (DNA-free™kit, Ambion®) of UV laser microdissected samples was included before complementary DNA synthesis (Liss and Roeper, 2004).
In vitro electrophysiological recordings from juvenile and adult mouse brain slices
Mouse brain slice preparations and in vitro electrophysiological patch clamp recordings from SN DA neurons in 250 µm coronal midbrain slices of juvenile and adult mice were carried out as described (Lammel et al., 2008). To block fast synaptic transmission, 10 µM DNQX (6,7 dinitroquinoxaline-2,3-dione, Tocris) and 10 µM gabazine (SR95531 hydrobromide, Tocris) were added to artificial CSF. To analyse dopamine auto-inhibition of SN DA neurons in vitro, a minimum of 5 min stable baseline activity either in gramicidine 100 µg/ml (Sigma) perforated patch or in cell-attached configuration were recorded. This was followed by application of dopamine hydrochloride (Sigma) in artificial CSF via bath-perfusion for 15 min and a 20 min washout. Whole-cell recordings were performed with patch pipettes containing 0.1 mM EGTA (Sigma) in internal solution (135 mM K-gluconate, 5 mM KCl, 10 mM HEPES, 2 mM MgCl2, 0.1 mM NaGTP, 5 mM MgATP, all from Sigma). To test the effects of dopamine antagonists on SN DA neurons, we performed cell-attached recordings of baseline activity for 10 min, followed by application of 100 µM sulpiride (Tocris) in artificial CSF for another 10 min via bath perfusion. For L-type-Ca2+ channel and NCS-1 pharmacology, slices were pre-incubated for at least 40 min in artificial CSF containing 300 nM isradipine (Tocris) or 10 µM D2R/NCS-1 interfering peptide (DNIP)/scrambled DNIP peptide (Genscript; Saab et al., 2009) before and during cell-attached recordings. GIRK currents in SN DA neurons were evoked by local application (10 s) of 100 µM dopamine hydrochloride (Sigma) and recorded in voltage-clamp whole-cell configuration (membrane potential clamped at −50 mV, analysed with digital filter 500 mHz) in the presence of 100 µM tolbutamide (Sigma) and 20 µM ZD 7288 (Tocris) to block ATP-sensitive potassium channels (K-ATP) and hyperpolarization-activated cyclic nucleotide-gated channels, respectively. Excitatory postsynaptic currents were recorded at −70 mV and +40 mV from SN DA neurons in whole-cell patch-clamp, and AMPAR/NMDAR ratios were calculated as described previously (Lammel et al., 2011). Half maximal effective concentrations (EC50 values) of dopamine for activity inhibition were calculated from individual cells by applying increasing dopamine concentrations (0.001–100 µM, Sigma), each for 3 min. Mean frequency at each concentration was normalized to the respective baseline frequency for each cell. Individual and mean concentration–response curves were fitted to the Hill equation (curves fitted with constraints; max = 1, min = 0) to obtain EC50 values. For functional SN DA cell-type identification, cells were re-patched, and SN DA neuron-specific basal electrophysiological properties (frequency, action potentials, and sag component) were recorded in whole-cell configuration (Lammel et al., 2008; Ungless and Grace, 2012).
Immunoelectron microscopy
Double-labelling immunoelectron microscopy was performed and relative abundance of GIRK2 and D2-autoreceptor immunoreactivity in TH-positive SN DA neurons in control and cocaine injected juvenile wild-type mice was determined by quantification of immunolabelling in 60 μm coronal slices, as described by Koyrakh et al. (2005). Primary monoclonal antibody anti-TH (1–2 µg/ml, Calbiochem) was visualized by immunoperoxidase reaction and primary guinea pig polyclonal anti-GIRK2 and rabbit polyclonal anti-D2R antibody (not splice variant specific, 1–2 µg/ml; Koyrakh et al., 2005; Narushima et al., 2006; Aguado et al., 2008) by the silver-intensified immunogold reaction. Secondary antibody mixtures included goat anti-rabbit (Fab fragment; diluted 1:100) coupled to 1.4 nm gold (Nanoprobes), goat anti-guinea pig (Fab fragment; diluted 1:100) coupled to 1.4 nm gold (Nanoprobes), and biotinylated goat anti-mouse (diluted 1:100; Vector Laboratories). Electron photomicrographs for ultrastructural analyses were captured with a CCD camera (Mega View III; Soft Imaging System) using a Jeol-1010 electron microscope (Jeol).
Data analysis
Data analysis and graphical representations were performed using FitMaster software (HEKA Electronics), GraphPad Prism 5 (GraphPad Software, Inc.), Igor Pro 6 (Wavemetrics Inc.) and Neuroexplorer (Nex Technologies) for electrophysiological experiments and SDS 2.3 (Applied Biosystems) for quantitative PCR. Statistical analysis was performed in Igor Pro 6, the R project for statistical computing and GraphPad Prism 5 (GraphPad Software, Inc.). Statistical significance was set at P < 0.05. Mann-Whitney tests were performed in GraphPad Prism 5 software and used to determine statistical differences (*P < 0.05, **P < 0.01 and ***P < 0.001). The coefficient of variation of the interspike interval was calculated using a Gaussian fit over the histogram with Neuroexplorer and Igor Pro 6. Coefficient of variation as [%] = (SD fit) / (mean fit) × 100. If not otherwise stated all data are given as mean ± SEM.
Results
Substantia nigra dopaminergic neurons of patients with Parkinson’s disease display elevated levels of D2-autoreceptors and GIRK2 messenger RNA compared with controls
Given the important therapeutic role of D2 receptor agonists in Parkinson’s disease, and of D2-autoreceptors and their main downstream target GIRK2 for D2-autoreceptor responses that control substantia nigra dopamine function in health and disease, we quantified messenger RNA levels of D2-autoreceptors and GIRK2 in laser-microdissected human SN DA neurons from post-mortem Parkinson’s disease brains and controls using reverse transcription real-time quantitative PCR (Fig. 1A and B, and Supplementary Table 1). We detected significantly higher messenger RNA levels for D2-autoreceptors and GIRK2 messenger RNA in remaining SN DA neurons from Parkinson’s disease brains compared to those of controls (Fig. 1B). D2-autoreceptor/GIRK2 ratios were not changed (Fig. 1C). These findings point to an altered, sensitized D2-autoreceptor/GIRK2 signalling control of activity of SN DA neurons in Parkinson’s disease. To address and define a possible dopamine-dependent homeostatic plasticity of D2-autoreceptor/GIRK2 pacemaker control in SN DA neurons, we analysed isolated somatodendritic dopamine D2-autoreceptor responses in mouse SN DA neurons. As age-dependent, postnatal differences in mouse substantia nigra dopamine pacemaker control have been described (Chan et al., 2007), we analysed juvenile (postnatal Day 13) as well as adult (postnatal Day 90) mice.
Figure 1.
