

Deubiquitination of Rad18 drives its localization to sites of DNA damage and formation of the Rad18–SHPRH complexes necessary for error-free lesion bypass.
Abstract
Deoxyribonucleic acid (DNA) lesions encountered during replication are often bypassed using DNA damage tolerance (DDT) pathways to avoid prolonged fork stalling and allow for completion of DNA replication. Rad18 is a central E3 ubiquitin ligase in DDT, which exists in a monoubiquitinated (Rad18•Ub) and nonubiquitinated form in human cells. We find that Rad18 is deubiquitinated when cells are treated with methyl methanesulfonate or hydrogen peroxide. The ubiquitinated form of Rad18 does not interact with SNF2 histone linker plant homeodomain RING helicase (SHPRH) or helicase-like transcription factor, two downstream E3 ligases needed to carry out error-free bypass of DNA lesions. Instead, it interacts preferentially with the zinc finger domain of another, nonubiquitinated Rad18 and may inhibit Rad18 function in trans. Ubiquitination also prevents Rad18 from localizing to sites of DNA damage, inducing proliferating cell nuclear antigen monoubiquitination, and suppressing mutagenesis. These data reveal a new role for monoubiquitination in controlling Rad18 function and suggest that damage-specific deubiquitination promotes a switch from Rad18•Ub–Rad18 complexes to the Rad18–SHPRH complexes necessary for error-free lesion bypass in cells.
Introduction
Cellular DNA is continuously damaged by a range of endogenous and exogenous sources. If not sensed and repaired efficiently, DNA damage leads to genome instability and eventually cancer. Cells are particularly susceptible to DNA damage during replication, as many lesions can stall the replication fork, ultimately causing fork collapse and genome rearrangements (Ciccia and Elledge, 2010). Therefore, cells have a system for bypassing DNA lesions, either directly at the replication fork or in gaps behind the fork (Daigaku et al., 2010; Karras and Jentsch, 2010; Ulrich, 2011; Diamant et al., 2012). Bypass can be accomplished using specialized translesion synthesis (TLS) polymerases, which can be error prone depending on the polymerase and the type of DNA lesion involved (Waters et al., 2009). Alternatively, cells can invoke an error-free template-switching process, which uses the newly replicated sister chromatid as a template for replication (Branzei, 2011). Together, these two bypass pathways allow for DNA damage tolerance (DDT) and repair of the lesion at a later time.
The DDT pathways are largely coordinated by mono- or polyubiquitination of the replicative clamp proliferating cell nuclear antigen (PCNA; Hoege et al., 2002; Moldovan et al., 2007). Although several E3 ubiquitin ligases control this modification, Rad18 is a central regulator, required for both types of PCNA ubiquitination (Kannouche et al., 2004; Watanabe et al., 2004; Chiu et al., 2006; Ulrich, 2009). Loss of Rad18 increases mutation rates in cells and sensitizes them to DNA damage, illustrating the importance of the DDT pathways in genome stability and cell survival (Friedl et al., 2001; Tateishi et al., 2003). However, overexpression of Rad18 is also deleterious, as it disrupts the proper assembly of some DNA repair foci (Helchowski et al., 2013) and leads to inappropriate PCNA ubiquitination and TLS polymerase recruitment in the absence of DNA damage (Bi et al., 2006). These events could perturb DNA repair or processive DNA replication and increase mutagenesis, consistent with the fact that Rad18 is up-regulated in certain cancers (Wong et al., 2012; Zhou et al., 2012; Xie et al., 2014). Thus, tight control of Rad18 levels and activity promotes genome maintenance.
Although Rad18-dependent PCNA ubiquitination is crucial to initiate DDT, how DDT pathways are fine-tuned to promote accurate bypass of different types of DNA lesions is poorly understood. In the TLS branch of DDT, the lesion-specific response is partially dictated by polymerase choice. There are five TLS polymerases in human cells, each of which can be error prone when replicating an undamaged DNA template, but some of which can be strikingly accurate when bypassing certain types of DNA lesions, making correct polymerase choice essential (Waters et al., 2009). Yet, how the correct polymerase is recruited to a DNA lesion is still unclear. Monoubiquitination of PCNA is a key step in TLS polymerase recruitment (Kannouche et al., 2004; Watanabe et al., 2004), but as the TLS polymerases all contain ubiquitin-binding domains and/or PCNA interacting motifs (Waters et al., 2009), this modification cannot dictate specificity. Therefore, other mechanisms must exist to help distinguish between DNA lesions and coordinate the appropriate response.
At least part of this damage-specific DDT response may be dictated by two additional E3 ubiquitin ligases, SNF2 histone linker plant homeodomain RING helicase (SHPRH) and helicase-like transcription factor (HLTF; Motegi et al., 2006, 2008; Unk et al., 2006, 2008, 2010). Our previous work showed that these proteins affect mutation frequency in a damage-specific manner: HLTF loss increases mutagenesis induced by UV irradiation, whereas SHPRH loss increases mutagenesis induced by the DNA-alkylating agent methyl methanesulfonate (MMS). These effects are at least partially caused by changes in TLS polymerase recruitment mediated by interactions between these proteins and POL η or POL κ. However, this is not the only role of SHPRH and HLTF in DDT. Indeed, HLTF is a DNA translocase that can induce replication fork reversal in vitro (Blastyák et al., 2010; Achar et al., 2011), and SHPRH may have similar activity (Sood et al., 2003; Blastyák et al., 2010). HLTF and SHPRH also have distinct domain architectures, suggesting that these proteins could have unique functions and properties that contribute to their ability to confer damage specificity to the DDT response.
The damage-specific responses of SHPRH and HLTF are partially mediated through competitive binding to Rad18. HLTF binding to Rad18 is constitutive and apparently unaffected by DNA damage, whereas the binding of SHPRH to Rad18 increases strikingly when cells are treated with MMS (Lin et al., 2011; Moldovan and D’Andrea, 2011). HLTF is also degraded by the proteasome after MMS treatment, and inhibition of this degradation prevents formation of the SHPRH–Rad18 complex. This suggests that SHPRH cannot compete for Rad18 binding when HLTF is present. Indeed, SHPRH is of relatively low abundance in the cell compared with HLTF and other known Rad18-interacting partners (Lin et al., 2011). Interestingly, however, although elimination of HLTF is necessary, it is not sufficient to induce the interaction between SHPRH and Rad18, raising the possibility that another damage-inducible step is required.
Here, we demonstrate that Rad18 in cells exists in at least two forms: an inactive, monoubiquitinated form (Rad18•Ub) and an active, nonubiquitinated form. Rad18•Ub does not interact with SHPRH or HLTF, and it does not form foci, promote PCNA monoubiquitination, or prevent mutagenesis after exposure to DNA damaging agents. Interestingly, Rad18•Ub also has a strong preference for binding to nonubiquitinated Rad18, suggesting that the ubiquitinated form may inhibit other Rad18 molecules in trans. Upon exposure to MMS or H2O2, Rad18 is deubiquitinated, promoting a switch from Rad18•Ub–Rad18 complexes to Rad18–SHPRH complexes and error-free bypass of DNA lesions. Thus, our data provide important mechanistic insights into the damage-specific regulation of DDT, identifying a new level of control that acts on Rad18 activity and function, upstream of HLTF, SHPRH, or the TLS polymerases.
Results
The Rad18–SHPRH interaction depends on modification of Rad18
Our previous work showed that the interaction between the E3 ubiquitin ligases Rad18 and SHPRH is significantly enhanced after exposure to MMS, but not UV irradiation, and that this interaction is blocked by HLTF (Lin et al., 2011). To better understand the requirements for this interaction, we expressed discrete fragments of SHPRH and tested the ability of each to interact with FLAG-Rad18 (Fig. 1 A). We found SHPRH353–628 was the only fragment that interacted with Rad18 when expressed in cells (Fig. 1 B). Within this region, a smaller fragment consisting of the H15 histone linker domain, homologous to a region found in H1 and H5 histone families (Kasinsky et al., 2001), was also sufficient to interact with Rad18 (Fig. S1 A). This finding demonstrates for the first time that the H15 domain in SHPRH is a protein–protein interaction motif that mediates its association with Rad18.
Figure 1.

MMS-induced modification of Rad18 promotes its interaction with SHPRH. (A) Domain structure of SHPRH. Helic-C, helicase C-terminal domain; PHD, plant homeodomain. (B) The SHPRH353–628 (SH353–628) fragment of SHPRH interacts with Rad18. GFP-tagged SHPRH fragments shown in A were expressed in cells with FLAG-tagged Rad18 and lysed under condition A. FLAG-Rad18 and interacting proteins were analyzed by Western blotting. (C) MMS alters Rad18, not SHPRH, to promote the Rad18–SHPRH interaction. Purified GST-tagged Rad18 or SHPRH353–628 were used to pull down full-length FLAG-SHPRH or FLAG-Rad18 (respectively) from transfected cell lysates, which were mock treated or exposed to 0.005% MMS for 4 h. Associated proteins were analyzed by Western blotting.
Next, we asked whether we could recapitulate the effect of MMS on the Rad18–SHPRH interaction using this minimal complex in a semi–in vitro pull-down system. To do so, GST-tagged Rad18 and SHPRH353–628 were purified from Escherichia coli to avoid mammalian posttranslational modifications. In parallel, SHPRH and Rad18 were FLAG-tagged and expressed in human cells, which were treated with or without MMS, and then lysed and incubated with the GST-tagged version of its binding partner. Surprisingly, exposure of cells expressing FLAG-Rad18 to MMS was sufficient to induce the interaction between Rad18 and SHPRH seen previously (Fig. 1 C, first and second lanes). A fragment containing the SHPRH H15 domain could also interact inducibly with endogenous Rad18 in MMS-treated cells (Fig. S1 B). In contrast, exposing cells expressing full-length FLAG-SHPRH to MMS did not induce the interaction (Fig. 1 C, third and fourth lanes). These findings suggest that posttranslational modification of Rad18 promotes its interaction with the H15 domain of SHPRH after MMS treatment and raise the possibility that this modification is the second damage-inducible step needed, in addition to HLTF degradation, to promote the Rad18–SHPRH interaction.
