

Abstract
Increasing evidence has revealed that the activation of the JNK pathway participates in apoptosis of nerve cells and neurological function recovery after traumatic brain injury. However, which genes in the JNK family are activated and their role in traumatic brain injury remain unclear. Therefore, in this study, in situ end labeling, reverse transcription-PCR and neurological function assessment were adopted to investigate the alteration of JNK1, JNK2 and JNK3 gene expression in cerebral injured rats, and their role in cell apoptosis and neurological function restoration. Results showed that JNK3 expression significantly decreased at 1 and 6 hours and 1 and 7 days post injury, but that JNK1 and JNK2 expression remained unchanged. In addition, the number of apoptotic nerve cells surrounding the injured cerebral cortex gradually reduced over time post injury. The Neurological Severity Scores gradually decreased over 1, 3, 5, 14 and 28 days post injury. These findings suggested that JNK3 expression was downregulated at early stages of brain injury, which may be associated with apoptosis of nerve cells. Downregulation of JNK3 expression may promote the recovery of neurological function following traumatic brain injury.
Keywords: neural regeneration, JNK1, JNK2, JNK3, traumatic brain injury, TdT-mediated dUTP nick end labeling, reverse transcription-PCR, cell apoptosis, neurological function recovery, neuroregeneration
Research Highlights
-
(1)
The downregulation of JNK3 expression in the peripheral area of the injured cerebral cortex in the early stages of traumatic brain injury may be associated with apoptosis of nerve cells. However, JNK1 and JNK2 expression remained unchanged.
-
(2)
Changes in JNK3 expression in rats with traumatic brain injury correlated well with nerve cell apoptosis and neurological function changes.
-
(3)
More apoptotic nerve cells were found at early stages when compared with late stage traumatic brain injury, which may be related to the induction of neural regeneration and repair and inhibition of cell apoptosis.
-
(4)
JNK3 is involved in nerve cell apoptosis, neurological function recovery and neural regeneration.
INTRODUCTION
Whether JNK is expressed in nerve cells after traumatic brain injury remains unclear. Moreover, which subtypes in the JNK family are involved in traumatic brain injury and how their expression contributes to nerve cell apoptosis and neurological function recovery remain unknown.
In this study, we focused on whether JNK3 expression participates in nerve cell apoptosis and neurological function recovery post traumatic brain injury. In addition, we investigated the possible role of JNK3 in nerve regeneration, so as to provide a novel target for traumatic brain injury therapy.
Due to cerebral cortex impairment post traumatic brain injury, patients can suffer from symptoms including headache, epilepsy, paralysis, behavioral and/or cognitive dysfunction, and in severe cases, a persistent vegetative state[1,2,3,4]. Nerve regeneration and nerve structure reconstruction are topics of great interest in the area of neuroscience research[5,6]. The relationship between activation of the JNK/SAPK pathway and neural injury, apoptosis and neurological function has gained the interest of neuroscientists. JNK[7], also called stress-activated protein kinase[8], is one of the critical signal transduction pathways of mitogen-activated protein kinases[9]. JNK can be activated by various stressors, thereby activating the phosphorylated serine/threonine protein kinase in the amino-activated region of c-Jun, a kind of nuclear factor[10,11,12]. The JNK family is encoded by JNK1, JNK2 and JNK3 genes, among which JNK1 and JNK2 are extensively expressed in all kinds of tissues and cells, while JNK3 is mainly expressed in the nervous system, heart and testis. The JNK/stress-activated protein kinase signal pathway is known to participate in various physiological processes[13], such as embryo development, cellular necrosis, regulation of cellular proliferation and apoptosis[14,15,16,17,18].
Studies using animal models of brain injury and in vitro cultures have shown that several signal pathways, including apoptosis associated genes (survival, Bcl-2, Bcl-xl and JNK), extracellular signal regulation associated kinases and death promoter genes (Bax, tumor-suppressor gene, p53 and caspase family) are tightly associated with traumatic brain injury[19,20]. Among them, JNK is an important member of the ERK, p38 and JNK signaling pathways, and plays a crucial role in cellular apoptosis[21]. Park and colleagues[22] revealed that activated JNK initiates cellular apoptosis mainly through triggering the mitochondrial pathway. It elevates the transcriptional activity of c-Jun by phosphorylating locus-Ser-63 and ser-73 in the serine residue of c-Jun, resulting in an increase of c-Jun protein expression. The effect of JNK in promoting the expression of c-Jun could induce the upregulation of FasL, which binds to corresponding cell membrane receptors, ultimately leading to the activation of the apoptotic pathway. However, the roles of JNK1, JNK2 and JNK3 in nerve cell apoptosis and neurological function recovery remain poorly understood.