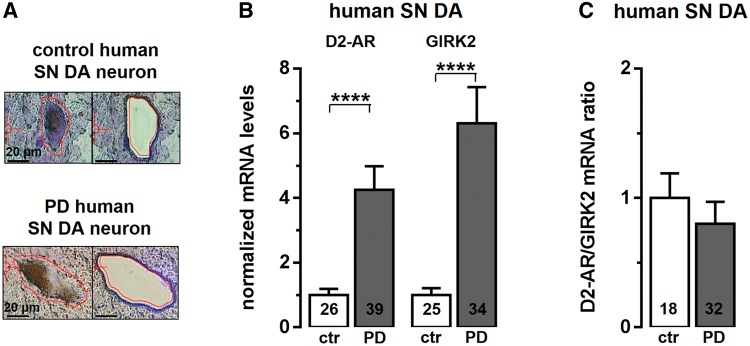
Elevated D2-autoreceptor (AR) and GIRK2 messenger RNA levels in remaining human SN DA neurons from patients with Parkinson’s disease (PD). (A) Neuromelanin-positive human SN DA neurons (SN DA) (before and after UV laser microdissection) from post mortem Parkinson’s disease and control midbrain sections (for details, see Supplementary Table 1). (B) Real-time quanititative PCR results identified significantly higher relative messenger RNA levels for D2-autoreceptors and GIRK2 in human SN DA neurons from Parkinson’s disease brains compared to controls (ctr) (D2-autoreceptors: controls, n = 26, 1 ± 0.19; Parkinson’s disease, n = 39, 4.25 ± 0.73; ****P < 0.0001; Mann-Whitney test; GIRK2: controls, n = 25, 1 ± 0.25; Parkinson’s disease, n = 34, 6.31 ± 1.12; ****P < 0.0001; Mann-Whitney test). (C) D2/GIRK2 ratios were not different in SN DA neurons from controls and Parkinson’s disease patients. All data are shown as the mean ± SEM.
Dopamine D2-autoreceptor-mediated pacemaker inhibition of SN DA neurons is pronounced after postnatal maturation
We compared the unperturbed kinetics and dopamine sensitivities of D2-autoreceptor-mediated pacemaker inhibition in SN DA neurons after postnatal maturation in in vitro brain slices of juvenile (postnatal Day 13) and adult (postnatal Day 90) mice (Fig. 2A and B). Stimulation of somatodendritic D2-autoreceptors through dopamine bath application reversibly inhibited spontaneous pacemaker activity after initial membrane potential hyperpolarization (Fig. 2A). To ensure full D2-autoreceptor activation, we applied 100 µM dopamine for 15 min. The kinetics of the dopamine-evoked D2-autoreceptor-mediated pacemaker inhibition differed significantly between adult and juvenile SN DA neurons (Fig. 2A and B). Adult SN DA neurons did not recover pacemaker activity in the presence of saturating dopamine concentrations. However, SN DA neurons from juvenile mice displayed a prominent desensitization of the dopamine-induced pacemaker inhibition, regaining 40% of their baseline firing frequency at the last minute of dopamine application (Fig. 2E and Supplementary Table 3). The sensitivity of SN DA neurons to the dopamine-induced inhibition was ∼2-fold higher in adult neurons compared to those of juvenile mice (dopamine EC50 values: juvenile, n = 10: 6.57 ± 1.09 μM; adult, n = 11: 3.29 ± 0.48 μM; P = 0.03). The maximal dopamine-induced membrane hyperpolarization was not significantly different in juvenile and adult SN DA neurons (Supplementary Table 4). Pacemaker frequency and its precision (expressed as coefficient of variation of the interspike interval, in %) were similar between adult and juvenile SN DA neurons under basal conditions in the absence of dopamine (Fig. 2C and D, and Supplementary Table 5). Inhibition of D2-autoreceptor function by the D2 anta gonist sulpiride (100 µM) had no effect on pacemaker frequency or precision of juvenile or adult SN DA neurons, arguing against an intrinsic dopamine tonus, and thus D2-autoreceptor activity in vitro under our recording conditions (Fig. 2C and D, and Supplementary Table 6).
Figure 2.

In vivo transient high-dopamine states (induced by l-DOPA or cocaine) cause adult-like non-desensitizing dopamine D2-autoreceptor responses in juvenile SN DA neurons (SN DA). (A) Representative SN DA neuron pacemaker activity recordings from in vitro brain slices of adult and juvenile wild-type mice, pretreated (4 or 3 days, respectively) in vivo with saline, l-DOPA, or cocaine (perforated/cell-attached patch-clamp). Dopamine bath application is indicated by red bars. (B) Normalized frequencies plotted against time for all analysed SN DA neurons revealed pronounced desensitization of D2-autoreceptor responses in juveniles (n = 14) compared to adults (n = 10), as well as adult-like, non-desensitizing D2-autoreceptor responses in juvenile SN DA neurons after in vivo l-DOPA (n = 15) or in vivo cocaine (coc; n = 12), compared to controls (ctr; saline, n = 14). (C) Bar graphs show no change in mean frequencies before and after sulpiride application in SN DA neurons from controls, as well as after saline, l-DOPA, or cocaine in vivo injections, before dopamine application. (D) Pacemaker precision [given as coefficient of variation of interspike interval values, (CV ISI)] of juvenile and adult SN DA neurons was also not altered after sulpiride application, as well as after saline, l-DOPA, or cocaine in vivo injections. (E) D2-autoreceptor responses of mouse substantia nigra and ventral tegmental area dopaminergic neurons represented as mean activity of respective dopaminergic neurons at the last minute of dopamine application (15 min). Data are shown as the mean ± SEM. Data values detailed in Supplementary Tables 3, 5 and 6.
A single dose of l-DOPA or cocaine in vivo induces adult-like, stable D2-autoreceptor responses in SN DA neurons in vitro
We next studied possible effects of pathophysiologically and Parkinson’s disease-therapeutically relevant transient high-dopamine states on mouse substantia nigra dopamine D2-autoreceptor responses by injecting them in vivo either with l-DOPA (the precursor of dopamine, and still the gold standard in Parkinson’s disease therapy), cocaine (enhancing extracellular dopamine levels by inhibiting dopamine re-uptake through the plasmalemma dopamine transporter; Ritz et al., 1987), or saline (for controls), and analysed D2-autoreceptor responses 4 or 3 days later, respectively. l-DOPA and cocaine pretreatment strongly affected D2-autoreceptor responses in juvenile SN DA neurons (Fig. 2A, B and E): it converted their desensitizing D2-autoreceptor responses into non-desensitizing D2-autoreceptor responses that resembled the adult phenotype (Fig. 2A, B and E, and Supplementary Table 3). This reduction of desensitization of D2-autoreceptor responses after in vivo induction of transient high-dopamine states was a hallmark of juvenile SN DA neurons and was not observed in juvenile (mesolimbic) ventral tegmental area dopaminergic neurons (Fig. 2E and Supplementary Table 3). Neither in vivo l-DOPA nor cocaine treatment significantly altered the basal spike-frequency or spike-timing precision (coefficient of variation of the interspike interval) of juvenile or adult substantia nigra dopamine pacemaker (Fig. 2C and D, and Supplementary Table 5). As l-DOPA and cocaine injections both induce transient high-dopamine states by a distinct molecular mechanisms, but had the same effects on juvenile substantia nigra dopamine D2-autoreceptor responses, our data strongly suggest that D2-autoreceptor responses show a dopamine-dependent plasticity. As the cocaine injection protocol, in contrast to the l-DOPA protocol, is well-established for mice (see ‘Materials and methods’ section for details), we have continued our study with the cocaine paradigm to induce transient in vivo high-dopamine states (Saal et al., 2003).