Damage-inducible Rad18–SHPRH interaction coincides with a change in Rad18 mobility
Next, we sought to identify the type of posttranslational modification involved in this damage-regulated interaction. As DNA damage activates the ataxia telangiectasia–mutated (ATM) and ATM- and Rad3-related (ATR) kinases (Ciccia and Elledge, 2010), we hypothesized one of these proteins may promote Rad18 binding to SHPRH after MMS treatment. However, chemical inhibitors of these kinases did not affect the interaction (Fig. 2 A), despite preventing ATM autophosphorylation or phosphorylation of the ATR substrate Chk1. Caffeine, a less specific inhibitor of both ATM and ATR, also had no effect on this interaction (unpublished data). Therefore, the Rad18–SHPRH interaction is not regulated by ATM or ATR.
Figure 2.
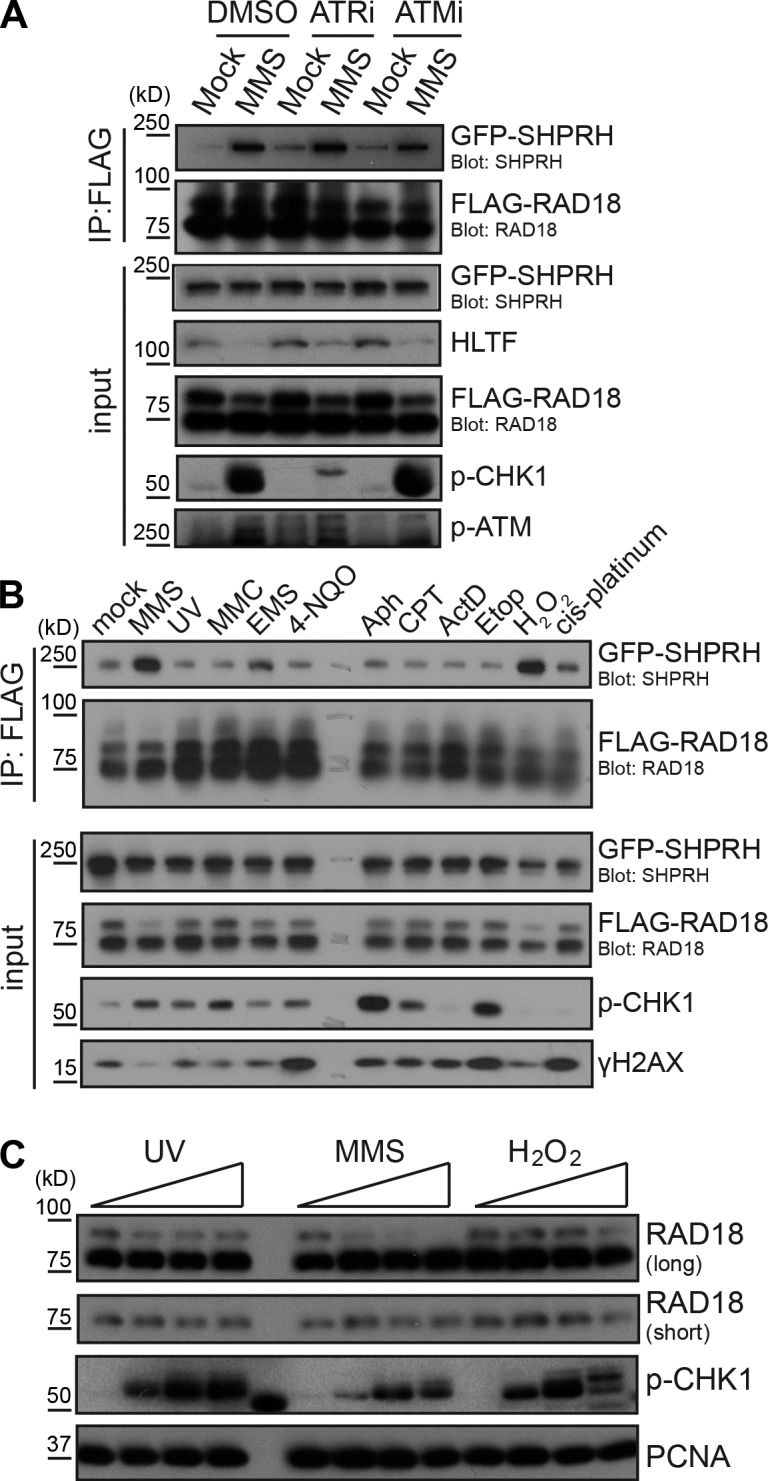
Damage-inducible Rad18–SHPRH binding coincides with loss of high molecular weight Rad18. (A) Inhibition of checkpoint kinases does not affect the Rad18–SHPRH interaction. Cells transfected with FLAG-Rad18 and GFP-SHPRH were mock treated or exposed to 0.005% MMS and the respective kinase inhibitors for 4 h before being lysed and analyzed as in Fig. 1 B. ATRi, ATR inhibition; ATMi, ATM inhibition. (B) Rad18 and SHPRH interact specifically after MMS or H2O2 treatment. Cells expressing FLAG-Rad18 and GFP-SHPRH were treated with 50 ppm MMS, 50 J/m2 UV, 30 µM mitomycin C (MMC), 0.1% ethyl methanesulfonate (EMS), 20 µM 4-NQO, 30 µM aphidicolin (Aph), 2 µM camptothecin (CPT), 1 µM actinomycin D (ActD), 20 µM etoposide (Etop), 0.1% hydrogen peroxide (H2O2), or 50 µM cis-platinum for 4 h before being lysed under condition B and analyzed by Western blotting. (C) Endogenous Rad18 is deubiquitinated after MMS and H2O2 treatment. Untransfected cells were treated with UV (0, 100, 200, and 400 J/m2), MMS (0, 25, 50, and 100 ppm), or H2O2 (0, 1, 2, and 4 mM) for 4 h before being lysed and analyzed by Western blotting.
Intriguingly, Rad18 is monoubiquitinated in several different mammalian cell lines (Miyase et al., 2005). Indeed, we consistently observed two forms of Rad18 in HEK 293T and U2OS cells, in a ratio that varied depending on the type of gel and the antibody used for detection (Fig. S2 A). Using optimized detection conditions, however, we consistently observed a 1:3 ratio of the higher to lower molecular weight Rad18 forms, suggesting that ∼25% of Rad18 is modified in asynchronous HEK 293T cells (Fig. S2 A). We also found that ∼20% of Rad18 is modified in U2OS cells, using the same conditions (Fig. S2 B). To test whether this modification is affected by DNA damage, we exposed cells to a range of genotoxic agents. Surprisingly, only MMS and H2O2 consistently triggered significant loss of the higher molecular weight form of both ectopically expressed (Fig. 2 B) and endogenous Rad18 (Fig. 2 C). These agents also strongly induced binding between SHPRH and Rad18, whereas ethyl methanesulfonate (EMS) had a weaker effect (Fig. 2 B). This is consistent with the previous effects of MMS and EMS on HLTF degradation (Lin et al., 2011). These data suggest that Rad18 changes in response to agents that induce DNA base damage and that loss of the higher molecular weight Rad18 species is important to induce the damage-specific interaction with SHPRH.
Rad18 is deubiquitinated after MMS treatment
To confirm that the higher molecular weight form of Rad18 was indeed ubiquitinated Rad18, we immunopreciptated the endogenous Rad18 and blotted for ubiquitin. Both mono- and diubiquitinated Rad18 were detected. This was confirmed by treating the precipitated protein with the catalytic core of the ubiquitin protease Usp2, which removes conjugated ubiquitin (Fig. 3 A; Ryu et al., 2006). To confirm that this ubiquitinated Rad18 is specifically lost after MMS treatment, we transfected cells with His-tagged ubiquitin and lysed cells under denaturing conditions. Isolation of the His-ubiquitin–conjugated proteins revealed that Rad18 is ubiquitinated in untreated, but not MMS-treated, cells (Fig. 3 B). Interestingly, both mono- and polyubiquitinated Rad18 species are lost after MMS treatment, suggesting that this loss is not specific to any one form of ubiquitinated Rad18. The increased level of polyubiquitinated Rad18 in this experiment, relative to that seen with endogenous protein, is likely caused by overexpression of ubiquitin and enrichment of the ubiquitinated Rad18 species.
Figure 3.
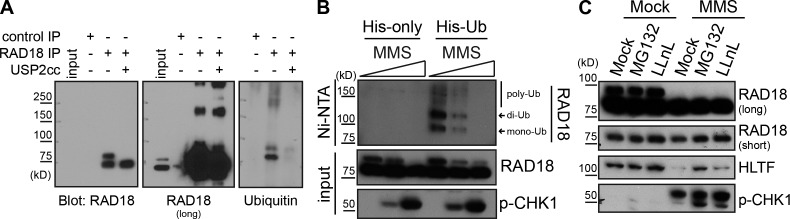
Rad18 is deubiquitinated after MMS treatment. (A) Endogenous Rad18 is monoubiquitinated. Rad18 was immunoprecipitated (RF antibody) from untransfected cells, and purified protein on beads was treated with or without the Usp2 catalytic core (USP2cc) before being analyzed by Western blotting for the presence of ubiquitin. (B) Rad18 is deubiquitinated after MMS treatment. Cells mock transfected or transfected with His-tagged ubiquitin were treated with MMS (0, 25, and 50 ppm) for 4 h before being lysed under denaturing conditions. His-ubiquitin (Ub) and covalently bound proteins were purified on nickel agarose beads (NTA, nitrilotriacetic acid) and analyzed by Western blotting. (C) Proteasome inhibition does not prevent Rad18 deubiquitination after MMS. Untransfected cells were mock or MMS (0.005%) treated in conjunction with 50 µM MG132 or 25 µM LLnL for 4 h and processed as in Fig. 2 C.