In this study, the modified Feeney method[23] was adopted to establish the rat traumatic brain injury model using the drop weight impact test. Reverse transcription-PCR was employed to detect JNK expression and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) was used to examine cellular apoptosis in the injured peripheral area of the cerebral cortex. Neurological function deficit score evaluation was performed as described by Chen et al[24] to assess neurological function in traumatic brain injury rats.
The present study aimed to preliminarily investigate the alteration of JNK expression in the peripheral area of the injured cerebral cortex and explore the relationship between JNK subtypes and nerve cell apoptosis as well as the possible role of JNK in nerve cell apoptosis and neurological function recovery.
RESULTS
Quantitative analysis of experimental animals
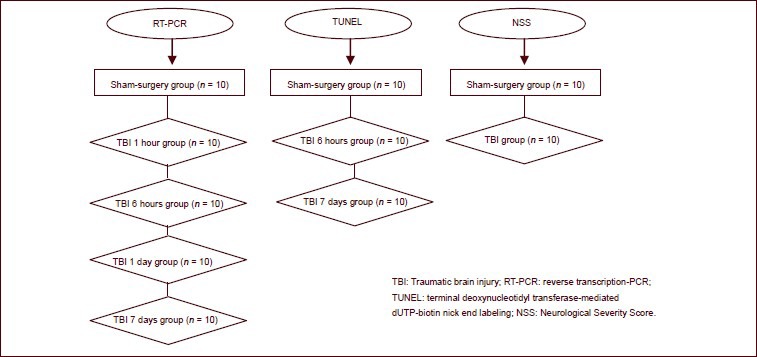
A total of 100 rats were randomly assigned to sham-surgery (n = 30) and traumatic brain injury (n = 70; weight drop impact) groups. Ten rats from the sham-surgery group and 10 rats from the traumatic brain injury group at 1 and 6 hours and 1 and 7 days post traumatic brain injury were subjected to reverse transcription-PCR detection. Tissue surrounding the injured cerebral cortex from sham-surgery rats and rats from the 6 hour and 7 day groups were harvested and subjected to TUNEL staining. Neurological Severe Score evaluation was used to assess neurological function changes in each rat from the sham-surgery group and traumatic brain injury group.
All 100 rats were included in the final analysis. The flow diagram of animal grouping is as follows:
JNK mRNA expression in the peripheral area of the injured cerebral cortex after traumatic brain injury
Reverse transcription-PCR at 1 and 6 hours and 1 and 7 days post injury revealed that all three JNK molecules were detected. However, only levels of JNK3 at 1 and 6 hours and 1 and 7 days significantly decreased in the traumatic brain injury group when compared with the sham-surgery group (P < 0.05), while levels of JNK1 and JNK2 remained unchanged between the traumatic brain injury and sham-surgery groups (P > 0.05; Figure 1).
Figure 1.
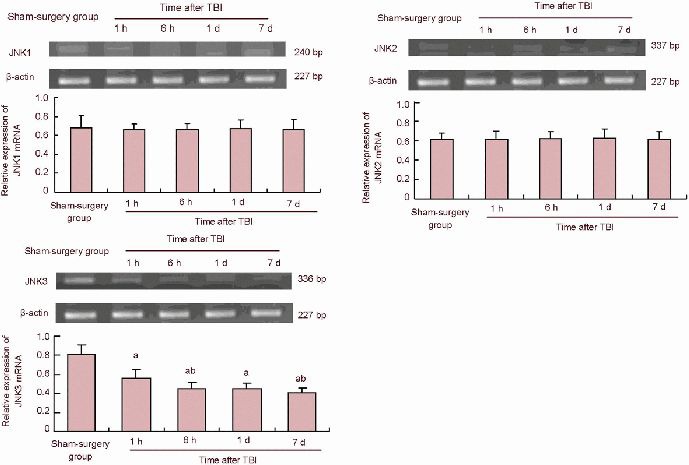
Reverse transcription-PCR results of JNK1, JNK2, JNK3 expression in the peripheral area of the injured cerebral cortex after TBI in rats.
aP< 0.05, vs. sham-surgery group; bP< 0.05, vs. previous time point group and 6 hours, 1 and 7 days groups. Data are expressed as mean ± SD of 10 rats from each group (one-way analysis of variance and least significant difference t-test). Sham-surgery group (gene expression expressed as the ratio of absorbance to the target gene/β-actin). TBI: Traumatic brain injury; h: hour; d: day.
Cell apoptosis in the peripheral area of the injured cerebral cortex in traumatic brain injury rats
TUNEL staining showed that the number of TUNEL-positive cells in the peripheral area of the injured cerebral cortex at 6 hours after injury significantly increased in the traumatic brain injury group compared with the sham-surgery group (P < 0.05); while at 7 days post injury, the number of TUNEL-positive cells was markedly less at 6 hours in the traumatic brain injury group (P < 0.05; Figure 2).