Functional expression of D2-auto- receptors and GIRK2 is not altered in juvenile SN DA neurons after in vivo induction of a transient high-dopamine state
We next addressed the underlying molecular mechanism of altered, adult-like D2 autoreceptor responses in juvenile SN DA neurons after in vivo induction of a transient high-dopamine state by cocaine. We confirmed that mouse SN DA neurons predominantly express GIRK2 messenger RNA alone, together with D2 dopamine receptor messenger RNA (Fig. 3A, D2 receptor and GIRK2 were co-expressed in 100% of analysed SN DA neurons, n = 85, without other GIRKs in ∼80%). Other dopamine receptor subunits were not detected or only in a small fraction of SN DA neurons (Fig. 3A; D1: n = 1, D5: n = 2, D3: n = 3, D4: n = 0 out of 39 TH- and D2-positive SN DA neurons). Although most SN DA neurons co-expressed both the short D2s and the long D2l receptor-splice variants (∼65%, Fig. 3A), 35% expressed only one splice variant (only D2l: n = 23 of 85; only D2s: n = 6 of 85). As all analysed SN DA neurons showed typical functional D2-autoreceptors responses, we conclude that both D2s and D2l alone can act as functional D2-autoreceptors in mouse SN DA neurons. To probe whether the relative expression of the D2-autoreceptor splice variants in SN DA neurons was altered after cocaine injection, we quantified D2 short and long variants, and calculated D2l/s ratios in SN DA neurons from juvenile mice with and without cocaine treatment. We found no change in D2s or D2l messenger RNA levels or D2l/s ratios in juvenile SN DA neurons after cocaine (Fig. 3A). We also quantified D3 and D4 messenger RNA in SN DA neurons from juvenile mice without and after cocaine. However, we detected no expression of D3 or D4 using real-time quantitative PCR, neither in controls or after cocaine (n = 6 probes each, data not shown). These data rule out that altered expression of D2 splice variants or other D2-type receptors (D3, D4) are causing the observed functional change in substantia nigra dopamine D2-autoreceptor desensitization after in vivo cocaine. The reduced desensitization of D2-autoreceptors in juvenile SN DA neurons after in vivo induced transient high-dopamine states was also not associated with an enhanced expression of either D2-autoreceptors or GIRK2 (Fig. 3). D2-autoreceptor messenger RNA and immunoreactivity (Fig. 3A and B) was unaffected after cocaine. GIRK2 messenger RNA (Fig. 3A) and its functional channel activity (Fig. 3C) were downregulated rather than upregulated. We also detected no change in the dopamine transporter and vesicular monoamine transporter (Vmat2, also known as Slc18a2) messenger RNA ratio in juvenile SN DA neurons after cocaine, the transporters that define dopamine uptake and release capacity, respectively (Fig. 3D).
Figure 3.

Functional expression of D2-autoreceptors (AR) [D2 short/long variants (s/l), D3, D4] and GIRK2 is not altered in juvenile SN DA neurons after in vivo induction of a transient high-dopamine state. (A, top) Agarose gel electrophoresis of multiplex nested reverse transcritpion PCR products identified D2 receptor (both splice variants) and GIRK2 as the molecular components of the D2-autoreceptors in SN DA neurons. Note the absence of D3 and D4 receptor, as well as GIRK1. Bottom: Quantitative analysis (reverse transcritpion quantitative PCR) of laser-microdissected (LMD) juvenile SN DA neurons showed no change in the ratio of D2 long and D2 short splice variant expression levels, after cocaine in single SN DA neurons (saline, n = 15, 1 ± 0.12; cocaine, n = 15, 0.85 ± 0.07; P = 0.29; Mann-Whitney test). Further, no changes in D2-autoreceptors (relative messenger RNA levels normalized to saline controls: saline, n = 18, 1 ± 0.14; cocaine, n = 19, 0.90 ± 0.08; P = 0.73; Mann-Whitney test), D3 or D4 receptor messenger RNA (data not shown, as no signal was detected. D3 receptor: saline, n = 6; cocaine: n = 6; D4 receptor: saline, n = 6; cocaine, n = 6) were detected. GIRK2 messenger RNA levels were also decreased rather than increased after in vivo cocaine (saline, n = 19, 1 ± 0.15; cocaine, n = 20, 0.62 ± 0.07; *P = 0.04; Mann-Whitney test; note the significantly altered D2-autoreceptor/GIRK2 ratios after cocaine due to decreased GIRK2-levels; saline, n = 18, 1 ± 0.08; cocaine, n = 19, 1.57 ± 0.12; **P = 0.002; Mann-Whitney test). All data given as mean ± SEM normalized to controls. (B, left) Pre-embedding double-immunolabelling at electron microscopy level for D2-autoreceptors and GIRK2 in TH-positive SN DA neurons 3 days after saline/cocaine in vivo injection of juvenile wild-type mice (n = 3 animals each; arrows point to immunogold labelling at extrasynaptic plasma membrane and crossed arrows at intracellular sites). Right: No significant differences in immunoreactivity or subcellular localization of D2-autoreceptor protein (n = 3 animals each; saline, 1107.3 ± 8.7; cocaine, 1034.3 ± 33.5; P = 0.2; Mann-Whitney test), and GIRK2 protein (n = 3 animals each; saline, 1408.3 ± 9.1; cocaine, 1396.3 ± 7.1; P = 0.4; Mann-Whitney test). (C, left) Representative traces of dopamine evoked GIRK currents in juvenile SN DA neurons recorded in whole-cell voltage-clamp in vitro in the presence of 100 µM tolbutamide and 20 µM ZD7288 to block ATP-sensitive potassium channels (K-ATP) and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Right: Mean dopamine-evoked GIRK current amplitudes were not increased, but significantly reduced after in vivo cocaine in juvenile SN DA neurons (saline, n = 12, 101.48 ± 7.69 pA; cocaine, n = 10, 75.88 ± 12.74 pA; *P = 0.03; Mann-Whitney test). (D) DAT/VMAT2 messenger RNA ratios in juvenile SN DA neurons, defining their individual dopamine-release capacity, were not altered after in vivo cocaine (normalized ratios: saline, n = 8, 1 ± 0.14; cocaine, n = 8, 1.23 ± 0.13; P = 0.29; Mann–Whitney test). Data are given as mean ± SEM.