As we observed polyubiquitinated Rad18 in these experiments, we considered the possibility that monoubiquitinated Rad18 is polyubiquitinated and degraded after MMS treatment. Intriguingly, however, the proteasome inhibitors MG132 and N-acetyl-l-leucyl-l-leucyl-l-norleucinal (LLnL) did not rescue loss of monoubiquitinated Rad18 after exposure to MMS (Fig. 3 C), nor did we see accumulation of polyubiquitinated Rad18 (Fig. S2 C), despite the fact that these treatments rescued HLTF levels. This suggests that ubiquitinated Rad18 is not degraded by the proteasome after MMS treatment. Collectively, these findings indicate that Rad18 is deubiquitinated after exposure to certain types of DNA damaging agents and that deubiquitination is correlated with the inducible, damage-specific interaction between Rad18 and SHPRH.
Rad18 ubiquitination regulates its interactions with SHPRH and other Rad18 molecules
To examine the functional significance of Rad18 deubiquitination, we sought to modulate its ubiquitination state in cells. Several groups have attempted to identify and mutate the ubiquitination site on Rad18 without success (Miyase et al., 2005; Notenboom et al., 2007; Inagaki et al., 2011). Therefore, we chose to promote Rad18 autoubiquitination instead, through ectopic expression of the E2 Rad6 (Miyase et al., 2005). Rad6 expression increased Rad18 ubiquitination and was able to prevent the MMS-inducible interaction between FLAG-Rad18 and GFP-SHPRH (Fig. 4 A), suggesting that Rad18 ubiquitination impedes its ability to interact with SHPRH after MMS treatment.
Figure 4.

Direct modulation of Rad18 ubiquitination affects Rad18–Rad18 and Rad18–SHPRH interactions. (A) Promoting Rad18 ubiquitination in vivo prevents damage-inducible binding to SHPRH. Cells were cotransfected with GFP-SHPRH, FLAG-Rad18, and either Rad6 or empty vector and then mock or MMS (0.005%) treated for 4 h before being lysed and processed as in Fig. 2 B. Quantification indicates ratio of GFP-SHPRH in the IP relative to the respective input sample, normalized to the mock-transfected, mock-treated control (cont.). (B) Deubiquitinating Rad18 affects its protein–protein interactions. FLAG-Rad18 was cotransfected with Rad6 and then purified from lysates under high-salt conditions. Beads were mock treated or deubiquitinated by Usp2 and used to pull-down GFP-SHPRH or GFP-Rad18 from transfected cells lysed as in Fig. 1 C. Pull-downs were analyzed by Western blotting. USP2cc, Usp2 catalytic core. (C) Rad18 shifts to a smaller complex after MMS treatment. Cells were mock treated or exposed to 0.01% MMS for 2 h and then lysed and separated on a 5–30% glycerol gradient. Gradient fractions were analyzed for the presence of Rad18 and PCNA by Western blotting. White lines indicate that intervening lanes have been spliced out.
To test this idea further, we asked whether Rad18 deubiquitination is sufficient to promote its interaction with SHPRH. To do so, we affinity purified FLAG-Rad18 coexpressed with Rad6 in cells and treated the immobilized proteins in vitro with or without the catalytic core of Usp2. Interestingly, when used to pull-down GFP-SHPRH from lysates, deubiquitinated Rad18 did not exhibit a significantly higher affinity for SHPRH than did untreated Rad18 (Fig. 4 B). This finding suggests that deubiquitination is a prerequisite for damage-inducible Rad18–SHPRH binding but is not sufficient. As Rad18 has been reported to dimerize in vivo and in vitro (Ulrich and Jentsch, 2000; Miyase et al., 2005; Notenboom et al., 2007; Masuda et al., 2012), we also tested the effect of Rad18 ubiquitination on its interaction with GFP-Rad18 expressed in cells. Surprisingly, the untreated, ubiquitinated FLAG-Rad18 pulled down significantly more GFP-Rad18 than the Usp2-treated FLAG-Rad18 (Fig. 4 B), suggesting that ubiquitination promotes interactions with additional Rad18 molecules.
As ubiquitination appears to alter Rad18 protein–protein interactions, and MMS induces Rad18 deubiquitination, we hypothesized that Rad18 complexes may change after MMS treatment. To test this, we analyzed lysates of mock- or MMS-treated cells on a glycerol gradient. After MMS treatment, Rad18 consistently appeared in lower molecular weight fractions of the gradient than Rad18 from undamaged cells (Fig. 4 C), suggesting that a significant portion of the protein is found in a smaller complex after MMS treatment. Collectively, our findings indicate that MMS-induced Rad18 deubiquitination alters Rad18 complexes and promotes interactions between Rad18 and other binding partners.
Rad18-Ub fusions preferentially bind to nonubiquitinated Rad18
Our results indicate that ubiquitin modulates Rad18 protein–protein interactions in vivo. To test this hypothesis further, we created linear chimeras of Rad18 and ubiquitin, with ubiquitin fused either to the N (Ub-Rad18) or C (Rad18-Ub) terminus of Rad18 (Fig. 5 A and Fig. S3, A and B). We also generated matched chimeras with an isoleucine-to-alanine point mutation at I44 in the ubiquitin domain of each fusion (Ub(I44A)). This mutation has been shown to disrupt binding between ubiquitin and the zinc finger (ZnF) ubiquitin-binding motif of POL η (Bomar et al., 2007; Bienko et al., 2010). We reasoned that this mutant should allow us to distinguish whether any effects of fusing Rad18 to ubiquitin are a result of simple steric hindrance and/or disruption of protein folding versus specific recognition of the ubiquitin by a ubiquitin-binding motif. Importantly, binding between Rad18 and Rad6 was unaffected by fusion to ubiquitin or Ub(I44A) (Fig. S3 C), suggesting the Rad18 chimeras are properly folded. The ubiquitin fusion also did not inhibit Rad18 ubiquitination in cells (Fig. S3 D, input) or in vitro (Fig. S3 D, in vitro ubiquitination).
Figure 5.

Rad18-Ub fusion proteins preferentially bind to nonubiquitinated Rad18. (A) Structure of GST–Rad18-Ub chimera proteins. GST (dark gray), ubiquitin (white), and Rad18 (light gray) were fused in frame. The I44A mutation in ubiquitin prevents recognition by a ZnF ubiquitin-binding domain (Bomar et al., 2007). (B) Fusing ubiquitin to Rad18 promotes binding to Rad18 but not to SHPRH. GST-Rad18 fusion proteins were incubated with whole-cell lysates transfected with FLAG-Rad18 or FLAG-SHPRH and analyzed as in Fig. 1 C. (C) The Rad18–Rad18 interaction is weaker than the Rad18-Ub–Rad18 interaction. GST-Rad18 fusion proteins were incubated with whole-cell lysates transfected with FLAG-Rad18 and analyzed as in Fig. 1 C. Ubiquitin blots were used to confirm the identity of the GST constructs. (D) Rad18-Ub chimeras bind preferentially to nonubiquitinated Rad18. GST-Rad18 fusion proteins were incubated with cell lysates cotransfected with FLAG-Rad18 and Rad6 to attain a ratio of ∼1:1 Rad18/Rad18•Ub, and analyzed as in Fig. 1 C. (E) SHPRH and ubiquitin compete for binding to Rad18. GST-tagged ubiquitin on beads was incubated with FLAG-Rad18 and increasing amounts of FLAG-SHPRH, both purified from human cells. Proteins bound to the beads were analyzed by Western blotting.
Next, we tested the ability of these ubiquitin fusions to bind to SHPRH and Rad18. Consistent with our previous observations, affinity-purified FLAG-SHPRH (Fig. S3 E) or FLAG-SHPRH in cell lysates (Fig. 5 B, lanes 11 and 13) bound more weakly to the GST–Rad18-Ub chimeras than to wild-type Rad18 or their respective Ub(I44A) counterparts (Fig. 5 B, lanes 10, 12, and 14). This preference was also observed when GFP-tagged Rad18 chimeras were coimmunoprecipitated with FLAG-SHPRH in cells (Fig. S3 F). Strikingly, however, all five GST-Rad18 ubiquitin fusions exhibited the opposite trend when we tested their ability to interact with FLAG-Rad18 expressed in cell lysates, with FLAG-Rad18 binding preferentially to the ubiquitin fusion proteins (Fig. 5 B, lanes 4 and 6). This ubiquitin-mediated binding was much stronger than the Rad18–Rad18 interaction alone (Fig. 5 C). Interestingly, fusing ubiquitin to either side of Rad18 inhibited its interaction with SHPRH and promoted its interaction with Rad18, although there were differences in the extent to which the interactions were altered (Fig. 5 B). The reasons for this are currently unknown, but it is clear that the presence or absence of ubiquitin dominates in these experiments.
While examining the differences between the Rad18–Rad18 and Rad18•Ub–Rad18 complexes in lysates, we noticed that only nonubiquitinated Rad18 consistently interacted with our Rad18-Ub fusion proteins (Fig. 5 B, input vs. lanes 4 and 6). To explore this preferential binding further, we coexpressed FLAG-Rad18 and Rad6 in a manner that achieved a nearly 1:1 ratio of ubiquitinated and nonubiquitinated FLAG-Rad18 in cells and then tested this interaction. Under these conditions, the GST–Rad18-Ub fusion protein interacted only with the nonubiquitinated form of FLAG-Rad18, whereas GST-Rad6 interacted with both ubiquitinated and nonubiquitinated forms equally (Fig. 5 D, lanes 4 and 6). Unexpectedly, GST-Rad18 did not pull down ubiquitinated FLAG-Rad18, suggesting that the preexisting Rad18•Ub–Rad18 complex in lysates is very stable and could not be disrupted by excess nonubiquitinated Rad18. Altogether, our findings suggest that ubiquitination of Rad18 promotes its interaction with nonubiquitinated Rad18, rather than SHPRH, but does not alter its interaction with Rad6.