Figure 2.
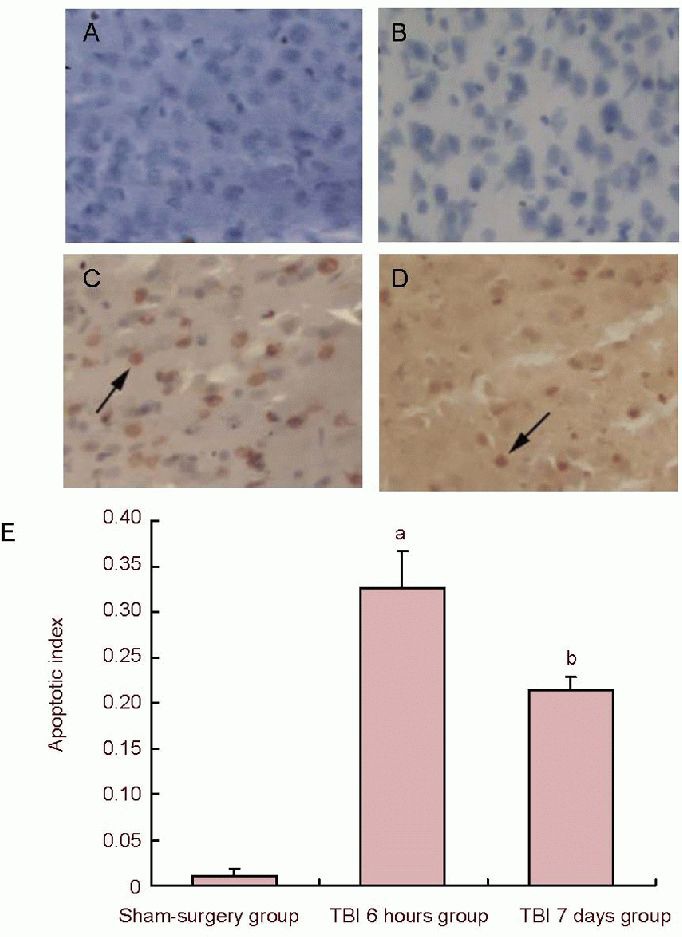
Cell apoptosis in the peripheral area of the cerebral cortex in traumatic brain injury (TBI) rats (TUNEL staining).
(A) Cell apoptosis of negative control (× 200); (B) morphology of apoptotic cells in the sham-surgery group (× 200); (C) cell apoptosis at 6 hours post injury (× 200); (D) morphology of apoptotic cells at 7 days post injury: the number of apoptotic cells was markedly reduced when compared to 6 hours post injury; arrows in C and D show brown positive apoptotic cells with thickening nuclei.
(E) Quantification of TUNEL-positive cells. Data represent the change in cellular apoptosis index in all experimental groups (apoptosis index = number of TUNEL-positive cells/total number of cells). aP< 0.05, vs. sham-surgery group; bP< 0.05, vs. TBI 6 hours group. Data are expressed as mean ± SD of 10 rats from each group (one-way analysis of variance and least significant difference t-test). TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling.
Behavioral changes in traumatic brain injury rats
Neurological Severe Score in the sham-surgery group slightly increased at 1 day post traumatic brain injury, suggesting mild neurological function impairment. Neurological Severe Scores in the traumatic brain injury group were prominently higher than in the sham-surgery group (P < 0.05), suggesting that there was serious impairment in neurological function resulting from traumatic brain injury. Comparison of 1, 3, 5, 7, 14, 28 days post traumatic brain injury showed that Neurological Severe Scores had a tendency to reduce over time post traumatic brain injury (P < 0.05), indicating that little recovery existed from 1 to 28 days after traumatic brain injury (Figure 3).
Figure 3.
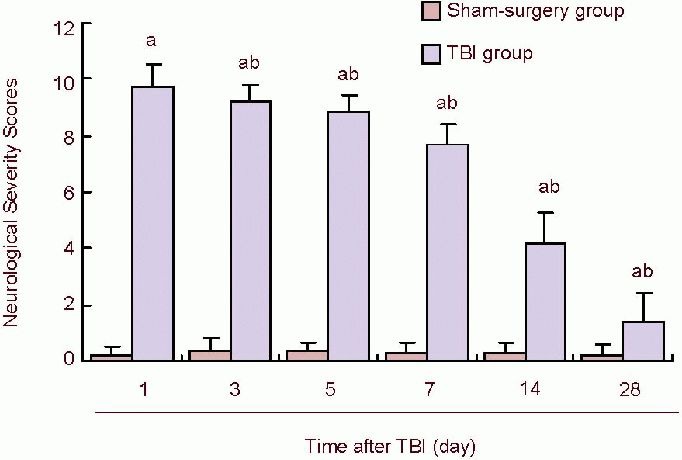
Neurological Severity Scores at different time points in TBI rats.