Cav1.3 L-type calcium channel function and internal Ca2+ is crucial for non-desensitizing D2-autoreceptor responses induced by high-dopamine states in vivo, in juvenile SN DA neurons
As calcium signalling is not only critical for the control of D2- receptor desensitization in general, but also triggers selective substantia nigra dopamine degeneration in Parkinson’s disease, and as L-type-Ca2+ channel-blockers protect for Parkinson’s disease in humans and mice, we evaluated a potential role of internal calcium ([Ca2+]i) and L-type-Ca2+ channels for the expression of the high dopamine-dependent, non-desensitizing D2-autoreceptors in juvenile SN DA neurons. We first tested if the cocaine effect on D2-autoreceptors depended on unperturbed [Ca2+]i by buffering [Ca]i with 0.1 mM EGTA using whole cell dialysis (Fig. 4A and Supplementary Table 3). EGTA buffering did not affect the kinetics of dopamine autoinhibition in juvenile substantia nigra dopamine neurons in saline treated animals compared to those recorded in perforated patch. However, after cocaine treatment, D2-autoreceptor responses still displayed prominent desensitization, in contrast with the non-desensitizing responses after cocaine recorded in perforated patch (Fig. 4A). These data indicate a complex dependence of cocaine-induced D2-autoreceptor potentiation on intracellular Ca2+. They also emphasize the importance of analysing D2-autoreceptors signalling in non-perturbed recording configurations, as the cocaine effect is absent in whole cell recordings.
Figure 4.

Ca2+and Cav1.3 L-type-Ca2+ channels are crucial for adult-like, non-desensitizing D2-autoreceptor responses, induced by a transient high-dopamine state in vivo, in juvenile SN DA neurons. (A) Mean activity of substantia nigra dopaminergic neurons at the last minute of dopamine application [15 min, recorded in perforated patch (pp) juvenile: saline, n = 14; cocaine, n = 12] compared to those recorded in whole cell (wc) configuration with 0.1 mM EGTA (whole cell: juvenile: saline, n = 6; cocaine, n = 7) to buffer free [Ca2+]i in juvenile SN DA neurons. Note the absence of cocaine-induced stabilization of D2-autoreceptor responses in whole cell compared to unperturbed perforated patch recordings. (B) Experiments as in Fig. 1 but in the presence of the L-type-Ca2+ channel blocker isradipine (300 nM, represented with blue bar). Isradipine completely blocked changes in kinetics of D2-autoreceptor desensitization of SN DA neurons, after transient high-dopamine state induced by cocaine, in wild-type mice (saline, n = 7; cocaine, n = 7). (C) Similar experiments as in B, but using Cav1.2 DHP−/− mice (with Cav1.2 L-type-Ca2+ channels, insensitive to isradipine; saline, n = 6; cocaine, n = 9). Application of 300 nM isradipine still abolished cocaine D2-autoreceptor response potentiation, pointing to the crucial role of Cav1.3 L-type-Ca2+ channels in this process. (D and E) Isradipine (300 nM) did not reduce basal pacemaker frequencies or activity pattern (coefficient of variation of interspike interval) of juvenile SN DA neurons. (F) Activity of SN DA neurons at the last minute of dopamine application. Data values detailed in Supplementary Tables 3 and 5. (G) Quantitative analysis (reverse transcription quantitative PCR) of laser-microdissected (LMD) mouse and human SN DA neurons detected no change in messenger RNA levels of Cav1.2 or Cav1.3, the pore-forming alpha subunit of L-type-Ca2+ channels in the brain, after in vivo cocaine (mice, left) or in Parkinson’s disease compared to controls (humans, right). Normalized expression levels: mouse: Cav1.2: saline, n = 18, 1 ± 0.01; cocaine, n = 18, 1.01 ± 0.02; P = 0.92; Cav1.3: saline, n = 17, 1 ± 0.17; cocaine, n = 18, 0.81 ± 0.16; P = 0.37; Mann-Whitney test. Human: Cav1.2: control, n = 4, 1 ± 0.01; Parkinson’s disease, n = 5, 0.90 ± 0.11; P = 0.43; Cav1.3: control, n = 16, 1 ± 0.16; Parkinson’s disease, n = 33, 1.14 ± 0.14; P = 0.64; Mann-Whitney test). Data are shown as mean ± SEM.
We next tested if L-type-Ca2+ channels could provide the Ca2+ source for Ca2+-dependent control of D2-autoreceptor responses, by continuous treatment of slices with the selective L-type Ca2+ channel blocker isradipine (300 nM), which is already in clinical trials as neuroprotective therapy for Parkinson’s disease (Striessnig et al., 2006; Simuni et al., 2010; Surmeier et al., 2012), before, during and after dopamine application. Isradipine completely abolished the effect of in vivo cocaine treatment on D2-autoreceptor desensitization in juvenile SN DA neurons (Fig. 4B and F, and Supplementary Table 3), revealing a crucial role of these channels for the expression of the here described cocaine effect.
Another important observation was that although 300 nM isradipine modified the cocaine response, it did not significantly reduce basal pacemaker frequency or its precision in juvenile or adult SN DA neurons (Fig. 4D and E, and Supplementary Table 5).
In SN DA neurons, the pore-forming α1-subunits of both brain L-type-Ca2+ channels, Cav1.2 and Cav1.3, are expressed (Olson et al., 2005; Sinnegger-Brauns et al., 2009). Since isradipine inhibits both isoforms, we cannot distinguish which of these channels mediate its pharmacological effect on SN DA neurons. To address this question, we took advantage of homozygous Cav1.2 DHP−/− mutant mice. These mice express Cav1.2 L-type-Ca2+ channels with normal function, but their high sensitivity to isradipine is eliminated by a single mutation within the DHP-binding pocket (Sinnegger-Brauns et al., 2004; Striessnig and Koschak, 2008). Thus, Cav1.2-mediated isradipine effects are absent in these mice, whereas drug effects through Cav1.3 are unaffected. In SN DA neurons from Cav1.2 DHP−/− mutant mice the effect of isradipine was indistinguishable from wild-type (Fig. 4C and F, and Supplementary Table 3). As the isradipine concentration used was very low (300 nM), and no other targets are known to be affected at this concentration, Cav1.3 remains the only plausible molecular transducer for the cocaine-induced, calcium-dependent, and isradipine-sensitive change in substantia nigra dopamine D2-autoreceptor responses.
We also assessed the role of Cav1.3 more directly in Cav1.3-deficient mice (Cav1.3 knockout mice) (Platzer et al., 2000). In this model, although basal substantia nigra dopamine pacemaker activity remained unaffected, the constitutive absence of Cav1.3 activity throughout development prevented the postnatal wild-type substantia nigra dopamine D2-autoreceptor phenotypes, as the non-desensitizing ‘adult’ D2-autoreceptor responses were present in SN DA neurons from both juvenile as well as adult Cav1.3 knockout mice (Supplementary Fig. 1 and Supplementary Table 7). Because of absence of the juvenile desensitizing D2-autoreceptor phenotype, we did not further study in vivo cocaine effects in Cav1.3 knockout mice.
Taken together the findings from both mouse models reveal a novel functional role of Cav1.3 L-type-Ca2+ channels for both the development of age-dependent D2-autoreceptor differences in SN DA neurons, and for the switch of juvenile desensitizing D2-autoreceptor response to those of adult substantia nigra dopamine neurons in response to a transient in vivo high-dopamine state.
