

Abstract
Background
The gut microbiota plays an important role in human health and disease by acting as a metabolic organ. Metagenomic sequencing has shown how dysbiosis in the gut microbiota is associated with human metabolic diseases such as obesity and diabetes. Modeling may assist to gain insight into the metabolic implication of an altered microbiota. Fast and accurate reconstruction of metabolic models for members of the gut microbiota, as well as methods to simulate a community of microorganisms, are therefore needed. The Integrated Microbial Genomes (IMG) database contains functional annotation for nearly 4,650 bacterial genomes. This tremendous new genomic information adds new opportunities for systems biology to reconstruct accurate genome scale metabolic models (GEMs).
Results
Here we assembled a reaction data set containing 2,340 reactions obtained from existing genome-scale metabolic models, where each reaction is assigned with KEGG Orthology. The reaction data set was then used to reconstruct two genome scale metabolic models for gut microorganisms available in the IMG database Bifidobacterium adolescentis L2-32, which produces acetate during fermentation, and Faecalibacterium prausnitzii A2-165, which consumes acetate and produces butyrate. F. prausnitzii is less abundant in patients with Crohn’s disease and has been suggested to play an anti-inflammatory role in the gut ecosystem. The B. adolescentis model, iBif452, comprises 699 reactions and 611 unique metabolites. The F. prausnitzii model, iFap484, comprises 713 reactions and 621 unique metabolites. Each model was validated with in vivo data. We used OptCom and Flux Balance Analysis to simulate how both organisms interact.
Conclusions
The consortium of iBif452 and iFap484 was applied to predict F. prausnitzii’s demand for acetate and production of butyrate which plays an essential role in colonic homeostasis and cancer prevention. The assembled reaction set is a useful tool to generate bacterial draft models from KEGG Orthology.
Keywords: Bifidobacterium adolescentis L2-32, Faecalibacterium prausnitzii A2-165, Genome-scale metabolic model, Metabolic modeling of gut microbiota
Background
Metagenomic sequencing facilitates the study of a large number of microorganisms in environmental samples [1]. This technique has been used to study the composition of gut microbiota [2], its role in human metabolism [3,4] and its relation to diseases such as atherosclerosis [5], obesity [6,7] and Crohn’s disease [8]. In functional metagenomic studies, it is common to use KEGG Orthology (KO) [9] to annotate gene functions [10]. KO can be used to predict the composition ratio of microbial gene families and pathways from the human microbiome project [11]. The functional annotation for a large number of sequenced bacteria, nearly 4,650 bacterial genomes, is stored in the Integrated Microbial Genomes (IMG) database, and the genomes are mapped to KEGG pathway images [12]. This tremendous new genomic information adds a new opportunity for systems biology, as it enables use of information about genome content for prediction of metabolic phenotypes of species in the gut [13], or to develop community systems [14] or supra-model organisms [15]. Therefore, it is relevant to reconstruct accurate genome scale metabolic models (GEMs) from KO annotated by metagenomic analysis.
Several methods have been developed to reconstruct genome scale models from GEMs of closely related organisms [16], KEGG [16-18], and the Model SEED [19]. The RAVEN toolbox [16] has been used to generate GEMs for the eukaryotic microorganisms Pichia stipitis and Pichia pastoris using iIN800, a GEM of Saccharomyces cerevisiae[20]. However, this method requires a GEM of a closely related organism. The RAVEN toolbox has another function to solve this problem by assigning gene to KO using MUSCLE [21] and HMMER [22]. Then it generates the draft model by mapping KO to KEGG reactions. The web-based methods, FAME [17] and MicrobesFlux [18], are able to produce draft models for ~750 and ~1,200 KEGG genomes, respectively. The disadvantage of both FAME and MicrobesFlux is that they are limited to organisms already annotated in KEGG. The Model SEED can generate a draft model for a desired organism based on RAST annotation of genes [23]. Even though some of these methods have computational gap filling methods, there is still a need for manual curation to obtain a functional model. Manual curation is generally cumbersome and time-consuming. The lack of visualization, such as organization and readability of reactions and genes names into the model Excel file and KEGG maps, can hamper manual curation of generated draft models.
To facilitate generation of a draft model and manual gap filling, we assembled an organized reference reaction data set consisting of common microbial reactions, where every reaction is assigned with KOs. The reactions were collected from high quality GEMs and from Rhea, a manually annotated database of chemical reactions [24], but not from KEGG reactions. In spite of the accuracy of KEGG reactions, some reactions need to substantially manual curation for substrate and co-factor usage, and the reactions in reconstructed GEMs generally have to be well annotated in terms of substrate and co-factor usage and elemental balancing. Our reaction data set can, in principle, be used to generate draft models for all 4,650 bacteria in the IMG database, KEGG organisms, or other user-defined organisms annotated by KO.
Here, we used this reaction data set to generate high quality GEMs for two bacterial genomes from the IMG database: B. adolescentis L2-32 and F. prausnitzii A2-165. Bifidobacterium is a dominating genus in the phyla Actinobacteria present in the human gut microbiota and Faecalibacterium is the most abundant genus among the Firmicutes. Firmicutes, Bacteroidetes and Actinobacteria are the most highly abundant phyla in the human gut microbiota [2]. Both Bifidobacterium and Faecalibacterium interact with Bacteroidetes[25,26]. Moreover, Bifidobacterium produces acetate to protect the host from infection [27], and Faecalibacterium has a relation with Crohn disease [28]. Furthermore, the production of butyrate by Faecalibacterium, among others, has been associated with a healthy state [5,29,30].