Data represent Neurological Severity Scores. Scores 13 to 18 indicate severe injury; 7 to 12, moderate injury; 1 to 6, mild injury. In TBI groups, Neurological Severe Scores gradually decreased over time, suggesting that after TBI, neurological function recovered to some extent. aP< 0.05, vs. sham-surgery group; bP< 0.05, vs. previous time points of TBI groups. Data are expressed as mean ± SD of 10 rats from each group (one-way analysis of variance and least significant difference t-test). TBI: Traumatic brain injury; d: day.
DISCUSSION
In this study, three main results were obtained. Firstly, JNK was expressed in nerve cells of rats after traumatic brain injury. The JNK signaling pathway appeared to play a crucial role in nerve cell apoptosis. Secondly, we found that among the three members of the JNK family, JNK3 had the strongest relationship in traumatic brain injury. Importantly, changes in JNK3 expression correlated well with nerve cell apoptosis and Neurological Severe Scores after traumatic brain injury. Finally, our results indicated that more apoptosis occurred in nerve cells at early stages than advanced stages of traumatic brain injury. This may attribute to the initiation of the repair mechanism involving nerve regeneration at the advanced stage of traumatic brain injury, ultimately resulting in the inhibition of cellular apoptosis. Therefore, we concluded that JNK3 participated in the regulation of nerve cell apoptosis, neurological functional recovery and nerve regeneration. We first reported that JNK3 participated in nerve cell apoptosis and neurological function recovery.
Until recently, there has been some controversy about whether JNK expression was initiated after nerve cell injury. John et al[25] found that JNK was activated following diffuse axonal injury, and Sugino et al[26] revealed that JNK expression was upregulated in the cerebral cortex in an animal model of ischemic cerebral injury. However, Mori et al[27] did not find any alteration in JNK expression in the cerebral cortex after cerebral injury by western blot analysis. Moreover, Otani et al[28] found that JNK was not activated in the cerebral cortex following cerebral injury. Given these controversies exist, we investigated whether gene expression upstream of JNK (initiated genes) changed in the cerebral cortex region following traumatic brain injury using an established traumatic brain injury rat model in which moderate and severe cerebral injury could be generated. Reverse transcription-PCR was employed to detect alterations in JNK expression in the peripheral area of cerebral injury. Results showed that the level of JNK3 expression was significantly downregulated in the surrounding area of the injured cortex at 1 and 6 hours and 1 and 7 days post traumatic brain injury, while as JNK1 and JNK2 expression did not change when compare to the sham-surgery group. Meanwhile, the number of TUNEL-positive cells in the surrounding area of the injured cerebral cortex began to markedly increase at 6 hours post traumatic brain injury, suggesting that when nerve cell apoptosis occurred in the injured cerebral cortex, there existed some changes in the regulation of gene expression upstream of JNK (gene-start). However, gene-start upstream of JNK did not represent JNK protein expression. Thus, these changes may be associated with the interaction of apoptosis inhibiting and promoting factors in vivo. The above findings on JNK3 expression alterations following traumatic brain injury will provide novel cues to further research the roles of JNK3 in the apoptotic signal pathway at the site of injury after traumatic brain injury.