At the messenger RNA level, we found no change in expression of Cav1.2 and Cav1.3 L-type-Ca2+ channel pore-forming α1-subunits in mouse SN DA neurons after in vivo cocaine (Fig. 4G). Also, we found no change in expression levels of Cav1.2 and Cav1.3 in remaining human SN DA neurons of patients with Parkinson’s disease compared to controls (Fig. 4G).
As Cav1.2 and Cav1.3 expression were not altered, we probed for altered AMPA/NMDAR ratios in juvenile cocaine-treated SN DA neurons after in vivo cocaine as an alternative calcium source, causing synaptic plasticity after cocaine in adult ventral tegmental area dopamine neurons (Ungless et al., 2001; Lammel et al., 2011). However, we detected no change in AMPAR/NMDAR-mediated excitatory postsynaptic currents after in vivo cocaine in juvenile SN DA neurons (Supplementary Fig. 2).
Cocaine induced, adult-like, D2-auto- receptor responses in juvenile substantia nigra dopaminergic neurons are mediated by NCS-1
We next explored a possible contribution of NCS-1 for Cav1.3 dependent control of D2-autoreceptor responses in juvenile SN DA neurons, in response to in vivo transient high-dopamine, induced by cocaine. NCS-1 can directly interact with postsynaptic D2 receptors in a Ca2+-dependent manner and inhibit receptor desensitization by preventing its phosphorylation through G protein-coupled receptor kinase 2 (GRK2, also known as ADRBK1), and thus beta-arrestin-mediated receptor internalization (Kabbani et al., 2002). We analysed mouse substantia nigra dopamine D2-autoreceptor responses in the presence of a membrane-permeable peptide, DNIP (D2R/NCS-1 interfering peptide), which physically blocks NCS-1 binding to D2 receptor (Saab et al., 2009). As control, a membrane-permeable, scrambled version of the peptide (scrambled DNIP) was used, which did not influence basal pacemaker activity or the kinetics of the D2-autoreceptor response of SN DA neurons after in vivo cocaine (Figs 5A, 5B and Supplementary Tables 3 and 5). Similar to 300 nM of the substantia nigra dopamine- and Parkinson’s disease-protective L-type-Ca2+ channel-blocker isradipine, 10 µM DNIP completely abolished the cocaine effect on D2-autoreceptor responses in juvenile SN DA neurons. With NCS-1/D2-autoreceptor interaction blocked, D2-autoreceptors displayed rapid desensitization, similar to that seen without cocaine pretreatment (Fig. 5A and B, and Supplementary Table 3). To prove that NCS-1 function is necessary and sufficient for modulating D2-autoreceptor desensitization in SN DA neurons, we analysed D2-autoreceptor responses in a general NCS-1−/− mouse, lacking NCS-1 in all cells. Substantia nigra dopaminergic neurons of NCS-1−/− mice displayed rapidly desensitizing D2-autoreceptors responses already under control conditions that were not converted to a sensitized response after in vivo cocaine, confirming that NCS-1 is necessary and sufficient for preventing D2-autoreceptor desensitization in mouse SN DA neurons (Fig. 5A and B ,and Supplementary Tables 3 and 5).
Figure 5.

Adult-like, non-desensitizing D2-autoreceptor responses in SN DA neurons are mediated by the neuronal calcium sensor 1 (NCS-1). (A, left) Normalized frequency plots show that non-desensitizing D2-autoreceptor responses of juvenile SN DA neurons after in vivo cocaine were completely blocked by 10 µM DNIP (cell-permeant D2R/NCS-1 interfering peptide, n = 8), whereas the scrambled version had no effect (10 µM srDNIP, n = 6). (Right) Respective analysis (as in A) of a general NCS-1 knockout mouse (NCS-1−/−) demonstrated that NCS-1 is crucial for non-desensitizing D2-autoreceptor responses of juvenile SN DA neurons. (B) Activity of SN DA neurons from (A) at the last minute (15 min) of dopamine application (note the significantly pronounced D2-autoreceptor desensitization in NCS-1−/− already under control conditions). Data values detailed in Supplementary Table 3. (C) Reverse transcrition quantitative PCR analysis of mouse and human SN DA neurons identified 2-fold higher Ncs-1 messenger RNA levels after cocaine (saline, n = 36, 1 ± 0.08; cocaine, n = 32, 1.94 ± 0.15; ***P = 0.0001; Mann-Whitney test), as well as in Parkinson’s disease, compared with controls (control, n = 27, 1 ± 0.20; Parkinson’s disease, n = 33, 2.60 ± 0.27; ****P < 0.0001; Mann-Whitney test). (D) Cartoon depicting molecular components of a novel Cav1.3/NCS-1/D2-autoreceptor/GIRK2 signalling network in SN DA neurons, which allows homeostatic adaptation of their inhibitory dopamine D2 responses, and thus their pacemaker activity.
To address why Ca2+ -dependent NCS-1/D2-autoreceptors interactions seem to be more prominent after cocaine (and thus D2-autoreceptors desensitization was reduced), we quantified NCS-1 messenger RNA. We found that NCS-1 messenger RNA levels were ∼2-fold higher in juvenile SN DA neurons after in vivo cocaine (P = 0.0001; Fig. 5C). This could explain enhanced L-type-Ca2+ channel-dependent Ca2+-stimulated NCS-1/D2-autoreceptor signalling, without increased expression of L-type-Ca2+ channels, but due to increased NCS-1 availability. This would allow SN DA neurons to adapt the degree of their dopamine D2-autoreceptor activity to homeostatic dopamine challenges, by tuning NCS-1 expression. To address if a similar increased NCS-1 expression is also present in human SN DA neurons in Parkinson’s disease, hinting to a corresponding homeostatic adaptive change in D2-autoreceptors signalling, we quantified messenger RNA levels of NCS-1 in human SN DA neurons from controls and endstage Parkinson’s disease brains (Fig. 5C, right). Indeed, similar to mice after cocaine, we detected ∼2-fold higher messenger RNA levels of NCS-1 in remaining SN DA neurons from Parkinson’s disease brains (Fig. 5C). Thus, increased NCS-1/D2-autoreceptor signalling, downstream of Cav1.3, may also fine-tune human substantia nigra dopamine neuron activity in Parkinson’s disease. It might (compensatory) allow remaining dopamine to reduce firing, activity-associated rhythmic cellular calcium load, and thus vulnerability of SN DA neurons.