As modification of Rad18 with ubiquitin blocks its ability to interact with SHPRH, it is possible that ubiquitin and SHPRH directly compete for Rad18 binding. To further test this idea, glutathione beads bound with GST-Ub were incubated with affinity-purified FLAG-SHPRH and FLAG-Rad18, and the concentration of SHPRH was varied, whereas that of Rad18 was held constant. Under these conditions, less Rad18 bound to the GST-Ub beads as the SHPRH concentration was increased (Fig. 5 E), indicating that SHPRH effectively competes with the GST-Ub to keep FLAG-Rad18 in solution. This finding, together with our observation that the Ub(I44A) fusion protein behaves similarly to nonubiquitinated Rad18 (Fig. 5 B and Fig. S3 F), suggests that recognition of the ubiquitin on Rad18 by a ubiquitin binding domain is key in regulating this switch in binding partners.
The UBZ-type ZnF domain of Rad18 binds to both SHPRH and HLTF
To investigate the mechanism by which ubiquitin interferes with the Rad18–SHPRH interaction, we asked whether any of the known functional domains in Rad18 were required for this association (Fig. 6 A). To do so, we coexpressed GFP-SHPRH with a panel of FLAG-Rad18 deletion mutants lacking the RING (ubiquitin ligase), SAP (DNA binding), ZnF (ubiquitin binding), or Rad6-binding domain. SHPRH reproducibly bound more strongly to the Rad18ΔRING protein than to wild-type Rad18 (Fig. 6 B), although the reasons for this are currently unclear. Interestingly, we also found that deletion of the UBZ (ubiquitin-binding ZnF)-type ZnF domain in Rad18 strongly reduced SHPRH binding (Fig. 6 B). Furthermore, a smaller bacterially expressed fragment of Rad18 containing the ZnF domain was able to interact with GFP-SHPRH in cell lysates (Fig. 6 C) and to bind directly to the previously mapped H15 domain of SHPRH (Fig. 6 D). These observations suggest that the ZnF domain of Rad18 is needed for binding to SHPRH.
Figure 6.

Rad18 binds SHPRH and HLTF through its ubiquitin-binding ZnF domain. (A) Domain structure of Rad18 showing wild-type, Rad18 ΔZnF(Δ200–224), and Rad18-ZnF(186–240) constructs (Huang et al., 2009). BD, binding domain. (B) The Rad18-ZnF is important to bind SHPRH. FLAG-Rad18 constructs lacking the indicated domains were cotransfected with GFP-SHPRH. FLAG-Rad18 and interacting proteins were analyzed as in Fig. 1 B. (C) The Rad18-ZnF contributes to SHPRH binding. GST–Rad18-ZnF or full-length Rad18 was used to pull-down GFP-SHPRH from lysates and analyzed as in Fig. 1 C. (D) The Rad18-ZnF interacts directly with the SHPRH-H15 domain. GST–SHPRH-H15 was used to pull-down FLAG–Rad18-ZnF from cell lysates and analyzed as in Fig. 1 C. (E) The Rad18-ZnF is important for binding HLTF. FLAG-Rad18 constructs lacking the indicated domains were cotransfected with untagged HLTF. FLAG-Rad18 and interacting proteins were analyzed as in Fig. 1 B. (F) HLTF binding is disrupted by the Rad18-Ub fusion. GST-Rad18 fusion proteins were incubated with cell lysates transfected with GFP-HLTF and analyzed as in Fig. 1 C.
Because we previously showed that SHPRH and HLTF compete for Rad18 binding in cells (Lin et al., 2011), we asked whether the ZnF domain of Rad18 was also required to bind HLTF. Indeed, deletion of the ZnF domain strongly reduced the interaction between Rad18 and HLTF (Fig. 6 E). HLTF expressed in cell lysates was also unable to interact with the GST–Rad18-Ub chimeras, similarly to SHPRH (Fig. 6 F). Interestingly, the ZnF domain of Rad18 has also been shown to bind to ubiquitin both in vitro and in vivo (Bish and Myers, 2007; Notenboom et al., 2007; Huang et al., 2009). Therefore, we conclude that the ZnF domain in Rad18 is important for binding to HLTF, SHPRH, and ubiquitin. This provides a possible explanation for why both HLTF degradation (Lin et al., 2011) and Rad18 deubiquitination (Fig. 4 A) are required for the formation of the Rad18–SHPRH complex after MMS treatment.
Rad18-Ub fusions interfere with accurate MMS-induced lesion bypass
Our results clearly show that Rad18 ubiquitination disrupts its interaction with SHPRH and promotes binding to nonubiquitinated Rad18. As SHPRH function is important to suppress MMS-induced mutagenesis (Lin et al., 2011), we hypothesized that ubiquitinated Rad18 may disrupt the ability of the Rad18–SHPRH complex to carry out accurate lesion bypass. To test this, we used the SupF reporter assay (Parris and Seidman, 1992) to measure mutation frequency in HEK 293T cells overexpressing either wild-type GFP-Rad18 or GFP–Ub-Rad18, which cannot interact with SHPRH (Fig. S3 F). Overexpression of the GFP–Ub-Rad18 fusion protein elevated the MMS-induced mutation frequency compared with expression of GFP-Rad18 (Fig. 7 A, first and third lanes). This suggests that the exogenous protein blocks the function of endogenous Rad18. To confirm that this effect is specifically mediated by the Rad18–SHPRH complex, we also knocked down SHPRH in these conditions. Interestingly, although knockdown of SHPRH increased MMS-induced mutagenesis in cells expressing GFP-Rad18 (Fig. 7 A, first and second lanes), the same knockdown did not further increase the mutagenesis in the presence of GFP–Ub-Rad18 (Fig. 7 A, third and fourth lanes). This supports our hypothesis that the SHPRH–Rad18 interaction induced by MMS treatment is important for accurate MMS lesion bypass and that Rad18 ubiquitination disrupts this response.
Figure 7.

Rad18-Ub fusions are unable to respond to DNA damage. (A) Knockdown of SHPRH and overexpression of Rad18-Ub is epistatic. SupF reporter plasmid (0.5% MMS) was recovered from HEK 293T cells 48 h after cotransfection with the indicated GFP-Rad18 constructs (Ub, ubiquitin) and siRNAs (siCtrl, control siRNA). Data represent mean and standard deviations from three independent experiments. At least 2,000 colonies were analyzed per condition. **, P < 0.01, relative to control knockdown. (B) Rad18-Ub chimeras are nuclear but fail to localize to sites of DNA damage. U2OS clones stably expressing different forms of GFP-Rad18 were treated with 0.005% MMS for 4 h before preextraction and fixation. Rad18 was visualized using direct GFP fluorescence, γ-H2AX was detected using immunofluorescence. Bars, 10 µm. (C) Rad18-Ub chimeras do not form foci after any type of DNA damage. Clones in B were treated with MMS for 4 h or allowed to recover for 4 h after exposure to the indicated doses of UV or ionizing radiation (IR). GFP-Rad18 foci were counted using ImageJ. Each bar represents a mean and standard deviation from three independent experiments. At least 100 cells were counted per condition. **, P < 0.01, relative to wild-type (WT) Rad18. (D) Rad18-Ub chimeras cannot induce PCNA ubiquitination. Rad18−/− HCT116 cells were transiently transfected with GFP-Rad18 chimera constructs and damaged with UV or MMS before being analyzed as in Fig. 2 C. (E) Rad18-Ub chimeras cannot suppress mutagenesis on MMS- or UV-damaged plasmids. SupF reporter plasmid (0.5% MMS or 1,000 J/m2 UV) was recovered from Rad18−/− HCT116 cells 48 h after cotransfection with the indicated GFP-Rad18 chimera constructs (Ub(I44A), ubiquitin-I44A). Data represent means and SEMs from four independent experiments. At least 2,000 colonies were analyzed per condition. *, P < 0.05, relative to GFP-only control. R18, Rad18.
Ubiquitinated Rad18 is unable to respond to DNA damage
As the ubiquitin-fused Rad18 is unable to interact with SHPRH or HLTF and interferes with endogenous Rad18 function, we hypothesized that it may be unable to perform other Rad18 functions as well. After treatment with DNA damaging agents, Rad18 is recruited to nuclear foci thought to mark stalled replication forks (Watanabe et al., 2004) or sites of DNA damage (Huang et al., 2009). To test the effect of ubiquitin on Rad18 localization in cells, we generated U2OS clones that stably express GFP-Rad18 or the Rad18-Ub chimeras (Fig. S4, A and B). We then tested the ability of these proteins to form detergent-resistant nuclear foci after different types of DNA damage, beginning with wild-type Rad18. Although wild-type Rad18 foci were observed after MMS treatment, the number of foci-positive cells decreased when Rad6 was transiently transfected into the cells, suggesting that ubiquitinated Rad18 is unable to form damage-inducible foci (Fig. S5 A). Next, we examined the cells stably expressing the Rad18-Ub fusions. Although previous cell fractionation data suggested that ubiquitinated Rad18 is localized predominantly in the cytosol (Miyase et al., 2005), we found that all of the stably expressed chimeras were enriched in the nucleus (Fig. 7 B, PFA-only column), with only a small portion in the cytosol. This difference may be caused by differences in the experimental approach. More importantly, the ubiquitin-fused proteins were not found in nuclear foci after treatment with MMS, UV, or ionizing radiation. In contrast, nuclear foci of wild-type Rad18 were observed under all of these conditions, and the Ub(I44A) mutant also formed foci (Fig. 7, B and C; and Fig. S4 C). These findings indicate that ubiquitinated Rad18 is not recruited efficiently to sites of DNA damage and suggest that ubiquitinated Rad18 is inactive, even when present in the nucleus.