Whether JNK3 plays a crucial role in nerve cell apoptosis remains unclear. To date, there are few reports about the roles of JNK1, JNK2 and JNK3 in nerve cell apoptosis, especially their role in traumatic brain injury. Choi and colleagues[29] found that activated JNK3 participated in the apoptosis of dopaminergic neurons in Parkinson's disease. Lee et al[30] demonstrated that the activation of JNK3 could promote apoptosis of glial cells in spinal cord injury. Some scholars[31,32] found that JNK1 and JNK2 regulated regiospecific cellular apoptosis at early stages of brain development. To determine the circumstances of JNK1, JNK2 and JNK3 expression following traumatic brain injury, injury of the zona rolandica, produced by weight drop impact, and JNK1, JNK2 and JNK3 expression in the area surrounding the injured cerebral cortex at 1 and 6 hours and 1 and 7 days post traumatic brain injury was detected. Results revealed that the level of JNK3 gene expression was significantly downregulated at early stages of traumatic brain injury, while JNK1 and JNK2 expression remained unchanged when compared with the sham surgery group. A number of TUNEL-positive cells were observed in the surrounding area of the injured cerebral cortex at 6 hours and 7 days post traumatic brain injury. Results indicated that the downregulation of JNK3 gene expression in the surrounding area of the injured cerebral cortex at early stages of traumatic brain injury may be involved in the apoptosis of nerve cells. Cellular apoptosis is known to play a crucial role in the pathophysiological progression of nerve cell degeneration after cerebral injury. So far, there exists variation in the time sequence of nerve cell apoptosis and necrosis onset following traumatic brain injury. Cervos-Navarro and Lafuente[33] showed that loss of nerve cells could be observed in the cerebral cortex and hippocampal neurons at early stages (within 48 hours) of cerebral injury. Several hours to days after cerebral injury, injured nerve cells appeared swollen. A number of nerve cells appeared apoptotic and atrophied over time (several days or several weeks post cerebral injury). Results from our study revealed that a large number of TUNEL-positive cells were present in the surrounding area of the injured cerebral cortex 6 hours after injury, indicating that cellular apoptosis was already initiated at early stages of traumatic brain injury, which was in accordance with results of Pravdenkova's study proposing nerve cell apoptosis may be initiated at early stages of cerebral injury[34]. The number of TUNEL-positive cells in the surrounding area of the injured cerebral cortex at 6 hours was less than that at 7 days post injury, but there existed many TUNEL-positive cells in this area. This result supports the notion proposed by Feng et al[35] that TUNEL-positive cells begin to emerge at 6 hours post traumatic brain injury and last as long as 2 weeks. These results further indicate that more apoptotic nerve cells exist at early stages of advanced stage cerebral injury, which may be associated with the initiation of nerve regeneration and repair, and the subsequent inhibition of cellular apoptosis.
JNK is closely associated with nerve regeneration, and neurological function recovery following cerebral injury is dependent on nerve regeneration. Nix et al[36] proposed that the activated JNK pathway ameliorates regenerative repair of injured neuritis. Results from our study revealed that the levels of JNK3 gene expression were downreguated at early stages of traumatic brain injury, and that Neurological Severe Scores on days 1, 3, 5, 7, 14 and 28 post traumatic brain injury reduced over time. This result is indicative that neurological function gradually recovers in advanced stages of traumatic brain injury and supports the discovery that JNK expression is associated with neurological function recovery after subarachnoid hemorrhage, as shown in Sozen's research[37]. Therefore, down regulation of JNK3 expression may participate in neurological functional recovery post traumatic brain injury. However, in this study the time points following traumatic brain injury may not have been enough to detect gene expression changes. In addition, we did not perform fluorescent TUNEL in combination with gene expression. Therefore, in future studies, the time duration for the observation of JNK expression post traumatic brain injury should be prolonged. In addition, the ultrastructure of nerve tissues should be monitored to obtain further insights on the role of the JNK signaling pathway.
MATERIALS AND METHODS
Design
A randomized, controlled animal study.
Time and setting
This experiment was performed at the Laboratory of Neurobiology, West China School of Preclinical and Forensic Medicine, Sichuan University, China from July to September 2011.
Materials
Experimental animals
A total of 100 healthy male 6-month-old Sprague-Dawly rats of specific pathogen-free grade, weighing 200–260 g, were provided by the Animal Experimental Center of Kunming Medical University (certificate No. SCXK (Dian) 2005-0008). Breeding conditions: Rats were bred in the animal laboratory room with individually ventilated cages, at 20–25°C at a humidity of 40–70%. Rats were kept in 12-hour day/night cycle, with an luminance of 150–300 Ix, and noise index of 60 dB.
Breeding administration: PVC plastic cages were used to breed the rats in clusters, with five rats bred in each cage. Whole nutrition granulated freed was used to breed the rats daily. Free access to food and water was allowed. The cages and padding were changed weekly. Following adaption breeding for 2 weeks, rats underwent experimentation.
Animal experiments and care were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology Department, Yunnan Province, China[38].
Methods
Establishment of traumatic brain injury models
The modified Feeney method[23] was adopted to establish the traumatic brain injury rat model using free weight drop impact. The rats were anesthetized by intraperitoneal injection of 3.6% (v/v) chloral hydrate (1 mL/100 g) and fixed on an animal bench. Following conventional sterilization, a “U” shaped incision, 5 mm away from the bregma along with coronal plane of the skull was made, and the scalp was turned open to the caudal side. The periosteum was isolated, and the right parietal bone was exposed. A skull pore 2.5 mm away from the sagittal suture and 1.5 mm posterior to the coronal suture was drilled and enlarged to a 5.0 mm × 5.0 mm bone window by removal of the skull to allow the cerebral dura mater to be exposed. Subsequently, a ferric cylinder weighing 50 g was freely dropped along with a metal pole from a height of 40 cm to impact a clout placed onto the motor cortex in the right parietal lobe. The diameter of the contact face between the metal lobe and surface of the cerebral cortex was 5.0 mm. This impact resulted in traumatic injury of the right motor cortex. After the operation, the rats were attentively cared, and administered with 5 IU penicillin daily by intraperitoneal injection so as to prevent infection. In the sham-surgery group, rats only underwent skull cut and removal, without impacting the brain.