Discussion
In summary, our data indicate a flexible D2-autoreceptor signalling in SN DA neurons, controlling their pacemaker activity, during maturation, and in response to pathophysiological in vivo dopamine challenges in mice, which could also be translated to human Parkinson’s disease pathology. Our findings describe the neuronal calcium sensor NCS-1 as a novel link between two major molecular players, modulating the activity of SN DA neurons as well as their vulnerability to degeneration in Parkinson’s disease: Cav1.3 L-type-Ca2+ channels and D2-autoreceptors (Beaulieu and Gainetdinov, 2011; Surmeier et al., 2012; Ford, 2014). We show that the degree of somatodendritic D2-autoreceptor desensitization in juvenile mouse SN DA neurons, in response to in vivo transient high-dopamine states, depends on Cav1.3 channels, as well as on the calcium dependent NCS-1 interaction with D2-autoreceptors. Our findings link flexible D2-autoreceptor desensitization with Cav1.3 L-type-Ca2+ channel function, through functional expression of NCS-1, and point to a role of this signalling network for adaptive changes in substantia nigra dopamine neuron activity due to altered extracellular dopamine levels. Our data also provide a fresh look at the molecular mechanisms involved in substantia nigra dopamine neuron activity control, as well as their pathophysiology in Parkinson’s disease and its dopamine-mimetic therapy.
Dopamine D2 receptors are important for proper function of dopamine midbrain neurons, and are important pharmacological targets for therapy of schizophrenia (antagonist; Ginovart and Kapur, 2012), and in particular Parkinson’s disease (agonist; Yamamoto and Schapira, 2008). Somatodendritic D2-autoreceptors are coupled to GIRK2 potassium channels, and reduce activity and excitability of SN DA neurons in response to locally released dopamine in the midbrain (Beckstead and Williams, 2007; Gantz et al., 2013; Ford, 2014). Dysfunction of these somatodendritic D2-autoreceptors is linked to Parkinson’s disease pathology (Ford, 2014); however their exact functions in health and disease are by far not as well examined as the function of pre- and particularly postsynaptic D2 receptors (Beckstead et al., 2007; Ford et al., 2010; Bello et al., 2011; Anzalone et al., 2012; Ford, 2014).
To date, only the short D2-autoreceptor splice variant, D2s has been identified as the D2-autoreceptor (Usiello et al., 2000; Beaulieu and Gainetdinov, 2011; Ford, 2014). Our combined functional and molecular single cell analysis strongly suggests that the long D2l splice variant alone can build functional D2-autoreceptors in SN DA neurons. Recent reports confirm this claim as expression of either D2s or D2l alone in SN DA neurons of D2 receptor knockout mice restored D2 agonist-induced activation of GIRKs (Neve et al., 2013). In accordance with the literature, reviewed in Ford (2014), we also show that the other D2-type receptors (D3, D4) are not important for D2-autoreceptor function, or its homeostatic plasticity in mouse SN DA neurons. We did not quantify excitatory acting D1-type dopamine receptors (D1 and D5), as their messenger RNAs were almost never detected in SN DA neurons (Fig. 3A), they are not coupled to inhibitory GIRK2 receptors, and have only been functionally described postsynaptically (Vallone et al., 2000; Beaulieu and Gainetdinov, 2011).
We have previously shown that D2-autoreceptor responses are not homogeneous within distinct types of substantia nigra and vental tegmental area dopaminergic neurons from adult mice (Lammel et al., 2008). Now we show significantly elevated messenger RNA levels of D2-autoreceptors, as well as GIRK2 in remaining human SN DA neurons in Parkinson’s disease, pointing to altered D2-autoreceptor function in the course of the disease. We addressed a general possibility of a non-static, but plastic, D2-autoreceptor response in SN DA neurons in mice, and found that somatodendritic D2-autoreceptor responses of SN DA neurons are not uniform during postnatal maturation, but display prominent desensitization in juvenile mice (postnatal Day 13), which is absent in adults (postnatal Day 90). Furthermore we show that adult-like, non-desensitizing D2-autoreceptor responses could be induced in juvenile SN DA neurons by a transient high-dopamine state in vivo (via l-DOPA or cocaine). Most importantly, we identified that Cav1.3 L-type-Ca2+ channels are crucial for controlling dopamine-dependent D2-autoreceptor desensitization in mouse SN DA neurons, as they provide the calcium source for a Ca2+ dependent NCS-1/D2-autoreceptor interaction, which prevents D2-receptor desensitization (Beaulieu and Gainetdinov, 2011). That defines, to our knowledge, a first physiological function of L-type-Ca2+ channels in SN DA neurons. Our Cav1.3 knockout and Cav1.2 DHP−/− analysis ruled out Cav1.2 and identified Cav1.3 as the crucial L-type-Ca2+ channel for this function.
The here identified role of Cav1.3 L-type-Ca2+ channels, for NCS-1 mediated modulation of D2-autoreceptors and activity of SN DA neurons, has not yet been described. Postsynaptically, in striatal medium spiny neurons, D2 receptors upstream of L-type-Ca2+ channels can reduce L-type-Ca2+ channel activity through a calcineurin signalling cascade (Hernandez-Lopez et al., 2000; Olson et al., 2005). Also postsynaptically, in hippocampal neurons, reduced desensitization of D2-receptors, due to NCS-1/D2-autoreceptor interaction and thereby prevention of phosphorylation through GRK2, and thus receptor internalization, has been reported (Jo et al., 2008; Saab et al., 2009). Accordingly, we hypothesize that after induction of transient elevated dopamine levels, by in vivo l-DOPA or cocaine, D2-autoreceptor responses desensitize less in juvenile SN DA neurons due to increased NCS-1 expression and its higher availability for binding to D2-autoreceptors, leading to reduced GRK2 phosphorylation and D2-autoreceptor internalization in response to prolonged dopamine exposure. The in vitro Ca2+ buffering experiments (EGTA) as well as the DNIP experiments and the analysis of the Ncs1−/− mice further support this hypothesis. We hypothesize accordingly that in remaining SN DA neurons in Parkinson’s disease, due to increased expression of NCS-1, D2 and GIRK2, inhibitory D2-autoreceptor responses are also sensitized.
At the messenger RNA level, Cav1.3 (or Cav1.2) expression was not increased, neither in mouse SN DA neurons after cocaine nor in human SN DA neurons in Parkinson’s disease. However, our data do not exclude altered Cav1.3 activity. Evidence for enhanced Cav1.3 protein expression in SN DA neurons in Parkinson’s disease compared to controls has been described in immunohistochemical experiments (Hurley et al., 2013). However, although L-type-Ca2+ channel activity is necessary, increased calcium entry through L-type-Ca2+ channels is not necessarily required to account for the functional D2-autoreceptor plasticity observed in mice, as the detected higher NCS-1 levels would provide enhanced calcium-dependent NCS-1/D2 signalling, preventing desensitization. Indeed, NCS-1 expression is stimulated by calcineurin (Hamasaki-Katagiri and Ames, 2010), a phosphatase that itself is stimulated by D2 receptor activity (Greengard et al., 1999). Thus, a transient increase in dopamine, that leads to increased D2-autoreceptors stimulation, would be sufficient to increase calcium-dependent NCS-1 expression, e.g. via calcineurin-stimulation, and thus to increased calcium-dependent NCS-1/D2 interaction, in a negative feedback loop.