Finally, we simulated and compared the interactions between the two organisms using two approaches for community modeling: Flux balance analysis [31-33] and OptCom [34].
Methods
Figure 1 shows a summary of the methods developed and employed in this study. Reconstruction was based on the assignment of genes to KOs and this was used to generate a draft model from the reaction data set. A KEGG map viewer is helpful in identifying gaps in the reconstruction.
Figure 1.

Method summary. (I) The gene assigned with KO for each studied organism was downloaded from the IMG database or KEGG. (II) The KO was mapped with the reaction data set. (III) A draft model was exported to MS-Excel format by our function saveDraftModel, the draft model was mapped to KEGG maps using our function DrawPathway. (IV) The draft was curated manually from literature and other gene annotations in IMG files such as TIGRFAMs and Pfam. After this, the model was simulated using RAVEN and MOSEK. (V) The community interaction design described how the organisms share growth medium components. (VII) Community interaction was converted to XML format. (VIII) Both optCom and FBA models were generated from XML files.
Model reconstructions
Step 1 (Collecting reference reaction data set)
We assembled a reference reaction data set containing reactions assigned with KO for each KEGG map (see Additional file 1: Figure S1). For organisms Escherichia coli K-12 MG1655, Staphylococcus aureus N315 and Saccharomyces cerevisiae, we mapped the genes from each KEGG map to the corresponding reactions contained in the respective GEMs iAF1260 [35], iSB619 [36] and iTO977 [37]. The GEM reactions, together with the associated KOs, were then added to the reference reaction data set. In the case the organism had no genes for the enzyme or all GEMs lacked the reaction, we downloaded it from Rhea by the Matlab function get_reaction_from_Rhea. This function downloads Rhea reactions as XML format and prints them. To avoid the mismatched metabolite names between Rhea reactions and the reference reaction set, we evaluated the metabolite names in the Rhea reaction using its corresponding KEGG reaction. We retained Rhea metabolite names if they did not exist in the reference reaction data set, and the Rhea reaction and its corresponding KO were subsequently inserted into the reference reaction data set. iAF1260 reactions not present in any of the KEGG maps, especially transporters and reactions occurring in the cell wall, were inserted into the reference reaction data set with the corresponding KO as the corresponding gene. Moreover, we added the capsular polysaccharide and teichoic acid biosynthesis reactions, which are required for cell wall biosynthesis, from the GEM of Lactobacillus plantarum WCFS [38], because cell wall could be a significant fraction of gram dry weight of Gram-positive bacteria. Also we added the methane metabolism from the GEM of Methanosarcina barkeri[39] and added the siderophore group biosynthesis from the GEM of Mycobacterium tuberculosis[40]. Finally, the reactions were organized, ordered and made readable to facilitate the manual gap filling process.
Step2 (Generating draft models)
We downloaded the gene annotation for each studied organism from the IMG database [12]. We extracted the set GK= (gene,KO) for each organism from the downloaded IMG file using the function get_gene_ko_from_img. For organisms available in KEGG, the set GK can be obtained directly using the function get_gene_ko_from_kegg_org_id, otherwise the users can build the set GK themselves. The set GK was passed to the function buildDraftModel to extract reactions using KO identifiers from the reaction data set. The draft model was exported to an Excel file by the function saveDraftModel. Finally, we removed the exchange reaction for metabolites that were not participating in any cytosolic reaction. All the described functions are provided in Additional file 2 and can be used in the RAVEN toolbox.
Step 3 (Gap filling)
The gaps in each model were filled manually by mapping the model to KEGG maps and inserting the required reactions to ensure full connectivity in the model. To find genes for the filled reactions or metabolic genes, we extended the search to other available gene annotation in the IMG database. Both studied organisms have genes annotated by Pfam [41], TIGRFAMs [42], TC families [43] and METACYC [44]. Moreover, B. adolescentis L2-32 has gene annotations by SEED. Annotation with TC families has no specific gene assignment, so we ran a bidirectional blast between the TC protein sequence and each organism sequence. Also, we searched for the residual filled reactions by their enzyme name or EC number in the Pfam database to get the corresponding Pfam identifiers, and the corresponding genes were searched in IMG file. Additional file 3: Table S1 contains the results of this analysis.