To test the activity of ubiquitinated Rad18, we next examined PCNA monoubiquitination, an important Rad18-dependent signal for TLS-mediated DDT (Hoege et al., 2002). Although PCNA monoubiquitination normally increases after DNA damage, overexpression of Rad6 suppressed this increase (Fig. S5 B), suggesting that ubiquitination of Rad18 inhibits its activity. As Rad6 interacts with and/or ubiquitinates several other proteins in the cell that could confound downstream readouts of Rad18 function, we also tested the effect of Rad18 ubiquitination more directly by examining the ability of the ubiquitin chimeras to induce PCNA ubiquitination in Rad18 knockout cells. In the presence of DNA damage that would normally activate Rad18, the Rad18-Ub proteins did not induce PCNA monoubiquitination (Fig. 7 D). In contrast, wild-type Rad18 and the Ub(I44A) fusion proteins were proficient (Fig. 7 D). Thus, ubiquitination plays a general role in negatively regulating Rad18’s ability to recognize and respond to DNA damage.
Finally, as Rad18 recruitment and PCNA ubiquitination are both important for DDT, we hypothesized that the Rad18-Ub fusion proteins would be unable to participate in DDT and suppress damage-induced mutagenesis. To test this model, we transiently complemented Rad18 knockout cells with wild-type Rad18 or Rad18-Ub fusions and tested MMS- or UV-induced mutation frequency using the SupF reporter assay. Although transfection of wild-type or the Ub(I44A) fusion proteins was able to rescue the naturally high mutation level seen in the Rad18-null cells, expressing the ubiquitin fusion proteins failed to suppress the MMS- or UV-induced mutagenesis (Fig. 7 E). Transient expression of Rad6 also increased MMS-induced mutation frequency in wild-type cells (Fig. S5 C). These findings are consistent with the observation that ubiquitinated Rad18 does not localize to DNA damage foci or induce PCNA ubiquitination after DNA damage. Collectively, we conclude from these findings that the ubiquitinated Rad18 is inactive and unable to respond to DNA damage or prevent mutagenesis.
Discussion
Our work demonstrates that Rad18 ubiquitination allows for dynamic control of Rad18 activity in cells. Specifically, we find that a portion of Rad18 is ubiquitinated and inactivated in unchallenged cells and that damage-inducible deubiquitination of Rad18 is important for accurate DDT after exposure to certain types of DNA damaging agents. We show that Rad18 ubiquitination blocks its recruitment to sites of DNA damage, disrupts interactions with the key DDT proteins SHPRH and HLTF, and prevents the induction of PCNA monoubiquitination and accurate DNA lesion bypass after treatment with several types of DNA damaging agents. Furthermore, ubiquitinated Rad18 preferentially binds nonubiquitinated Rad18 over another ubiquitinated Rad18 molecule. This suggests that ubiquitinated Rad18 could bind nonubiquitinated Rad18 in cells, preventing an otherwise active molecule from being recruited to damaged DNA and possibly inactivating it in trans (Fig. 8).
Figure 8.

Ubiquitination of Rad18 controls its interactions and functions. In undamaged cells, Rad18 exists in equilibrium between the ubiquitinated and nonubiquitinated state and forms complexes with distinct binding partners. Upon treatment with MMS, HLTF is degraded, and Rad18 is deubiquitinated, freeing the ZnF domain of Rad18 to interact with the H15 domain of SHPRH and inducing formation of SHPRH–Rad18 complexes, which prevents MMS-induced mutagenesis.
The effects of deubiquitination on Rad18 activity after MMS treatment constitute a new damage-specific role for ubiquitin in DDT signaling, complementing the emerging and varied usage of ubiquitin for a variety of signaling events in DDT (Jackson and Durocher, 2013). For example, mono- and polyubiquitination of the replicative clamp, PCNA, alters the spectrum of proteins that interact with PCNA at the replication fork and helps determine the type of DDT performed. Monoubiquitination of the TLS polymerases, particularly POL η and POL ι, also controls their localization and activity in DDT (Bienko et al., 2010; McIntyre et al., 2013). Here, we define a third role for ubiquitin in DDT, demonstrating that ubiquitination controls Rad18 by holding it in an inactive state, until deubiquitination is triggered by exposure to certain types of DNA damage.
We hypothesize that monoubiquitination and inactivation of Rad18 serves as an additional layer of regulation on Rad18 activity, which may prevent use of the error-prone TLS polymerases on undamaged DNA. Rad18 is already known to be regulated during the cell cycle through Cdc7-dependent phosphorylation (Day et al., 2010), and our data now add a mechanism to regulate Rad18 after DNA damage. Autoinhibition through ubiquitination could serve as an efficient and rapid negative feedback mechanism for constraining Rad18 DDT activity in unchallenged cells, while maintaining a pool of Rad18 that can be readily accessed if a rapid influx of active molecules is required after certain types of DNA damage.
Intriguingly, although Rad18-Ub fusion proteins are unable to be recruited to all types of DNA lesions tested, deubiquitination is only observed after MMS/EMS and H2O2 treatment. This suggests that the available pool of nonubiquitinated Rad18 that naturally exists in cells is sufficient for some types of DDT, such as after exposure to UV light, but insufficient after other types of damage. The fact that the cell tightly controls the formation of the Rad18–SHPRH complex, through HLTF degradation, Rad18 deubiquitination, and possibly other mechanisms, suggests that this complex may have detrimental roles when formed under inappropriate contexts. Indeed, unregulated SHPRH activity has been previously shown to induce mutations (Lin et al., 2011), suggesting that SHPRH is also tightly controlled in the cell.
How Rad18 deubiquitination is induced after MMS treatment is still unclear. As loss of monoubiquitinated Rad18 is not caused by proteasomal degradation (Fig. 3 C and Fig. S2 C), one possibility is that Rad18’s ligase activity is redirected to other substrates after MMS treatment, reducing autoubiquitination. Alternatively, ubiquitinated Rad18 may be actively targeted by one or more deubiquitinating enzymes (DUBs). These DUBs could be activated by specific types of DNA damage or may continuously deubiquitinate Rad18 in cells unless the ubiquitin is protected. One way this could occur is through formation of the ubiquitin-mediated Rad18 oligomer that we observe (Fig. 4 and Fig. 5). Indeed, there are some reported differences between the ubiquitination state of the Rad18 ZnF mutant (Rad18-C207F) in vitro and in vivo that support this idea. The Rad18-C207F mutant (which cannot bind to ubiquitin) is not ubiquitinated in cells (Miyase et al., 2005) but has been shown to be capable of ubiquitinating itself in vitro (Notenboom et al., 2007). Indeed, we observe the same in our experiments (Fig. S3 D). This suggests that Rad18-C207F is not actually ligase deficient but that its inability to bind to ubiquitin affects its ubiquitination state in cells. Based on our data, we hypothesize that the autoubiquitinated Rad18-C207F species is not maintained in cells because of the inability of this mutant to bind and protect the ubiquitin on itself or another Rad18 molecule from DUBs.
Importantly, our data clearly demonstrate that the Rad18 complexes found in untreated cells are distinct from those in MMS-treated cells. Previous work has shown that purified Rad18 and Rad6 coexpressed in bacteria form an active heterotrimer (Rad182Rad6; Masuda et al., 2012) or dimer of heterodimers (Rad182Rad62; Notenboom et al., 2007), which is capable of ubiquitinating PCNA in vitro. As this purified Rad18 is not ubiquitinated in these experiments, we hypothesize that this complex reflects the active form of Rad18 in cells, whereas our larger, ubiquitin-mediated complex is more likely inactive. This Rad18 complex almost certainly contains other binding partners that could affect Rad18 function and localization (Fig. 4 C). Some of these proteins may contribute to Rad18 inactivation in the cell, whereas others may themselves be sequestered and held inactive by Rad18-Ub, adding additional importance to Rad18 deubiquitination after MMS treatment. Indeed, we find that the ubiquitin-fused Rad18 preferentially interacts with nonubiquitinated Rad18 (Fig. 5 D), suggesting that ubiquitin-mediated Rad18 complexes could sequester Rad18 molecules that would otherwise be active. This would increase the impact of Rad18 ubiquitination on the entire Rad18 population, allowing the 25% of molecules that are ubiquitinated in HEK 293T cells (Fig. S2 A) to inactivate an additional 25% or more of the remaining Rad18, depending on the Rad18•Ub/Rad18 binding ratio. Though we currently favor this hypothesis, intramolecular binding between the ZnF domain and ubiquitin may also play a role in Rad18 inactivation.
Our observation that Rad18 interacts with several binding partners through its ZnF domain also suggests that Rad18 exists in several distinct complexes in the cell, including a self-associating oligomer. This is similar to what has been observed in yeast, where Rad5, the proposed yeast homologue of SHPRH and HLTF, as well as yeast Rad18 itself competes to interact with Rad18 through its ZnF domain (Ulrich and Jentsch, 2000). Interestingly, however, endogenous Rad18 does not appear to be ubiquitinated in yeast (Miyase et al., 2005), indicating this particular regulatory mechanism might only be found in higher eukaryotes. Considering the additional complexity in these cells, including multiple Rad5 homologues and a damage-specific DDT response, we hypothesize that the ubiquitination of Rad18 provides an extra level of control in higher eukaryotes that is needed to fine tune the composition and activity of DDT complexes.