Following anabiosis from the anesthesia, traumatic brain injury rats were subjected to neurological function evaluation using Neurological Severe Score. Rats with scores > 6 were considered successful traumatic brain injury models. Neurological Severe Score was performed on days 1, 3, 5, 7, 14 and 28 post injury by professional observers who were blind to the details of animal grouping (blind method), referring to the Neurological Severe Score Rating Scales reported by Chen et al[24] (0: normal; 1–6: minor injury; 7–12: moderate injury; 13–18: severe injury; 18: complete loss of neurological function).
Standards of Neurological Severity Score (including five items: A, B, C, D, E; full mark: 18):

TUNEL of cellular apoptosis surrounding the injured cerebral cortex in traumatic brain injury rats
Rats were anesthetized by intraperitoneal injection of 3.6% (v/v) chloral hydrate (1 mL/100 g). The surrounding brain tissues of the injured cerebral cortex in all groups were harvested and post-fixed. Cryosections of these samples of brain tissues were obtained using a freezing microtome (Leica Dmirb, Wetzlar, Germany). A total of 50 μL fresh Proteinase K 20 μg/mL (10 mmol/L Tris-HCI 147 μL +1 mg/mL Proteinase K 3 μL) was added to each slice, followed by digestion at 37°C for 15 minutes and washed in 0.01 mol/L PBS three times for 5 minutes each. Following soaking in the 0.1% (v/v) diethylpyrocarbonate, the slices were placed under room temperature for 30 minutes, followed by three washes with 0.01 mol/L PBS, each for 5 minutes. The remnant fluid was removed from the slices. Solution 1 and solution 2 (1:9) from the TUNEL-POD reagent kit (Roche Company, Basel, Switzerland, No.11684817910; solution 1: terminal deoxyribose transferase; solution 2: the reaction fluid containing nucleotide mixed liquor, which were incorporated on PE gloves for well distribution on ice) was added on each slice. A negative control was treated with solution 2 alone. Subsequently, the slices were incubated with freshly prepared 3% (v/v) H2O2-methanol at room temperature for 15 minutes, followed by three washes with 0.01 mol/L PBS, each for 5 minutes. A total of 50 μL bovine serum albumin (5% (v/v)) was added onto each slice and incubated at 37°C for 30 minutes, followed by removal of the remnant fluid without washing. A drop of POD transforming agent (enzyme labeling fluorescein antibody, Roche Company, No. 11684817910) was added onto each slice. The slices were placed into a wet box and incubated at 37°C for 40 minutes, followed by three washes with 0.01 mol/L PBS, each for 5 minutes. Diaminobenzidine coloration reagent kit (Beijing Zhongshan Jinqiao Biotechnology Company, China, No. ZL1-9031) was used for coloration at room temperature. Following observation under the light microscope for 5–10 minutes, distilled water was used to stop the coloration reaction. Slices were subjected to flow water flush so as to clear the diaminobenzidine granules adhered on the slices. Conventional dehydration, transparency and mounting were performed. The light microscope (Olympus, Tokyo, Japan) was used to observe TUNEL results. In the positive control, Dnase I was added onto slices and incubated at room temperature for 10 minutes, followed by three washes with 0.01 mol/L PBS, each for 5 minutes. Then the slices were covered with a plastic cap and placed in the wet box, and left at 37°C for 1 hour. Sections were then placed at 4°C overnight (over 20 hours). The labeled apoptotic nucleus exhibited dark staining with brown and yellow color. The mean apoptotic index = number of TUNEL-positive cells/number of total cell counted (200 ×, five slices randomly selected from each group and five fields of view).
Reverse transcription-PCR of cerebral cortex tissue surrounding the injury site in traumatic brain injury rats
Primer Premier 5.0 software (Palo Alto, CA, USA) was employed to design the sequences of the target genes JNK1, JNK2 and JNK3. β-actin served as an internal reference. The primers in reverse transcription-PCR were synthesized by Takara Biotechnology Company (Dalian, China).
The total RNA derived from the surrounding cerebral cortex of the injured area in traumatic brain injury rats was extracted by referring to the instructions of the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Canada, K1621). RNA underwent reverse transcription to generate 20 μL of cDNA template. The PCR reaction system was set up according to the conventional PCR method, which consisted of 2 × PCR Master Mix (Fermentas, Canada, K0171) 12. 5 μL, PCR water nuclease-free 10.5 μL, upstream primer (Takara Biotechnology) 0.5 μL, downstream primer (Takara Biotechnology) 0.5 μL, and brain tissue-sample 1 μL (25 μL in total). The PCR was conducted using the Gene Cycle™ PCR (Biorad) under the following conditions: degeneration at 94 °C for 5 minutes, degeneration at 94°C for 30 seconds, reannealing for 30 seconds; elongation at 72°C for 30 seconds, 34 cycles in total, followed by total elongation at 72°C for 10 minutes.