Flux balance analysis
Flux Balance Analysis (FBA) [45,46] was used to reconstruct and validate both models using the RAVEN toolbox and MOSEK (MOSEK Inc.) as a linear programming solver under the Matlab programming environment (Mathwork Inc.). Equation (1) describes the main formulation of FBA, where S is a stoichiometric matrix, V is a vector of flux values for all reactions and C is a weight vector for each flux in vector V. Typically all values in C are zero except the flux of the biomass reaction (V biomass ) which is fixed to one. UB is the upper bound for the flux, and LB is the lower bound for the flux. In FBA, we assumed that the model grows in a growth medium (GM) which represents a set of metabolites d, such as a carbon source, ammonia, phosphate, sulfur. Certain exchange reactions, which carry flux from the medium to the model (V d ), were fixed to the model uptake rate g d , for the metabolite d. For example 1 mmol/gDW/h of glucose uptake rate is fixed to flux (V glc ) which carries glucose from the medium to the model.
| (1) |
Both B. adolescentis L2-32 and F. prausnitzii A2-165 models grow anaerobically in rich media. We assumed that each model consumed ammonia as a source of nitrogen, phosphate, H2S or cysteine as a source of sulfur, nicotinate and all amino acids having transporter reactions. In addition, The F. prausnitzii A2-165 model consumed folic acid from the medium. Xanthine, uracil and urea transporters were closed.
The compositions of protein, RNA and DNA in the biomass were estimated from Neidhardt et al. [47]. The compositions of peptidoglycan and capsular polysaccharide are the same as in the GEM of Lactobacillus plantarum WCFS [38].
We used flux variability analysis [48] to evaluate the predicted fluxes by the COBRA toolbox function fluxVariability[49] (see Additional file 4: Table S2). To determine if a studied organism is able to grow on different carbon sources such as galactose, xylose or fructose, the transporter flux was fixed to 1 mmol/gDW/h and the biomass formation was optimized.
Community modeling
In community studies each model was allowed to take up as much glucose as possible to maximize growth. In FBA simulations, g d splits into two kinds of reactions with the first being a distribution of metabolite d to the different microorganisms in the community and the second being different transport reactions, where each reaction represents transport of glucose to one organism [31]. In OptCom, g d became two variables: uptake variable for growth medium metabolite d, uval d k , or/and export variable for growth media, eval d k , where k = bif or fap [34] and where bif is B. adolescentis L2-32 and fap is F. prausnitzii A2-165. Equation (2) gives the general OptCom problem formulation for this community. This problem has nonlinear constraint, so it cannot be solved using MOSEK. We therefore wrote a function generateOptComModel to convert RAVEN Matlab models to GAMS language (GAMS Development Corporation) and used the BARON solver hosted on the NEOS servers [50], a free optimization server, to solve the optimization problem.
| (2) |
Description of community using XML
We described a community structure without details of each model as XML format (see Additional file 5), because LibSBML fails to read an SBML containing a user defined attribute or XML tag for community features [51]. Both iBif452 and iFap484 competed for glucose while iFap484 consumed acetate produced by iBif452. The functions generateOptComModel and generateComModel used XML files to generate OptCom and FBA models.
Results and discussion
Reconstruction of reference reaction data set
GEMs elucidate how organisms consume nutrients, carbon source, ammonia, phosphate and autotrophic metabolites to build their biomass precursors and produce chemical byproducts [52]. The biochemical reactions included in a GEM are based on experimental or predicted function of enzymes contained by the studied organism [53]. Reconstructed GEMs share many components: exchange flux, transport, central metabolism, nucleotide, amino acids, cofactor biosynthesis, cell wall and lipid. Most GEMs use KEGG maps and literature to illustrate the content of each component, where it contains one or more KEGG maps. Additional file 1: Figure S1 (adapted from [54]) shows these components and the KEGG map name for each component and how flux distributes to each component and builds the necessary biomass precursors.
To cover the reactions in Additional file 1: Figure S1, we built a reference reaction data set from published GEMs and a manually curated reaction database Rhea. This reaction data set contained reactions for the central carbon metabolism (glycolysis, PP pathway, TCA, pyruvate), amino acids and nucleotide biosynthesis, cell wall (peptidoglycan, capsular polysaccharide and teichoic acid biosynthesis) and cofactors (folate, CoA and NAD+). We adopted the fatty acids biosynthesis and glycerophospholipd metabolism in iAF1260. Then we included reactions that connect other carbon resources, such as galactose and maltose to the main network.
The reactions were organized to facilitate manual revision and editing of newly reconstructed GEMs. Each reaction was assigned with KOs obtained from KEGG maps. The reference reaction data set comprises 2,340 reactions out of which 214 came from Rhea, 1256 unique metabolite and 2146 KOs (see Additional file 6: Table S3). The reference reaction data set was used to generate a draft model with an input file containing a gene and its KO. Additional file 7: Figure S2 shows how the reaction set covers KEGG maps.
GEMs description
Table 1 shows statistics for the reconstructed GEMs and comparison with draft models generated by Model SEED. The iBif452 model comprises 699 reactions, 611 unique metabolites and 452 genes constituting 18.62% of the total number of genes. It contains 6 genes from TIGRFAMs and one gene from Pfam. It needed 9 reactions to become a connected model. Model iFap484 comprises 713 reactions, 621 unique metabolites and 484 genes, constituting 13.93% of the total number of genes. It contains 6 genes from TIGRFAMs and two genes from Pfam. It needed 16 reactions to become a connected model. Additional file 8 contains both models as Excel and SBML format.
Table 1.