Ultimately, our data provide insight into the regulation of Rad18 activity through posttranslational modification and protein–protein interactions. As Rad18 is only deubiquitinated after certain types of DNA damage, there may be quantitative differences in the requirements for Rad18 activity under different DNA damage conditions, a question that will be interesting to investigate in the future. Additionally, the ratio of ubiquitinated to nonubiquitinated Rad18 has been shown to vary dramatically across different cell types (Miyase et al., 2005), suggesting that Rad18 ubiquitination may also be a mechanism for controlling Rad18 activity in different phases of the cell cycle or in altered cell states, such as cancer.
Materials and methods
Antibodies
The commercial antibodies used in these experiments are as follows: FLAG (mouse; M5 F4042; Sigma-Aldrich; Fig. 5, B and E), FLAG (mouse; M2 F3165; Sigma-Aldrich), GAPDH (mouse; 8245; Abcam), GFP (rabbit; sc8334; Santa Cruz Biotechnology, Inc.; Fig. 1, Fig. 4, Fig. 7, Fig. S3 F, and Fig. S4), GFP (mouse; sc9996; Santa Cruz Biotechnology, Inc.), p53 (mouse; sc126; Santa Cruz Biotechnology, Inc.), p-ATM (mouse; 4526; Cell Signaling Technology), PCNA (mouse; sc56; Santa Cruz Biotechnology, Inc.), Rad18 (rabbit; 79763; Abcam; Fig. S2 A), Rad18 (mouse; H00056852-M01; Novus Biologicals), SHPRH (mouse; TA501443; OriGene), ubiquitin (mouse; BML-PW8810-0500; Enzo Life Sciences; Fig. S4 B), ubiquitin (mouse; sc8017; Santa Cruz Biotechnology, Inc.), and γ-H2AX (mouse; 05-636; EMD Millipore).
S. Tateishi (Institute of Molecular Embryology and Genetics, Kumamoto University, Kumamoto, Japan) provided a rabbit Rad18 antibody generated against recombinant human GST-Rad18383–495 (Fig. S2 A, ST; Tateishi et al., 2000). The HLTF antibody was generated in rabbits immunized with human GST-HLTF38–589 (Lin et al., 2011), and rabbit p-CHK1 was raised against a peptide derived from human Chk1 phosphorylated at S345 (QGISFpSQPT; Lupardus and Cimprich, 2006). The Rad6 antibody was generated in rabbits at a commercial facility (Josman LLC) from recombinant full-length His-GST-Rad6 protein. The Rad18 (RF [R. Freire]) antibody (Fig. 2 B, Fig. 3 A, Fig. 4, Fig. S2, and Fig. S3) was generated in rabbits against recombinant His-Rad18300–495 (Fig. S2 D). Two different human SHPRH antibodies were used in this project. One, the SHPRH (KAC [K.A. Cimprich]) antibody (Fig. 2 A and Fig. 6), was generated in rabbits immunized with recombinant human GST-SHPRH1–250 (Lin et al., 2011). The SHPRH (RF) antibody (Fig. 4 B and Fig. S3) was generated in rabbits against recombinant human His-SHPRH1–300 (Fig. S2 E). Quantified Western blots (Fig. 3 and Fig. S2) were captured on an imaging system (AlphaView FluorChem HD2; ProteinSimple) and analyzed using the accompanying software, using local background subtraction.
Plasmids and cloning
The plasmids for pcDNA3.1-FLAG-Rad18 (cytomegalovirus [CMV] promoter), pGEX-4T-GST-Rad18 (tac promoter), EGFP-C-GFP-SHPRH (CMV promoter), pcDNA3.1-His-ubiquitin (CMV promoter), and untagged pCMV-Sport6-HLTF (CMV promoter; Lin et al., 2011) and pGEX-4T-GST-ubiquitin (tac promoter; Chang et al., 2006) have been described previously. Human pcDNA3.1-FLAG-SHPRH (CMV promoter) was subcloned from GFP-SHPRH. Rad6 was purchased from GE Healthcare (mouse Rad6A) and used directly (pCMV-Sport6; CMV promoter) or subcloned into pET28-His-GST (T7 promoter).
The Rad18 ubiquitin chimeras were generated by PCR from GST-Rad18 and His-ubiquitin and cloned into the pGEX-4T1 vector (tac promoter). These were subcloned into pEGFP-C1 and pcDNA3.1-FLAG (both CMV promoters) for the GFP- and FLAG-tagged constructs, respectively. The Ub(I44A) mutation was introduced by standard site-directed mutagenesis using the following primer sequences: forward, 5′-CTCCGGACCAGCAGCGTCTCGCCTTCGCTGGAAAGCAGCTTGA-3′; and reverse, 5′-TCAAGCTGCTTTCCAGCGAAGGCGAGACGCTGCTGGTCCGGAG-3′. In brief, wild-type plasmids were replicated with Phusion polymerase (Thermo Fisher Scientific) for 15 cycles in parallel reactions, each with only one primer. The individual reactions were mixed, and an additional 30 cycles were performed. Plasmids were transformed after Dpn1 digestion and ethanol precipitation.
S protein–FLAG-streptavidin-binding peptide-Rad18 domain deletion constructs, S protein–FLAG-streptavidin-binding peptide-Rad18-ZnF, and GST–Rad18-ZnF in Gateway-compatible vectors were provided by J. Huang (Life Sciences Institute, Zhejiang University, Zhejiang, China) and J. Chen (University of Texas MD Anderson Cancer Center, Houston, TX; Huang et al., 2009). SHPRH fragments were amplified from human SHPRH cDNA and cloned into the pEGFP-C1 (CMV promoter) or pGEX-4T (tac promoter) vectors (Table 1).
Table 1.
Primers used for the generation of SHPRH fragments
| SHPRH fragment | Forward primer (5′→3′) | Reverse primer (5′→3′) |
| nt | ||
| 1–352 | GAATTCATGAGCAGCCGAAGGAAACG | CCCTCGAGGATGCAGCCTGTATATGGAT |
| 353–628 | CCGAATTCCTCTACTATAATCCATATACAGGC | CCCTCGAGAGAGATACATGTACTTTTAGAC |
| 629–984 | CCGAATTCAACCAAGAACATGAAACA | GGCTCGAGCTGTGGGTGACAGCAGGC |
| 985–1,294 | CCGAATTCGCCTGCTGTCACCCACAG | GGCTCGAGCTCACTTATTGCCCAGAGACC |
| 1,295–1,683 | CCGAATTCGGTCTCTGGGCAATAAGT | CCTCGAGTTCAAGCTCTTCAGTTTCTTTGGT |
| H15-only 447–521 | GGGTCGACGAATTTGAACCAAAAGAA | TCGCGGCCGCTCATTTATCACATATATCCTC |
Cell culture and reagents
HEK 293T, Rad18−/− HCT116 (Shiomi et al., 2007), and U2OS cells were grown in high glucose DMEM (Gibco) with penicillin/streptomycin (Gibco), glutamine (Gibco), and 10% fetal bovine serum. Cells were damaged with MMS (Sigma-Aldrich), mitomycin C (Sigma-Aldrich), EMS (Sigma-Aldrich), 4-nitroquinoline 1-oxide (Sigma-Aldrich), aphidicolin (Sigma-Aldrich), camptothecin (Sigma-Aldrich), actinomycin D (EMD Millipore), etoposide (Sigma-Aldrich), hydrogen peroxide (Sigma-Aldrich), or cis-platinum (Sigma-Aldrich) at the indicated concentrations and times. Cells were also treated with ATR inhibitor (ATR-45; Charrier et al., 2011), ATM inhibitor (KU55933; Abcam), MG132 (Sigma-Aldrich), or LLnL (EMD Millipore) at the indicated concentrations and times.
GST pull-downs and immunoprecipitations (IPs)
GST pull-downs from lysates.
Cells were lysed in high-salt Hepes-Triton buffer (50 mM Hepes, pH 7.6, 400 mM NaCl, 1 mM EDTA, 0.75% Triton X-100, 8% glycerol, 2 mM NaF, 0.4 mM NaPPi, 10 µM leupeptin, 1 mM sodium vanadate, and 1 mM PMSF) for 1 h at 4°C and then diluted 1:1 with no-salt Hepes-Triton buffer (100 mM Hepes, pH 7.6, 2 mM EDTA, 1.5% Triton X-100, and 16% glycerol) before being clarified by centrifugation. The soluble fraction was incubated with the indicated GST proteins, prebound to glutathione beads, for 1 h at 4°C. After three washes with the diluted Hepes-Triton buffer, the bead-bound proteins were resuspended in Laemmli sample buffer and analyzed by Western blotting. GST proteins were visualized by staining the polyvinylidene difluoride membrane with Coomassie brilliant blue.
Co-IP.
For lysis condition A (Fig. 1, Fig. 2 A, Fig. 6, and Fig. S3), cells were lysed in 200 mM NaCl Hepes-Triton buffer for 1 h at 4°C and then clarified by centrifugation. For lysis condition B (Fig. 2 B and Fig. 4), cells were lysed in 500 mM NaCl Hepes-Triton buffer for 1 h at 4°C and then diluted 1:1 with no-salt Hepes-Triton buffer and clarified by centrifugation. The supernatant was incubated with anti-FLAG M2 agarose beads (Sigma-Aldrich) at 4°C overnight. After three washes with the diluted Hepes-Triton buffer, bead-bound proteins were resuspended in Laemmli sample buffer and analyzed by Western blotting.
Endogenous Rad18 IP.
Cells were lysed in 500 mM NaCl Hepes-Triton buffer for 1 h at 4°C and clarified by centrifugation. Supernatant was incubated with protein G–Sepharose (GE Healthcare) and 2 µl preimmune rabbit serum or anti-Rad18 serum (RF) at 4°C overnight. After three washes, bead-bound proteins were treated with or without the Usp2 catalytic domain (Boston Biochem) for 30 min at 37°C. After three washes, beads were resuspended in Laemmli sample buffer and analyzed by Western blotting.
His-ubiquitin pull-down.