Our finding that pharmacological L-type-Ca2+ channel-block did not significantly reduce basal pacemaker frequency or its precision in juvenile or adult mouse SN DA neurons clearly argues—at least under our recording conditions—against a crucial role of L-type-Ca2+ channels in SN DA neurons for pacemaker activity. This disputes a previously reported switch from HCN channel to a metabolically more challenging L-type-Ca2+ channel-driven pacemaker in SN DA neurons, that would render them particularly vulnerable to degeneration during postnatal maturation (Bean, 2007; Chan et al., 2007; Puopolo et al., 2007; Drion et al., 2011; Surmeier et al., 2012)
We also detected no cocaine-induced changes in AMPA/NMDA ratios in juvenile SN DA neurons, as described for adult ventral tegmental area dopamine neurons (Ungless et al., 2001; Lammel et al., 2011). In contrast, we observed a cocaine-induced, Cav1.3, calcium and NCS-1-dependent substantia nigra-specific ‘enhancement’ of D2-autoreceptor function in vitro, which could act as an excitability brake and thus counteract or prevent excitotoxicity and degeneration of substantia nigra dopamine neurons (Dong et al., 2009). The finding that SN DA neurons from PARK mice (with particularly high vulnerable SN DA neurons) display D2-autoreceptor responses with significantly enhanced desensitization (Goldberg et al., 2005; Tong et al., 2009) would support this interpretation.
Although L-type-Ca2+ channels seem not to be crucial for substantia nigra dopamine pacemaking, there is accumulating evidence that the pacemaker activity of SN DA neurons drives somatodendritic Ca2+ entry through L-type-Ca2+ channels, thus generating a neurotoxic Ca2+ load that triggers their degeneration (Chan et al., 2007; Puopolo et al., 2007; Khaliq and Bean, 2010; Surmeier and Schumacker, 2013). Reduction of substantia nigra dopamine activity through a sensitized D2-autoreceptor response could therefore counteract this neurotoxic Ca2+ load. This would especially occur in response to transient high dopamine states, induced e.g. by dopamine-mimetic therapy. Thus, Cav1.3/Ca2+ dependent NCS-1 modulation of D2-autoreceptor desensitization may emerge as a homeostatic response in human SN DA neurons in Parkinson’s disease, reducing electrical activity and protecting surviving SN DA neurons from acute overexcitability (to compensate the progressive dopamine loss) and excitotoxicity (Dong et al., 2009; Liss and Roeper, 2010; Callaghan et al., 2012).
In this view, our data also raise an important issue regarding the Parkinson’s disease protective effects of L-type-Ca2+ channel blockers in humans (Ritz et al., 2010; Marras et al., 2012; Pasternak et al., 2012). We have no information about the therapy of our patients with Parkinson’s disease; however, it is likely that they were treated with l-DOPA (and/or D2 agonist). If in Parkinson’s disease, pathophysiology and/or dopamine-mimetic treatment induces—potentially protective—sensitized D2-autoreceptor responses in aged SN DA neurons, then this may be inhibited by treatment with L-type-Ca2+ channel blockers. The resulting enhancement of substantia nigra activity would counteract their reduction of overall Ca2+ load and related metabolic stress, believed to be the neuroprotective mechanism of L-type-Ca2+ channel blockers in Parkinson’s disease (Surmeier et al., 2012; Surmeier and Schumacker, 2013). However, if in adult SN DA neurons, another calcium source, distinct from L-type-Ca2+ channels, supports the NCS-1-dependent D2-autoreceptor responses (as we have first evidence for Cav1.3 knockout mice), this would not be the case, and it could offer another potential drug target for Parkinson’s disease therapy.
As blood–brain barrier permissive L-type-Ca2+ channel blockers are already in clinical trials for neuroprotective Parkinson’s disease therapy (ClinicalTrials.gov Identifier: NCT00909545; Simuni et al., 2010; Parkinson Study, 2013), but as their general neuroprotective mechanism, as well as the functional roles of L-type-Ca2+ channels in SN DA neurons are still unclear, our data provide an important first molecular step towards a better understanding of L-type-Ca2+ channel function in SN DA neurons, and thus for tailored pharmacological neuroprotective therapy. All molecular components of the Cav1.3/NCS-1/D2-autoreceptor signalling network are already accessible in humans to pharmacological modulation. However, they are all expressed, in addition to in SN DA neurons, in a variety of other neuronal and non-neuronal cells. Thus, future studies need to address the specific acute and chronic pathophysiological consequences, as well as side-effects of pharmacological modulation of Cav1.3/NCS-1/D2-autoreceptor-signalling in SN DA neurons.
Supplementary Material
Acknowledgements
We thank Desiree Spaich and Stefanie Schulz for excellent technical support, Olaf Pongs for providing the NCS-1 KO mice, and Leica Microsystems for providing the LMD6000. We thank Ema Illijc and D James Surmeier for neurotoxical pilot experiments and discussions. We thank Frances M. Ashcroft, Dirk Isbrandt, and Jochen Roeper for discussion and/or critical reading the manuscript. ED, CP, BL contributed in vitro electrophysiology. SL, RM AMPA/NMDA ratios. JuS provided pilot in vivo electophysiological experiments.. FS, ED, MF, JD, AH, BL RT-PCR and analysis. MW provided D2R and GIRK2 antibodies, RL provided immunogold histochemistry. JS provided Cav1.2 DHP-/- and Cav1.3 KO mice. ED, JS, and BL designed the study and wrote the manuscript. The authors declare no competing financial, personal, or professional interests that could be construed to have influenced our paper.
Funding
The study was supported by the DFG, SFB497 and LI 1745/1 (BL), the Austrian Science Fund (FWF SFB F4412, F4402; BL, JS), the BMBF (NGFN, 01GS08134, BL), the Spanish Ministry of Education and Science (BFU-2009-08404), and the CONSOLIDER program (CSD2008-00005) to RL, and the Alfried Krupp prize (BL).
Supplementary material
Supplementary material is available at Brain online.