Description of GEMS: iBif452 and iFap484 of B. adolescentis L2-32 and F. prausnitzii A2-165 in comparison with draft model generating by Model SEED
|
B. adolescentis L2-32 |
F. prausnitzii A2-165 |
|||
|---|---|---|---|---|
| iBif452 | SEED draft | iFap484 | Seed draft | |
| Reactions |
699 |
663 |
713 |
787 |
| Unique metabolite |
611 |
691 |
621 |
798 |
| Genes |
452 |
543 |
484 |
586 |
| Reactions without genes |
84 |
|
90 |
|
| Exchange |
59 |
|
60 |
|
| Transporter |
12 |
|
10 |
|
| Spontaneous |
4 |
|
4 |
|
| FILL |
9 |
|
16 |
|
| Genes from other annotations |
7 |
|
8 |
|
| Coding genes | 2428 | 3475 | ||
Both iBif452 and iFap484 have reactions for the central carbon metabolism and can utilize other sole carbon sources than glucose, as reported in in vivo studies. iBif452 can utilize galactose, fructose and maltose, which is consistent with in vivo studies [55]. The iFap484 can also utilize galactose and maltose but cannot utilize xylose, which is also consistent with in vivo studies [56]. iBif452 features the bifid shunt pathway or the F6PPK pathway representing a special Bifidobacteria pathway converting glucose to pyruvate (see Additional file 9: Figure S3) [57-59]. The F6PPK pathway includes fructose-6-phosphate phosphoketolase converting D-Fructose 6-phosphate to Acetyl phosphate and D-Erythrose 4-phosphate, compared to the common part of glycolysis with 6-phosphofructokinase and fructose-1,6-bisphosphate aldolase. iFap484 has a Faecalibacterium prausnitzii butyrate producing pathway (see Additional file 10: Figure S4) [60]. Neither iBif452 nor iFap484 produces anything when the glucose uptake rate is 0 mmol/gDW/h and the objective function is biomass or ATP non-growth association maintenance, so the models did not generate energy or matter from nothing. In spite of the two models were validated with FBA in the following two sections, the comprehensive validation of the GEMs needs extensive experiments. Since these bacteria are not yet well-studied, we think the two models based on recently sequenced, may assist a lot to overcome some important questions, computing phenotypic states and describing the genotype-phenotype relationships.
Bifidobacterium adolescentis adolescentis L2-32 validation
Bifidobacterium has predicted genes for biosynthesis of all 20 amino acids, purines and pyrimidines [57]. However, Bifidobacterium only grows in complex media, probably because some of the genes in the biosynthetic pathway for amino acids are non-functional [57]. We assumed that iBif452 grows in media containing 12 amino acids for which it has transporter reactions.
The iBif452 model did not produce lactate when biomass or ATP production was optimized, while Bifidobacterium produces acetate, lactate, formate and ethanol in vivo. Under glucose limitation, Bifidobacterium does not produce lactate because it tries to maximize energy production by cleaving pyruvate to acetyl phosphate and formate [61]. Furthermore, the specific rate of sugar consumption affects the amount of lactate production. For example, the organism produces a large amount of lactate when it has a rapid sugar consumption, but produces a small amount of lactate when it consumes a less preferred sugar like oligofructose [62].
To study the ability of the model iBif452 to produce lactate, we maximized ATP production for non-growth association maintenance, i.e., the reaction ATP + H2O = > ADP + Phosphate + H. The model produced 3 mmol of ATP per 1 mmol of glucose and produced only acetate, formate and ethanol. When the model was constrained to produce 1 mmol of lactate per 1 mmol of glucose, it produced 2.5 mmol of ATP and 1.5 mmol of acetate per 1 mmol of glucose.
Figure 2 shows the effect of lactate production on the iBif452 model when the biomass is an objective function. In Figure 2A, the model achieved maximum biomass when there was no production of lactate. In Figure 2B, with increased production of lactate, the production rate of acetate, ethanol and formate decreased. The model was still able to produce acetate without formate because Bifidobacterium has the fructose-6-phosphate phosphoketolase enzyme, which converts fructose 6-phosphate into erythrose 4-phosphate and acetyl phosphate and the latter can yield an ATP when metabolized to acetate. Flux variability analysis showed the differences between maximum and minimum fluxes for acetate, formate and ethanol were 0.0003, 0.011 and 0.011 mmol per 1 mmol of glucose.
Figure 2.
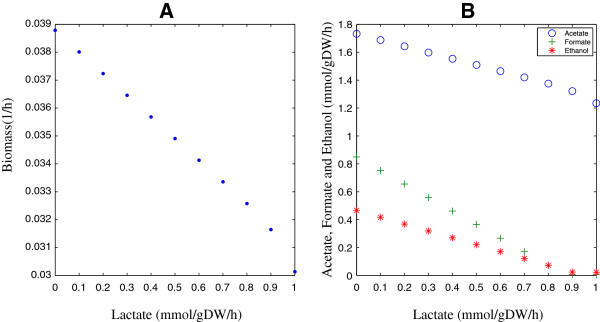
The effect of lactate production on the iBif452 Model. (A) The biomass decreases with increasing the lactate production. (B) The production of acetate, formate, and ethanol decrease with increasing the lactate production.