The sequences of primers, annealing temperature and the length of the JNK1, JNK2 and JNK3 PCR products are shown as follows:
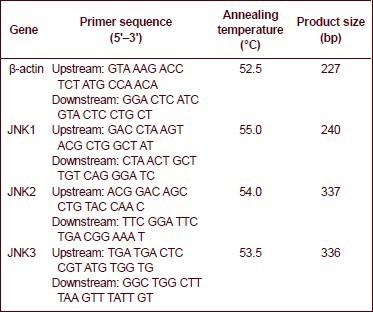
The PCR amplified products underwent agarose gel electrophoresis in which the DNA marker DL2000 (TAKARA, No.D513A) was used. The outcome of agarose gel electrophoresis was observed under an inverted fluorescence microscope (Leica Dmirb, Wetzlar, Germany). The GelDoc-gel imaging formation analyzing apparatus (Bio-Rad, CA, USA) was used for imaging. Image J software (National Institutes of Health, USD) was used to detect the scale of absorbance following PCR. The ratio of the absorbance scales to β-actin was used for statistical analysis.
Statistical analysis
Experimental data were expressed as mean ± SD and statistically analyzed using SPSS software (SPSS, Chicago, IL, USA). One-way analysis of variance and least significant difference t-test were performed. A level of P < 0.05 was considered statistically significant.
Footnotes
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Medical Ethical Committee of Kunming Medical University, China.
(Reviewed by Diwakarla S, Frenchman B, Fan B, Lv J)
(Edited by Wang J, Su LL, Li CH, Song LP)
REFERENCES
- [1].Theadom A, Barker-Collo S, Feigin VL, et al. The spectrum captured: a methodological approach to studying incidence and outcomes of traumatic brain injury on a population level. Neuroepidemiology. 2012;38(1):18–29. doi: 10.1159/000334746. [DOI] [PubMed] [Google Scholar]
- [2].Kirov II, Tal A, Babb JS, et al. Diffuse axonal injury in mild traumatic brain injury: a 3D multivoxel proton MR spectroscopy study. J Neurol. 2013;260(1):242–252. doi: 10.1007/s00415-012-6626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dash PK, Kobori N, Moore AN. A molecular description of brain trauma pathophysiology using microarray technology: an overview. Neurochem Res. 2004;29(6):1275–1286. doi: 10.1023/b:nere.0000023614.30084.eb. [DOI] [PubMed] [Google Scholar]
- [4].Brown R, Thompson HJ, Imran SA, et al. Traumatic brain injury induces adipokine gene expression in rat brain. Neurosci Lett. 2008;432(1):73–78. doi: 10.1016/j.neulet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stiefel MF, Tomita Y, Marmarou A. Secondary ischemia impairing the restoration of ion homeostasis following traumatic brain injury. J Neurosurg. 2005;103(4):707–714. doi: 10.3171/jns.2005.103.4.0707. [DOI] [PubMed] [Google Scholar]
- [7].Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22(6):631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- [8].Atzori C, Ghetti B, Piva R, et al. Activation of the JNK/P38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphory lation but not to apoptosis. J Neuropathol Exp Neurol. 2001;60(12):1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- [9].Schafers M, Svensson CI, Sommer C, et al. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23(7):2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, andp38 protein kinases. Science. 2002;298(5600):1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- [11].Shi S, Li QS, Li H, et al. Anti-apoptotic action of hydrogen sulfide is associated with early JNK inhibition. Cell Biol Int. 2009;33(10):1095–1101. doi: 10.1016/j.cellbi.2009.06.029. [DOI] [PubMed] [Google Scholar]
- [12].Repici M, Wehrlé R, Antoniou X, et al. c-Jun N-terminal kinase (JNK) and p38 play different roles in age-related Purkinje cell death in murine organotypic culture. Cerebellum. 2011;10(2):281–290. doi: 10.1007/s12311-010-0244-z. [DOI] [PubMed] [Google Scholar]
- [13].Hughes R, Gilley J, Kristiansen M, et al. The MEK-ERK pathway negatively regulates bim expression through the 3’ UTR in sympathetic neurons. BMC Neurosci. 2011;12(7):69. doi: 10.1186/1471-2202-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xia Z, Dickens M, Raingeaud J, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- [15].Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010;70(11):4260–4264. doi: 10.1158/0008-5472.CAN-10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu Y, Wang H, Zhu Y, et al. The protective effect of nordihydroguaiaretic acid on cerebral ischemia/reperfusion injury is mediated by the JNK pathway. Brain Res. 2012;1445(3):73–81. doi: 10.1016/j.brainres.2012.01.031. [DOI] [PubMed] [Google Scholar]