References
- Aguado C, Colon J, Ciruela F, Schlaudraff F, Cabanero MJ, Perry C, et al. Cell type-specific subunit composition of G protein-gated potassium channels in the cerebellum. J Neurochem. 2008;105:497–511. doi: 10.1111/j.1471-4159.2007.05153.x. [DOI] [PubMed] [Google Scholar]
- Anzalone A, Lizardi-Ortiz JE, Ramos M, De Mei C, Hopf FW, Iaccarino C, et al. Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J Neurosci. 2012;32:9023–34. doi: 10.1523/JNEUROSCI.0918-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. Neurophysiology: stressful pacemaking. Nature. 2007;447:1059–60. doi: 10.1038/4471059a. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Ford CP, Phillips PE, Williams JT. Presynaptic regulation of dendrodendritic dopamine transmission. Eur J Neurosci. 2007;26:1479–88. doi: 10.1111/j.1460-9568.2007.05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Williams JT. Long-term depression of a dopamine IPSC. J Neurosci. 2007;27:2074–80. doi: 10.1523/JNEUROSCI.3251-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noain D, Shin JH, Low MJ, et al. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2-autoreceptors. Nat Neurosci. 2011;14:1033–8. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson's disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug and alcohol dependence. 2012;120:35–40. doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, et al. ‘Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–6. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12:359–66. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–87. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drion G, Massotte L, Sepulchre R, Seutin V. How modeling can reconcile apparently discrepant experimental results: the case of pacemaking in dopaminergic neurons. PLoS Comput Biol. 2011;7:e1002050. doi: 10.1371/journal.pcbi.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience. 2014 doi: 10.1016/j.neuroscience.2014.01.025. 23: pii: S0306-4522(14)00037-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PE, Williams JT. Control of extracellular dopamine at dendrite and axon terminals. J Neurosci. 2010;30:6975–83. doi: 10.1523/JNEUROSCI.1020-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR, Williams JT. Spontaneous inhibitory synaptic currents mediated by a G protein-coupled receptor. Neuron. 2013;78:807–12. doi: 10.1016/j.neuron.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T, Hardy J, Mizuno Y. Milestones in PD genetics. Mov Disord. 2011;26:1042–8. doi: 10.1002/mds.23637. [DOI] [PubMed] [Google Scholar]
- Gazewood JD, Richards DR, Clebak K. Parkinson disease: an update. Am Fam Physician. 2013;87:267–73. [PubMed] [Google Scholar]
- Ginovart N, Kapur S. Role of dopamine D(2) receptors for antipsychotic activity. Handb Exp Pharmacol. 2012;212:27–52. doi: 10.1007/978-3-642-25761-2_2. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–96. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–47. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Grundemann J, Schlaudraff F, Liss B. UV-laser microdissection and mRNA expression analysis of individual neurons from postmortem Parkinson's disease brains. Methods Mol Biol. 2011;755:363–74. doi: 10.1007/978-1-61779-163-5_30. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in SN DA neurons. J Neurosci. 2009;29:11011–9. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki-Katagiri N, Ames JB. Neuronal calcium sensor-1 (Ncs1p) is up-regulated by calcineurin to promote Ca2+ tolerance in fission yeast. J Biol Chem. 2010;285:4405–14. doi: 10.1074/jbc.M109.058594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–95. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley MJ, Brandon B, Gentleman SM, Dexter DT. Parkinson's disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain. 2013;136(Pt 7):2077–97. doi: 10.1093/brain/awt134. [DOI] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis. 2011;43:364–71. doi: 10.1016/j.nbd.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Heon S, Kim MJ, Son GH, Park Y, Henley JM, et al. Metabotropic glutamate receptor-mediated LTD involves two interacting Ca(2+) sensors, NCS-1 and PICK1. Neuron. 2008;60:1095–111. doi: 10.1016/j.neuron.2008.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbani N, Negyessy L, Lin R, Goldman-Rakic P, Levenson R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J Neurosci. 2002;22:8476–86. doi: 10.1523/JNEUROSCI.22-19-08476.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–13. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, et al. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–78. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Hackel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–73. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–62. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren HS, Rylander D, Ohlin KE, Lundblad M, Cenci MA. The “motor complication syndrome” in rats with 6-OHDA lesions treated chronically with L-DOPA: relation to dose and route of administration. Behav Brain Res. 2007;177:150–9. doi: 10.1016/j.bbr.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Liss B, Roeper J. Correlating function and gene expression of individual basal ganglia neurons. Trends Neurosci. 2004;27:475–81. doi: 10.1016/j.tins.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Liss B, Roeper J. Ion channels and regulation of dopamine neuron activity. In: Iversen LL, Iversen SDI, Dunnett SB, Björklund A, editors. Dopamine handbook. Oxford, UK: Oxford University press; 2010. pp. 118–38. [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2004;16:110–23. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Usiello A, Carta M, Hakansson K, Fisone G, Cenci MA. Pharmacological validation of a mouse model of l-DOPA-induced dyskinesia. Exp Neurol. 2005;194:66–75. doi: 10.1016/j.expneurol.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Luscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci. 2010;11:301–15. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C, Gruneir A, Rochon P, Wang X, Anderson G, Brotchie J, et al. Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann Neurol. 2012;71:362–9. doi: 10.1002/ana.22616. [DOI] [PubMed] [Google Scholar]
- Narushima M, Uchigashima M, Hashimoto K, Watanabe M, Kano M. Depolarization-induced suppression of inhibition mediated by endocannabinoids at synapses from fast-spiking interneurons to medium spiny neurons in the striatum. Eur J Neurosci. 2006;24:2246–52. doi: 10.1111/j.1460-9568.2006.05119.x. [DOI] [PubMed] [Google Scholar]
- Neve KA, Ford CP, Buck DC, Grandy DK, Neve RL, Phillips TJ. Normalizing dopamine D2 receptor-mediated responses in D2 null mutant mice by virus-mediated receptor restoration: comparing D2 and D2. Neuroscience. 2013;248C:479–87. doi: 10.1016/j.neuroscience.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Schapira AH. Therapeutic prospects for Parkinson disease. Ann Neurol. 2013;74:337–47. doi: 10.1002/ana.24011. [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, et al. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–62. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson's disease (STEADY-PD) Mov Disord. 2013;28:1823–31. doi: 10.1002/mds.25639. [DOI] [PubMed] [Google Scholar]
- Pasternak B, Svanstrom H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol. 2012;175:627–35. doi: 10.1093/aje/kwr362. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Prinz A, Selesnew LM, Liss B, Roeper J, Carlsson T. Increased excitability in serotonin neurons in the dorsal raphe nucleus in the 6-OHDA mouse model of Parkinson's disease. Exp Neurol. 2013;248:236–45. doi: 10.1016/j.expneurol.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–56. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–37. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. 2010;67:600–6. doi: 10.1002/ana.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Saab BJ, Georgiou J, Nath A, Lee FJ, Wang M, Michalon A, et al. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron. 2009;63:643–56. doi: 10.1016/j.neuron.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schultz W. Multiple functions of dopamine neurons. F1000 Biol Rep. 2010;2:2. doi: 10.3410/B2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, De Jager PL, Feany MB. Parkinson's disease: genetics and pathogenesis. Annu Rev Pathol. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- Simuni T, Borushko E, Avram MJ, Miskevics S, Martel A, Zadikoff C, et al. Tolerability of isradipine in early Parkinson's disease: a pilot dose escalation study. Mov Disord. 2010;25:2863–6. doi: 10.1002/mds.23308. [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, et al. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca 2+ channels. J Clin Invest. 2004;113:1430–9. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, et al. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol. 2009;75:407–14. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2008;2:233–51. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, et al. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34(Pt 5):903–9. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord. 2013;28:715–24. doi: 10.1002/mds.25187. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009290. doi: 10.1101/cshperspect.a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J Biol Chem. 2013;288:10736–41. doi: 10.1074/jbc.R112.410530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, et al. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA. 2009;106:14622–7. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Grace AA. Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 2012;35:422–30. doi: 10.1016/j.tins.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Vallone D, Picetti R, Borrelli E. Structure and function of dopamine receptors. Neurosci Biobehav Rev. 2000;24:125–32. doi: 10.1016/s0149-7634(99)00063-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Schapira AH. Dopamine agonists in Parkinson's disease. Expert Rev Neurother. 2008;8:671–7. doi: 10.1586/14737175.8.4.671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.