The last results showed that the model aimed to generate ATP by converting pyruvate to acetate through acetyl-CoA and acetyl-phosphate and it therefore has to regenerate NAD + by forming ethanol, as this is only way this co-factor can be balanced when there is formation of acetate (See Additional file 9: Figure S3). Although the model predicts a flux distribution for the theoretical ratio between acetate and lactate in Bifidobacterium, it fails to predict the amount of lactate just like previous GEMs of lactic acid bacteria [63]. To overcome this problem, Oliveira et al. constrained the pyruvate formate lyase reaction to an interval to deal with lactate production in a GEM of L. Lactic[64]. Bas Teusink et al. fixed the measured flux in the GEM of L. plantarum WCFS1 [38]. Milan et al. added new enzyme turnover parameter to avoid metabolism overflow in GEM of L.lactis[65] based on flux balance analysis with molecular crowding [66]. Finally, Bas Teusink et al. showed that L. plantarum optimizes its yield when it grows with glycerol to support the prediction of GEM in lactic acid bacteria [67].
In the present work, we constrained the lactate flux in the model. When we constrain lactate production with a yield of 1 (mmol/mmol of glucose), the model produces acetate with a yield of 1.23 (mmol/mmol glucose), as listed in Table 2. Flux variability analysis shows acetate yield having a 0.007 (mmol/mmol glucose) difference between maximum and minimum fluxes. When the ATP production for non-growth association maintenance was used as an objective function, it produced acetate with a yield of 1.5 (mmol/mmol glucose). Flux variability analysis showed that the acetate yield was not different between the maximum and minimum fluxes. Finally, when the model was constrained to produce 0.21 mol of lactate per mol of glucose, it had a growth yield of 37.2 gDW per mol of glucose, which was very close to the in vivo. Bifidobacterium growth yield of 37.4 gDW per mol of glucose [68].
Table 2.
Comparison between in-silco prediction of short chain fatty acids of iBif452 and iFap484 with experimental data
Faecalibacterium prausnitzii A2-165 validation
To study the effect of external acetate on butyrate production in Faecalibacterium prausnitzii A2-165, biomass production was used as an objective function. The model produces butyrate with a yield of 1.62 (mmol butyrate/mmol glucose) and co-consumes 1.39 mmol of acetate per mmol of glucose. The ratio of acetate uptake to butyrate production was 85.8%, which is close to the 85-90% observed in in vivo studies of F. prausnitzii[69]. Flux variability analysis shows that the acetate, butyrate, and formate have a difference of 0.0005, 0.014, and 0.014 mmol/gDW/h between maximum and minimum fluxes, respectively. Table 2 shows the comparison between these values with in vivo studies [60], where F. prausnitzii consumed 10 mM of glucose and 9.55 ± 1.2 mM of acetate to produce 10.45 ± 1.53 mM of butyrate.
Figure 3 shows a sensitivity analysis of the effect of acetate and glucose on iFap484 with biomass as an objective function. The acetate and glucose uptake rate varied from 0 to 1 mmol/gDW/h. iFap484 cannot grow without glucose and grows poorly without acetate. This result is similar to in vivo studies of F. prausnitzii A2-165, that shows poor growth without acetate in the medium [69] and the F. prausnitzii strains L2-6 and ATCC 27766 cannot grow without acetate [70]. The model predicted increase in growth rate with acetate supplied was 3.93-fold, close to the in vivo value, where the increase in growth rate is 3.6-fold [60].
Figure 3.
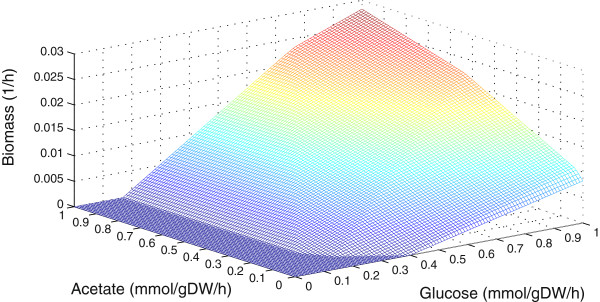
The effect of glucose and acetate on the growth of iFap484. Glucose and acetate uptake rate varied between 0 to 1 mmol/gDW/h.
Community simulation
Both OptCom and FBA methods were applied to iBif452 and iFap484 to simulate how B. adolescentis L2-32 and F. prausnitzii A2-165 co-culture together. Both organisms compete for 1 mmol/gDW/h of glucose to maximize their growth. The model iBif452 generates acetate and iFap484 consumes acetate to produce butyrate, which plays a critical role in colonic homeostasis and cancer prevention [71-73].
Figure 4 depicts the prediction of fluxes calculated using both methods: OptCom and FBA, where biomass was an objective function. iFap484 consumed 0.57 mmol/gDW/h of glucose and all the acetate produced by iBif452 to produce 0.92 mmol/gDW/h of butyrate. With flux variability analysis, the differences between maximum and minimum yield of acetate and butyrate were 0.00004 and 0.007 (mmol/mmol of glucose) respectively. There was no significant difference between OptCom and FBA simulations. However, for this simple system, FBA provided faster simulation than OptCom.
Figure 4.