Cells were transfected with His-ubiquitin or empty vector and treated with the indicated doses of MMS for 4 h and then lysed in 4 M urea buffer (50 mM Tris, pH 8, 200 mM NaCl, 1 mM EDTA, and 0.5% NP-40) for 1 h at 4°C. Lysates were clarified by centrifugation and incubated with nickel agarose at 4°C overnight. Beads were washed and resuspended in Laemmli sample buffer for analysis by Western blotting.
Glycerol gradients
HEK 293T cells were treated with 0.01% MMS for 2 h and then washed and lysed in glycerol-free lysis buffer (10 mM Hepes, pH 7.9, 10 mM KCl, 400 mM NaCl, 2 mM MgCl2, 0.5% NP-40, 0.5 mM EDTA, 10 µM leupeptin, 1 mM sodium vanadate, 1 mM PMSF, and 1 mM DTT) at 4°C for 1 h and then diluted 1:1 with no-NaCl, glycerol-free lysis buffer and rotated for an additional hour. Alternatively, cells were lysed in 150 mM NaCl, glycerol-free lysis buffer in the presence of 100 U/ml Benzonase (Sigma-Aldrich) with similar results. Lysates were clarified by centrifugation and loaded onto a 5–30% glycerol gradient. Gradients were spun in an ultracentrifuge at 50,000 rpm for 15 h at 4°C, and fractions were collected from the top of the tube for subsequent analysis by Western blotting.
Protein purification
GST protein purification.
GST fusion protein constructs were expressed in E. coli BL21 (DE3) at 16°C overnight. Bacterial pellets were resuspended in NETN (50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM PMSF, and 1 mM DTT) and treated with 1 mg/ml lysozyme (Sigma-Aldrich) for 1 h. Cells were lysed with three bursts of 15 s of sonication at 40% amplitude, and then, the lysozyme/sonication cycle was repeated. Supernatant was clarified via centrifugation and incubated with glutathione–Sepharose beads (Sigma-Aldrich) for 2 h at 4°C. After three washes in NETN buffer, beads were stored as a 50% slurry at 4°C.
FLAG protein purification.
Cells were lysed in 500 mM Hepes-Triton buffer and clarified via centrifugation. Supernatant was incubated with anti-FLAG M2 preconjugated beads at 4°C overnight. Beads were washed three times and either used directly in the in vitro ubiquitination assay (Fig. S3 D) or treated with Usp2 catalytic domain (Boston Biochem) for 30 min at 37°C, washed three times, and stored at 4°C as 50% slurry (Fig. 4 B). When used for the latter, FLAG-Rad18 was purified in the presence of 10 mM N-ethylmaleimide. Alternatively, FLAG protein was eluted for 30 min at 4°C with 200 µg/ml of 3×-FLAG peptide in 500 mM salt buffer (Fig. 5 E and Fig. S3 E). Elution was diluted 1:1 with 80% glycerol and stored at −20°C.
In vitro ubiquitination assay
Cells were cotransfected with FLAG-Rad18 constructs and untagged Rad6, and then, Rad18 was purified as described in the previous paragraph. The in vitro ubiquitination assay was adapted from Notenboom et al. (2007). In brief, the reaction mix (75 nM E1 [Boston Biochem], 100 µM ubiquitin, 10 mM ATP, 10 mM MgCl2, 25 mM Tris, pH 8, 150 mM NaCl, 2 µM ZnCl2, and 5 mM β-mercaptoethanol) was incubated for 5 min at room temperature, and then, 10 µl of the FLAG-Rad18 bead slurry was added. The reaction was incubated at 30°C for 1 h and stopped by the addition of 4× Laemmli sample buffer. Rad18 ubiquitination was assessed by Western blotting.
SupF mutagenesis assay
The protocol and reagents used for the SupF assay have been previously described (Lin et al., 2011). In brief, the pSP189 reporter plasmid (SV40 promoter) was damaged with 0.5% MMS or 1,000 J/m2 UV, purified, and then cotransfected into HEK 293T or Rad18-null HCT116 with DNA and/or siRNA as indicated. The replicated plasmid was retrieved after 48 h and analyzed in MBM7070 E. coli. At least 2,000 colonies were counted per experiment, and three to four independent experiments were used to calculate the mean and standard deviation.
Stable cell clones and immunofluorescence
U2OS cells were transiently transfected with individual GFP-Rad18 constructs and selected with 1 mg/ml G418. Surviving cells were grown up as individual cell colonies, and clones that had no free GFP expression were selected for further analysis.
Stable cell clones were plated on glass coverslips and damaged with 0.005% MMS for 4 h. Cells were treated with 0.5% NP-40 on ice for 3.5 min and then fixed in 4% paraformaldehyde for 15 min at room temperature. Slides were blocked with 2% BSA at 4°C overnight and mounted using ProLong gold antifade reagent (Life Technologies). γ-H2AX (EMD Millipore) was detected using a secondary antibody conjugated to Alexa Fluor 594 (594-nm excitation/617-nm emission). GFP (488-nm excitation/509-nm emission) and Hoechst 33342 (360-nm excitation/461-nm emission; Invitrogen) were detected directly. Images were captured at room temperature using a 40×, 0.75 NA objective (Carl Zeiss) on a microscope (Axioskop 2; Carl Zeiss) with a cooled mono 12-bit charge-coupled device camera (QICAM; QImaging) and QCapture Pro software (QImaging) at 8 bits/1,392 × 1,024 pixels. The input highlight level of the GFP foci images was lowered uniformly across all samples in each experiment using Photoshop CS4 (Adobe) and quantified with ImageJ (National Institutes of Health) using macro scripts (supplemental material). At least 100 cells were counted per condition per experiment, and three independent experiments were used to calculate the mean and SEM.
Statistical analysis
Two-tailed p-values were obtained using Student’s t test.
Online supplemental material
Fig. S1 shows that the H15 domain of SHPRH binds to Rad18. Fig. S2 provides details on the detection and characterization of Rad18 ubiquitination in cells. Fig. S3 shows the characterization of the Rad18-Ub chimeras, and Fig. S4 shows the characterization of GFP–Rad18-Ub chimera stable cell clones. Fig. S5 demonstrates that overexpression of Rad6 is able to recapitulate many of the functional defects observed for the Rad18-Ub chimeras in Fig. 7. Two Word (Microsoft) files are also provided that contain macro scripts: word file 1 (nuclear mask) is an ImageJ macro script for identifying cells in a microscopic image using the DAPI channel, and word file 2 (Rad18 foci) is a macro script for counting Rad18 foci within previously defined nuclear masks. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201311063/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201311063.dv.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jun Huang, Dr. Junjie Chen, and Dr. Satoshi Tateishi for providing reagents. We also thank Dr. Andrew Kile for reagents and the Cimprich laboratory for thoughtful discussion.
This work was supported by the National Defense Science and Engineering Graduate Fellowship and National Cancer Institute training grant (PHS CA09302) to M.K. Zeman, a Department of Defense (Breast Cancer Research Program) Predoctoral Fellowship (W81XWH-09-1-0026) and National Institutes of Health training grant (R90 DK071499) to J.-R. Lin, Ministry of Economy and Competitiveness grants (SAF2010-22357 and CONSOLIDER-Ingenio 2010 CDS2007-0015) to R. Freire, and an National Institutes of Health grant (ES016486) to K.A. Cimprich.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3 related
- CMV
- cytomegalovirus
- DDT
- DNA damage tolerance
- DUB
- deubiquitinating enzyme
- EMS
- ethyl methanesulfonate
- HLTF
- helicase-like transcription factor
- IP
- immunoprecipitation
- LLnL
- N-acetyl-l-leucyl-l-leucyl-l-norleucinal
- MMS
- methyl methanesulfonate
- PCNA
- proliferating cell nuclear antigen
- SHPRH
- SNF2 histone linker plant homeodomain RING helicase
- TLS
- translesion synthesis
- ZnF
- zinc finger
References
- Achar, Y.J., Balogh D., and Haracska L.. 2011. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA. 108:14073–14078 10.1073/pnas.1101951108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, X., Barkley L.R., Slater D.M., Tateishi S., Yamaizumi M., Ohmori H., and Vaziri C.. 2006. Rad18 regulates DNA polymerase κ and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell. Biol. 26:3527–3540 10.1128/MCB.26.9.3527-3540.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko, M., Green C.M., Sabbioneda S., Crosetto N., Matic I., Hibbert R.G., Begovic T., Niimi A., Mann M., Lehmann A.R., and Dikic I.. 2010. Regulation of translesion synthesis DNA polymerase η by monoubiquitination. Mol. Cell. 37:396–407 10.1016/j.molcel.2009.12.039 [DOI] [PubMed] [Google Scholar]
- Bish, R.A., and Myers M.P.. 2007. Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J. Biol. Chem. 282:23184–23193 10.1074/jbc.M701042200 [DOI] [PubMed] [Google Scholar]
- Blastyák, A., Hajdú I., Unk I., and Haracska L.. 2010. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 30:684–693 10.1128/MCB.00863-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomar, M.G., Pai M.-T., Tzeng S.-R., Li S.S.-C., and Zhou P.. 2007. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase η. EMBO Rep. 8:247–251 10.1038/sj.embor.7400901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei, D.2011. Ubiquitin family modifications and template switching. FEBS Lett. 585:2810–2817 10.1016/j.febslet.2011.04.053 [DOI] [PubMed] [Google Scholar]
- Chang, D.J., Lupardus P.J., and Cimprich K.A.. 2006. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J. Biol. Chem. 281:32081–32088 10.1074/jbc.M606799200 [DOI] [PubMed] [Google Scholar]
- Charrier, J.-D., Durrant S.J., Golec J.M.C., Kay D.P., Knegtel R.M.A., MacCormick S., Mortimore M., O’Donnell M.E., Pinder J.L., Reaper P.M., et al. . 2011. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 54:2320–2330 10.1021/jm101488z [DOI] [PubMed] [Google Scholar]