- [17].Zhang ZB, Li ZG. Cathepsin B and phospo-JNK in relation to ongoing apoptosis after transient focal cerebral ischemia in the rat. Neurochem Res. 2012;37(5):948–57. doi: 10.1007/s11064-011-0687-8. [DOI] [PubMed] [Google Scholar]
- [18].Mazzitelli S, Xu P, Ferrer I, et al. The loss of c-Jun N-terminal protein kinase activity prevents the amyloidogenic cleavage of amyloid precursor protein and the formation of amyloid plaques in vivo. J Neurosci. 2011;31(47):16969–16976. doi: 10.1523/JNEUROSCI.4491-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nathoo N, Narotam PK, Agrawal DK, et al. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J Neurosurg. 2004;101(2):233–240. doi: 10.3171/jns.2004.101.2.0233. [DOI] [PubMed] [Google Scholar]
- [20].Martínez-Lucas P, Moreno-Cuesta J, García-Olmo DC, et al. Relationship between the Arg72Pro polymorphism of p53 and outcome for patients with traumatic brain injury. Intensive Care Med. 2005;31(9):1168–1173. doi: 10.1007/s00134-005-2715-0. [DOI] [PubMed] [Google Scholar]
- [21].Huang T, Solano J, He D, et al. Traumatic injury activates MAP kinases in astrocytes: mechanisms of hypothermia and hyperthermia. J Neurotrauma. 2009;26(9):1535–1545. doi: 10.1089/neu.2008.0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Park MT, Song MJ, Lee H, et al. β-Lapachone significantly increases the effect of ionizing radiation to cause mitochondrial apoptosis via JNK activation in cancer cells. PLoS One. 2011;6(10):e25976. doi: 10.1371/journal.pone.0025976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Feeney DM, Boyeson MG, Linn RT, et al. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211(1):67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- [24].Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow strumal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- [25].John E, Greer, Melissa J, et al. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci. 2011;31(13):5089–5105. doi: 10.1523/JNEUROSCI.5103-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sugino T, Nozaki K, Takagi Y, et al. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20(12):4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mori T, Wang X, Jung JC, et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22(4):444–452. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- [28].Otani N, Nawashiro H, Fukui S, et al. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22(3):327–334. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- [29].Choi WS, Klintworth HM, Xia Z. JNK3-mediated apoptotic cell death in primary dopaminergic neurons. Met Mol Biol. 2011;758:279–292. doi: 10.1007/978-1-61779-170-3_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee JY, Choi SY, Oh TH, et al. 17β-Estradiol inhibits apoptotic cell death of oligodendrocytes by inhibiting RhoA-JNK3 activation after spinal cord injury. Endocrinology. 2012;153(8):3815–3827. doi: 10.1210/en.2012-1068. [DOI] [PubMed] [Google Scholar]
- [31].Toyoda H, Zhao MG, Xu H, et al. Requirement of extracellular signal-regulated kinase/mitogen-activated protein kinase for longterm potentiation in adult mouse anterior cingulate cortex. Mol Pain. 2007;3(1):36. doi: 10.1186/1744-8069-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song B, Xie B, Wang C, et al. c-Jun induction is independent of early growth response factor during cerebellar granule neuron apoptosis. Neuroreport. 2012;23(2):67–72. doi: 10.1097/WNR.0b013e32834e7d69. [DOI] [PubMed] [Google Scholar]
- [33].Cervos-Navarro J, Lafuente JV. Traumatic brain injuries: structural changes. J Neurol Sci. 1991;103(7):S3–14. doi: 10.1016/0022-510x(91)90002-o. [DOI] [PubMed] [Google Scholar]
- [34].Pravdenkova SV, Basnakian AG, James SJ, et al. DNA fragmentation and nuclear endonuclease activity in rat brain after severe closed head injury. Brain Res. 1996;729(2):151–155. [PubMed] [Google Scholar]
- [35].Feng HL, Guo LD, Huang GF, et al. A study on nerve cells apoptosis after traumatic brain injury in rats. Zhonghua Chuangshang Zazhi. 2000;16(2):97–99. [Google Scholar]
- [36].Nix P, Hisamoto N, Matsumoto K, et al. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108(26):10738–10743. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sozen T, Tsuchiyama R, Hasegawa Y, et al. Role of interleukin-1 beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40(7):2519–2525. doi: 10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]