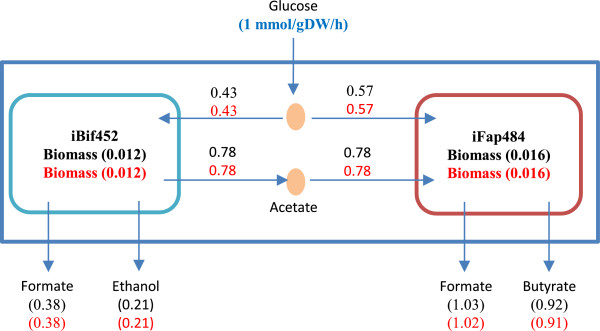
Simulation summary results using OptCom and FBA methods when iBif452 and iFap484 grow together on glucose. The black numbers are fluxes predicted by OptCom, the red numbers are fluxes predicted by the FBA method. ATP non-growth association maintenance was fixed at 0.4 and 0.5 mmol/gDW/h in the iFap484 and iBif452 models respectively. The unit of ATP, acetate, lactate, ethanol, formate and butyrate is mmol/gDW/h. The unit of biomass is (1/h).
Finally, FBA was used to simulate how a consortium of iBif452 and iFap484 interact. The composition of the consortia was varied from 0% to 100% iFap484, while keeping the total biomass constant, and the objective function was minimization of glucose uptake rate in both iBif452 and iFap484. The biomass production rate was fixed at × % in iFap484 and (1-×) % in iBif452 of total biomass, where the total biomass growth rate was 0.1 (1/h). Figure 5 shows the increase in butyrate production as iFap484 composition increases.
Figure 5.
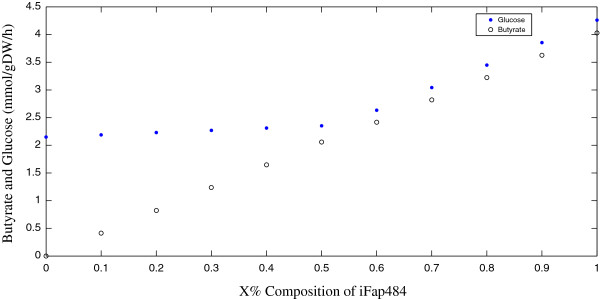
Abundances analysis of iFap484 and iBif452. How the amount of butyrate and the total glucose consumption change with different compositions of iFap484 and iBif452.
Conclusion
We assembled a reaction set from published GEMs, where each reaction is assigned with KO. This reaction set was used to generate draft GEMs for each non-KEGG organisms. It represented a simple method to generate bacterial draft models from KEGG KO, instead of generating it from KGML [74]. The description of a community as a XML format can be used together with the two community simulation methods Optcom and FBA. This saves time and effort when performing community modeling.
Community simulations of an acetate producer B. adolescentis adolescentis L2-32 and an acetate consumer F. prausnitzii A2-165 provided insights into metabolic cross talk between these two members of the gut microbiota. It shows the importance of acetate supply to butyrate production, since the growth and production of Faecalibacterium prausnitzii is severely hampered by limited acetate supply. This is an initial attempt to approach the very complex ecosystem and metabolic organ that the gut microbiota constitutes.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IEE-L developed the methods, reconstructed the two genome-scale metabolic models, performed the simulations and drafted the manuscript. FK and SS assisted in the process of model reconstruction and edited the manuscript. IN, THS and JN supervised the work and edited the manuscript. JN conceived and designed the project. All authors read and approved the final manuscript.
Supplementary Material
Mapping the main component of GEM to KEGG pathway maps (adapted from [54]).
Matlab funtions used in the paper.
Genes with annotations: TIGRFAMs, KOs, SEED, TC number.
Flux variability analysis for prediction fluxes in the study.
The structure of community as XML format.
Reference reaction data set.
Mapping the assembled reaction set to KEGG metabolic pathways maps.
The two models in SBML and MS Excel format. The two models are also available through the Human Metabolic Atlass http://www.metabolicatlas.com/downloads/micro.
Main carbon metabolism in Bifidobacterium adolescentis adolescentis L2-32.
Main carbon metabolism in Faecalibacterium prausnitzii A2-165.
Contributor Information
Ibrahim E El-Semman, Email: elsemman@aun.edu.eg.
Fredrik H Karlsson, Email: frekar@chalmers.se.
Saeed Shoaie, Email: saeeds@chalmers.se.
Intawat Nookaew, Email: intawat@chalmers.se.
Taysir H Soliman, Email: taysser.soliman@fci.au.edu.eg.
Jens Nielsen, Email: nielsenj@chalmers.se.
Acknowledgements
We thank Ali Zomorrodi and Prof. Costas Maranas for providing us with OptCom implementation. Ibrahim E. El-Semman appreciates the Egyptian Cultural Affairs and Missions Sector for their financial support. We acknowledge funding from the Knut and Alice Wallenberg Foundation. We thank Chalmers Library for funding the open access charge.