- Chiu, R.K., Brun J., Ramaekers C., Theys J., Weng L., Lambin P., Gray D.A., and Wouters B.G.. 2006. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2:e116 10.1371/journal.pgen.0020116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia, A., and Elledge S.J.. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell. 40:179–204 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku, Y., Davies A.A., and Ulrich H.D.. 2010. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 465:951–955 10.1038/nature09097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, T.A., Palle K., Barkley L.R., Kakusho N., Zou Y., Tateishi S., Verreault A., Masai H., and Vaziri C.. 2010. Phosphorylated Rad18 directs DNA polymerase η to sites of stalled replication. J. Cell Biol. 191:953–966 10.1083/jcb.201006043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant, N., Hendel A., Vered I., Carell T., Reissner T., de Wind N., Geacinov N., and Livneh Z.. 2012. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 40:170–180 10.1093/nar/gkr596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, A.A., Liefshitz B., Steinlauf R., and Kupiec M.. 2001. Deletion of the SRS2 gene suppresses elevated recombination and DNA damage sensitivity in rad5 and rad18 mutants of Saccharomyces cerevisiae. Mutat. Res. 486:137–146 10.1016/S0921-8777(01)00086-6 [DOI] [PubMed] [Google Scholar]
- Helchowski, C.M., Skow L.F., Roberts K.H., Chute C.L., and Canman C.E.. 2013. A small ubiquitin binding domain inhibits ubiquitin-dependent protein recruitment to DNA repair foci. Cell Cycle. 12:3749–3758 10.4161/cc.26640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege, C., Pfander B., Moldovan G.-L., Pyrowolakis G., and Jentsch S.. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 419:135–141 10.1038/nature00991 [DOI] [PubMed] [Google Scholar]
- Huang, J., Huen M.S.Y., Kim H., Leung C.C.Y., Glover J.N.M., Yu X., and Chen J.. 2009. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 11:592–603 10.1038/ncb1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki, A., Sleddens-Linkels E., van Cappellen W.A., Hibbert R.G., Sixma T.K., Hoeijmakers J.H.J., Grootegoed J.A., and Baarends W.M.. 2011. Human RAD18 interacts with ubiquitylated chromatin components and facilitates RAD9 recruitment to DNA double strand breaks. PLoS ONE. 6:e23155 10.1371/journal.pone.0023155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S.P., and Durocher D.. 2013. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell. 49:795–807 10.1016/j.molcel.2013.01.017 [DOI] [PubMed] [Google Scholar]
- Kannouche, P.L., Wing J., and Lehmann A.R.. 2004. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 14:491–500 10.1016/S1097-2765(04)00259-X [DOI] [PubMed] [Google Scholar]
- Karras, G.I., and Jentsch S.. 2010. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 141:255–267 10.1016/j.cell.2010.02.028 [DOI] [PubMed] [Google Scholar]
- Kasinsky, H.E., Lewis J.D., Dacks J.B., and Ausió J.. 2001. Origin of H1 linker histones. FASEB J. 15:34–42 10.1096/fj.00-0237rev [DOI] [PubMed] [Google Scholar]
- Lin, J.-R., Zeman M.K., Chen J.-Y., Yee M.-C., and Cimprich K.A.. 2011. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell. 42:237–249 10.1016/j.molcel.2011.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus, P.J., and Cimprich K.A.. 2006. Phosphorylation of Xenopus Rad1 and Hus1 defines a readout for ATR activation that is independent of Claspin and the Rad9 carboxy terminus. Mol. Biol. Cell. 17:1559–1569 10.1091/mbc.E05-09-0865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda, Y., Suzuki M., Kawai H., Suzuki F., and Kamiya K.. 2012. Asymmetric nature of two subunits of RAD18, a RING-type ubiquitin ligase E3, in the human RAD6A-RAD18 ternary complex. Nucleic Acids Res. 40:1065–1076 10.1093/nar/gkr805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre, J., Vidal A.E., McLenigan M.P., Bomar M.G., Curti E., McDonald J.P., Plosky B.S., Ohashi E., and Woodgate R.. 2013. Ubiquitin mediates the physical and functional interaction between human DNA polymerases η and ι. Nucleic Acids Res. 41:1649–1660 10.1093/nar/gks1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyase, S., Tateishi S., Watanabe K., Tomita K., Suzuki K., Inoue H., and Yamaizumi M.. 2005. Differential regulation of Rad18 through Rad6-dependent mono- and polyubiquitination. J. Biol. Chem. 280:515–524 10.1074/jbc.M409219200 [DOI] [PubMed] [Google Scholar]
- Moldovan, G.L., and D’Andrea A.D.. 2011. DNA damage discrimination at stalled replication forks by the Rad5 homologs HLTF and SHPRH. Mol. Cell. 42:141–143 10.1016/j.molcel.2011.03.018 [DOI] [PubMed] [Google Scholar]
- Moldovan, G.L., Pfander B., and Jentsch S.. 2007. PCNA, the maestro of the replication fork. Cell. 129:665–679 10.1016/j.cell.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Motegi, A., Sood R., Moinova H., Markowitz S.D., Liu P.P., and Myung K.. 2006. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 175:703–708 10.1083/jcb.200606145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi, A., Liaw H.J., Lee K.Y., Roest H.P., Maas A., Wu X., Moinova H., Markowitz S.D., Ding H., Hoeijmakers J.H.J., and Myung K.. 2008. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA. 105:12411–12416 10.1073/pnas.0805685105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notenboom, V., Hibbert R.G., van Rossum-Fikkert S.E., Olsen J.V., Mann M., and Sixma T.K.. 2007. Functional characterization of Rad18 domains for Rad6, ubiquitin, DNA binding and PCNA modification. Nucleic Acids Res. 35:5819–5830 10.1093/nar/gkm615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parris, C.N., and Seidman M.M.. 1992. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 117:1–5 10.1016/0378-1119(92)90482-5 [DOI] [PubMed] [Google Scholar]
- Ryu, K.-Y., Baker R.T., and Kopito R.R.. 2006. Ubiquitin-specific protease 2 as a tool for quantification of total ubiquitin levels in biological specimens. Anal. Biochem. 353:153–155 10.1016/j.ab.2006.03.038 [DOI] [PubMed] [Google Scholar]
- Shiomi, N., Mori M., Tsuji H., Imai T., Inoue H., Tateishi S., Yamaizumi M., and Shiomi T.. 2007. Human RAD18 is involved in S phase-specific single-strand break repair without PCNA monoubiquitination. Nucleic Acids Res. 35:e9 10.1093/nar/gkl979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood, R., Makalowska I., Galdzicki M., Hu P., Eddings E., Robbins C.M., Moses T., Namkoong J., Chen S., and Trent J.M.. 2003. Cloning and characterization of a novel gene, SHPRH, encoding a conserved putative protein with SNF2/helicase and PHD-finger domains from the 6q24 region. Genomics. 82:153–161 10.1016/S0888-7543(03)00121-6 [DOI] [PubMed] [Google Scholar]
- Tateishi, S., Sakuraba Y., Masuyama S., Inoue H., and Yamaizumi M.. 2000. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl. Acad. Sci. USA. 97:7927–7932 10.1073/pnas.97.14.7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi, S., Niwa H., Miyazaki J., Fujimoto S., Inoue H., and Yamaizumi M.. 2003. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol. Cell. Biol. 23:474–481 10.1128/MCB.23.2.474-481.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich, H.D.2009. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst.). 8:461–469 10.1016/j.dnarep.2009.01.006 [DOI] [PubMed] [Google Scholar]
- Ulrich, H.D.2011. Timing and spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett. 585:2861–2867 10.1016/j.febslet.2011.05.028 [DOI] [PubMed] [Google Scholar]
- Ulrich, H.D., and Jentsch S.. 2000. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 19:3388–3397 10.1093/emboj/19.13.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk, I., Hajdú I., Fátyol K., Szakál B., Blastyák A., Bermudez V., Hurwitz J., Prakash L., Prakash S., and Haracska L.. 2006. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA. 103:18107–18112 10.1073/pnas.0608595103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk, I., Hajdú I., Fátyol K., Hurwitz J., Yoon J.H., Prakash L., Prakash S., and Haracska L.. 2008. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA. 105:3768–3773 10.1073/pnas.0800563105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk, I., Hajdú I., Blastyák A., and Haracska L.. 2010. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst.). 9:257–267 10.1016/j.dnarep.2009.12.013 [DOI] [PubMed] [Google Scholar]
- Watanabe, K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., and Yamaizumi M.. 2004. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23:3886–3896 10.1038/sj.emboj.7600383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, L.S., Minesinger B.K., Wiltrout M.E., D’Souza S., Woodruff R.V., and Walker G.C.. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73:134–154 10.1128/MMBR.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, R.P.C., Aguissa-Touré A.H., Wani A.A., Khosravi S., Martinka M., Martinka M., and Li G.. 2012. Elevated expression of Rad18 regulates melanoma cell proliferation. Pigment Cell Melanoma Res. 25:213–218 10.1111/j.1755-148X.2011.00948.x [DOI] [PubMed] [Google Scholar]
- Xie, C., Wang H., Cheng H., Li J., Wang Z., and Yue W.. 2014. RAD18 mediates resistance to ionizing radiation in human glioma cells. Biochem. Biophys. Res. Commun. 445:263–268 10.1016/j.bbrc.2014.02.003 [DOI] [PubMed] [Google Scholar]
- Zhou, J., Zhang S., Xie L., Liu P., Xie F., Wu J., Cao J., and Ding W.-Q.. 2012. Overexpression of DNA polymerase iota (Polι) in esophageal squamous cell carcinoma. Cancer Sci. 103:1574–1579 10.1111/j.1349-7006.2012.02309.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.