References
- Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Comput Biol. 2010;6(2):e1000667. doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S. et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint HJ. Obesity and the gut microbiota. J Clin Gastroenterol. 2011;45:S128–S132. doi: 10.1097/MCG.0b013e31821f44c4. 110.1097/MCG.1090b1013e31821f31844c31824. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55(2):205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(suppl 1):D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methé B, Schloss PD, Gevers D, Mitreva M, Huttenhower C. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8(6):e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(D1):D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson FH, Nookaew I, Petranovic D, Nielsen J. Prospects for systems biology and modeling of the gut microbiome. Trends Biotechnol. 2011;29(6):251–258. doi: 10.1016/j.tibtech.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Zengler K, Palsson BO. A road map for the development of community systems (CoSy) biology. Nat Rev Micro. 2012;10(5):366–372. doi: 10.1038/nrmicro2763. [DOI] [PubMed] [Google Scholar]
- Borenstein E. Computational systems biology and in silico modeling of the human microbiome. Brief Bioinform. 2012;13(6):769–780. doi: 10.1093/bib/bbs022. [DOI] [PubMed] [Google Scholar]
- Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for <italic>Penicillium chrysogenum</italic>. PLoS Comput Biol. 2013;9(3):e1002980. doi: 10.1371/journal.pcbi.1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boele J, Olivier B, Teusink B. FAME, the flux analysis and modeling environment. BMC Syst Biol. 2012;6(1):8. doi: 10.1186/1752-0509-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Xu Y, Chen Y, Tang Y. MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Syst Biol. 2012;6(1):94. doi: 10.1186/1752-0509-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry M, DeJongh C, Best A, Frybarger P, Linsay B, Stevens R. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- Caspeta L, Shoaie S, Agren R, Nookaew I, Nielsen J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst Biol. 2012;6(1):24. doi: 10.1186/1752-0509-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23(1):205–211. [PubMed] [Google Scholar]
- Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass E, Kubal M, Meyer F, Olsen G, Olson R, Osterman A, Overbeek R, McNeil L, Paarmann D, Paczian T, Parrello B, Pusch G, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara R, Axelsen KB, Morgat A, Belda E, Coudert E, Bridge A, Cao H, de Matos P, Ennis M, Turner S, Owen G, Bougueleret L, Xenarios I, Steinbeck C. Rhea—a manually curated resource of biochemical reactions. Nucleic Acids Res. 2012;40(D1):D754–D760. doi: 10.1093/nar/gkr1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Calmeyn T, Leroy F, De Vuyst L. Coculture fermentations of bifidobacterium species and bacteroides thetaiotaomicron reveal a mechanistic insight into the prebiotic effect of inulin-type fructans. Appl Environ Microbiol. 2009;75(8):2312–2319. doi: 10.1128/AEM.02649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzosek L, Miquel S, Noordine M-L, Bouet S, Chevalier-Curt M, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, Langella P, Thomas M. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013;11(1):61. doi: 10.1186/1741-7007-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469(7331):543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J, Blugeon S, Bridonneau C, Furet J-P, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci. 2008;105(43):16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O/'Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O/'Sullivan O, Fitzgerald GF, Deane J, O/'Connor M, Harnedy N, O/'Connor K, O/'Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP, O/'Toole PW. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488(7410):178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Klitgord N, Segrè D. Environments that induce synthetic microbial ecosystems. PLoS Comput Biol. 2010;6(11):e1001002. doi: 10.1371/journal.pcbi.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh JA, Stahl DA. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol. 2007;3:92. doi: 10.1038/msb4100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S, Karlsson F, Mardinoglu A, Nookaew I, Bordel S, Nielsen J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep. 2013;3:2532. doi: 10.1038/srep02532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomorrodi AR, Maranas CD. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 2012;8(2):e1002363. doi: 10.1371/journal.pcbi.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Palsson B. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol. 2005;5(1):8. doi: 10.1186/1471-2180-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst Biol. 2013;7(1):36. doi: 10.1186/1752-0509-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teusink B, Wiersma A, Molenaar D, Francke C, de Vos WM, Siezen RJ, Smid EJ. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J Biol Chem. 2006;281(52):40041–40048. doi: 10.1074/jbc.M606263200. [DOI] [PubMed] [Google Scholar]
- Gonnerman MC, Benedict MN, Feist AM, Metcalf WW, Price ND. Genomically and biochemically accurate metabolic reconstruction of Methanosarcina barkeri Fusaro, iMG746. Biotechnol J. 2013;8(9):1070–1079. doi: 10.1002/biot.201200266. [DOI] [PubMed] [Google Scholar]
- Jamshidi N, Palsson B. Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol. 2007;1(1):26. doi: 10.1186/1752-0509-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40(D1):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31(1):371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Yen MR, Noto K, Tamang DG, Elkan C. The transporter classification database: recent advances. Nucleic Acids Res. 2009;37(suppl 1):D274–D278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40(D1):D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ND, Papin JA, Schilling CH, Palsson BO. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21(4):162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Park JM, Kim TY, Lee SY. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol Adv. 2009;27(6):979–988. doi: 10.1016/j.biotechadv.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Ingraham J, Schaechter M. Physiology of the Bacterial Cell: A Molecular Approach. Sunderland, MA: Sinauer Associates; 1990. [Google Scholar]
- Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5(4):264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Schellenberger J, Que R, Fleming R, Thiele I, Orth J, Feist A, Zielinski D, Bordbar A, Lewis N, Rahmanian S, Kang J, Hyduke D, Palsson B. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzyk J, Mesnier MP, More JJ. The NEOS Server. Computational Science & Engineering, IEEE. 1998;5(3):68–75. doi: 10.1109/99.714603. [DOI] [Google Scholar]
- Bornstein BJ, Keating SM, Jouraku A, Hucka M. LibSBML: an API Library for SBML. Bioinformatics. 2008;24(6):880–881. doi: 10.1093/bioinformatics/btn051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Agren R, Bordel S, Nielsen J. Use of genome-scale metabolic models for understanding microbial physiology. FEBS Lett. 2010;584(12):2556–2564. doi: 10.1016/j.febslet.2010.04.052. [DOI] [PubMed] [Google Scholar]
- Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protocols. 2010;5(1):93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos GN, Aristidou AA, Nielsen J. Metabolic Engineering. San Diego: Academic Press; 1998. Chapter 2 - Review of Cellular Metabolism; pp. 21–79. [Google Scholar]
- Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6(8):1535–1543. doi: 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Siles M, Khan TM, Duncan SH, Harmsen HJM, Garcia-Gil LJ, Flint HJ. Cultured representatives of two major phylogroups of human colonic faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl Environ Microbiol. 2012;78(2):420–428. doi: 10.1128/AEM.06858-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, O’Sullivan DJ. Genomic insights into bifidobacteria. Microbiol Mol Biol Rev. 2010;74(3):378–416. doi: 10.1128/MMBR.00004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NP, Whitehead TR, Côté GL. Gas chromatography–mass spectrometry (GC-MS) techniques for metabolic flux analysis of the Bifido shunt pathway. Biocatal Biotransformation. 2006;24(1):95–98. [Google Scholar]
- Fandi KG, Ghazali HM, Yazid AM, Raha AR. Purification and N-terminal amino acid sequence of fructose-6-phosphate phosphoketolase from Bifidobacterium longum BB536. Lett Appl Microbiol. 2001;32(4):235–239. doi: 10.1046/j.1472-765X.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA):Acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol. 2002;68(10):5186–5190. doi: 10.1128/AEM.68.10.5186-5190.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan B, Macfarlane G. Effect of dilution rate and carbon availability on bifidobacterium breve fermentation. Appl Microbiol Biotechnol. 1994;40(6):800–805. doi: 10.1007/BF00173978. [DOI] [Google Scholar]
- Van der Meulen R, Adriany T, Verbrugghe K, De Vuyst L. Kinetic analysis of bifidobacterial metabolism reveals a minor role for succinic acid in the regeneration of NAD+ through its growth-associated production. Appl Environ Microbiol. 2006;72(8):5204–5210. doi: 10.1128/AEM.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teusink B, Bachmann H, Molenaar D. Systems biology of lactic acid bacteria: a critical review. Microb Cell Factories. 2011;10(Suppl 1):S11. doi: 10.1186/1475-2859-10-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A, Nielsen J, Forster J. Modeling lactococcus lactis using a genome-scale flux model. BMC Microbiol. 2005;5(1):39. doi: 10.1186/1471-2180-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoek M, Merks R. Redox balance is key to explaining full vs. partial switching to low-yield metabolism. BMC Syst Biol. 2012;6(1):22. doi: 10.1186/1752-0509-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Beg Q, de Menezes M, Ernst J, Bar-Joseph Z, Barabasi A-L, Boros L, Oltvai Z. Impact of the solvent capacity constraint on E. coli metabolism. BMC Syst Biol. 2008;2(1):7. doi: 10.1186/1752-0509-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teusink B, Wiersma A, Jacobs L, Notebaart R, Smid E. Understanding the adaptive growth strategy of Lactobacillus plantarum by in silico optimisation. PLoS Comput Biol. 2009;5:e1000410. doi: 10.1371/journal.pcbi.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries W, Stouthamer AH. Fermentation of glucose, lactose, galactose, mannitol, and xylose by bifidobacteria. J Bacteriol. 1968;96(2):472–478. doi: 10.1128/jb.96.2.472-478.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Holtrop G, Lobley GE, Calder AG, Stewart CS, Flint HJ. Contribution of acetate to butyrate formation by human faecal bacteria. Br J Nutr. 2004;91(06):915–923. doi: 10.1079/BJN20041150. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Hold GL, Harmsen HJM, Stewart CS, Flint HJ. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J Syst Evol Microbiol. 2002;52(6):2141–2146. doi: 10.1099/ijs.0.02241-0. [DOI] [PubMed] [Google Scholar]
- Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Dore J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- Wrzodek C, Buchel F, Ruff M, Drager A, Zell A. Precise generation of systems biology models from KEGG pathways. BMC Syst Biol. 2013;7(1):15. doi: 10.1186/1752-0509-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mapping the main component of GEM to KEGG pathway maps (adapted from [54]).
Matlab funtions used in the paper.
Genes with annotations: TIGRFAMs, KOs, SEED, TC number.
Flux variability analysis for prediction fluxes in the study.
The structure of community as XML format.
Reference reaction data set.
Mapping the assembled reaction set to KEGG metabolic pathways maps.
The two models in SBML and MS Excel format. The two models are also available through the Human Metabolic Atlass http://www.metabolicatlas.com/downloads/micro.
Main carbon metabolism in Bifidobacterium adolescentis adolescentis L2-32.
Main carbon metabolism in Faecalibacterium prausnitzii A2-165.