

Abstract
We report the identification and characterization of a five-carbon protein post-translational modification (PTM) called lysine glutarylation (Kglu). This protein modification was detected by immunoblot and mass spectrometry (MS), and then comprehensively validated by chemical and biochemical methods. We demonstrated that the previously annotated deacetylase, sirtuin 5 (SIRT5), is a lysine deglutarylase. Proteome-wide analysis identified 683 Kglu sites in 191 proteins and showed Kglu is highly enriched on metabolic enzymes and mitochondrial proteins. We validated carbamoyl phosphate synthase 1 (CPS1), the rate-limiting enzyme in urea cycle, as a glutarylated protein and demonstrated that CPS1 is targeted by SIRT5 for deglutarylation. We further showed that glutarylation suppresses CPS1 enzymatic activity in cell lines, mice, and a model of glutaric academia type I disease, the last of which has elevated glutaric acid and glutaryl-CoA. This study expands the landscape of lysine acyl modifications and increases our understanding of the deacylase SIRT5.
Keywords: Acylation, lysine acetylation, lysine succinylation, lysine glutarylation, glutaryl-CoA, mass spectrometry, protein post-translational modification, SIRT5, succinyl-CoA
Introduction
Under physiological conditions, cells are constantly exposed to diverse stressors and variations in energy supply, leading to fluctuations in cellular energy status. Therefore, cells require adaptive strategies to respond to these dynamic changes in the extracellular environment in order to maintain metabolic homeostasis, including regulation of energy producing pathways. Indeed, several mechanisms have been described by which cells adapt to such environmental changes, such as transcriptional regulation by alterations of epigenetic marks, modulation of metabolic enzyme activities by cellular metabolites, as well as protein post-translational modifications (PTMs) (Furuya and Uyeda, 1980; Lu and Thompson, 2012). Cellular metabolites play a key role in the regulation of epigenetic modifications. For example, acetyl-CoA, SAM, NAD+, and alpha-ketoglutarate are donors/substrates or cofactors for histone lysine modifying enzymes, i.e. those modulating levels of acetylation and methylation (Imai et al., 2000; Takahashi et al., 2006; Tsukada et al., 2006; Wellen et al., 2009). These PTM enzymes have important roles in modulation of chromatin structure and transcriptional regulation (Berger, 2007; Chi et al., 2010).
In addition to histones, PTM-regulatory enzymes, such as AMP-activated protein kinase (AMPK) and lysine deacetylases (KDACs or HDACs), can control activity of their substrates (Chalkiadaki and Guarente, 2012; Hardie et al., 2012), many of which are metabolic enzymes. Importantly, the activity of these PTM enzymes can be modified depending on cellular energy status. For example, sirtuin-family HDACs (or class III HDACs) can be regulated by the cellular NAD+ levels (Imai et al., 2000; Schmidt et al., 2004). SIRT1 is the homolog to Sir2, the founding member of this deacetylase subfamily, and has been shown to play an important role in cellular metabolism (Chalkiadaki and Guarente, 2012). Lysine acetylation levels are coupled to metabolism, and play key roles in regulating mitochondrial enzymes, chromatin biology, and other cellular processes (Chalkiadaki and Guarente, 2012; Verdin et al., 2010; Xiong and Guan, 2012). Recently, we identified lysine succinylation (Ksucc) and malonylation (Kmal) as protein PTMs (Peng et al., 2011; Xie et al., 2012; Zhang et al., 2011). We and others showed that SIRT5, a sirtuin family HDAC, has potent desuccinylase and demalonylase activities (Du et al., 2011; Peng et al., 2011). A recent study using quantitative proteomics demonstrated that SIRT5 ablation in MEFs induces a dramatic increase of lysine succinylation, demonstrating SIRT5 is the major enzyme for lysine desuccinylation in the cells (Park et al., 2013). In addition, lysine succinylation is an abundant protein post-translational modification in diverse model organisms (Weinert et al., 2013). While little is known about the mechanisms of acylation, succinyl-CoA and malonyl-CoA could serve as the donors for these two PTM reactions (Du et al., 2011; Peng et al., 2011; Wagner and Payne, 2013). In addition to these metabolites, NAD+ is required for SIRT5-dependent desuccinylation and demalonylation reactions (Du et al., 2011; Peng et al., 2011). Together, association of three key cellular metabolites in these pathways strongly suggests that succinylation and malonylation PTMs have roles in metabolism (Park et al., 2013). It remains unknown if additional PTMs exist that could control cellular metabolic circuits.
In this study, we report a new type of lysine modification called lysine glutarylation (Kglu). First, we identified and comprehensively validated Kglu as an evolutionarily conserved PTM. We then showed that SIRT5 catalyzes lysine deglutarylation both in vitro and in vivo. We further validated CPS1, the rate-limiting enzyme important for ammonia detoxification in urea cycle, as a glutarylated substrate. Glutarylation of CPS1 inhibits its activity, but can be reserved by SIRT5. We further showed that in a glutaric academia type I (glutaryl-CoA dehydrogenase deficiency) disease model of elevated glutaryl-CoA, mice have elevated glutarylation of mitochondrial proteins, including CPS1, and reduced CPS1 enzymatic activity, suggesting protein glutarylation as one possible alternative fate of elevated glutaryl-CoA in this disease. We demonstrate that lysine glutarylation is a SIRT5- and nutrient-regulated PTM, which impacts metabolic processes and other mitochondrial functions.
Results
We recently described two acyl modifications, lysine succinylation and malonylation (Peng et al., 2011; Xie et al., 2012; Zhang et al., 2011). Succinyl-CoA and malonyl-CoA can be cofactors for lysine succinylation and lysine malonylation, respectively (Du et al., 2011; Peng et al., 2011; Wagner and Payne, 2013). Glutaryl-CoA is structurally similar to succinyl-CoA and malonyl-CoA (Figure 1A), and is an important metabolite of amino acid metabolism (Figure 1B and Figure S1). We predicted glutaryl-CoA could serve as the donor molecule for the lysine glutarylation reaction (Koeppen et al., 1979; Mitzen and Koeppen, 1984), an analogous to a role for acetyl-CoA in lysine acetylation. Indeed, we have shown that a few other short-chain acyl-CoAs can serve as precursors of lysine acylation, such as lysine propionylation, lysine butyrylation, and lysine crotonylation (Chen et al., 2007; Tan et al., 2011).
Figure 1. Lysine deglutarylation and glutaryl-CoA pathways.

(A) Structures of glutaryl-lysine, succinyl-lysine and malonyl-lysine.
(B) Illustration of glutaryl-CoA synthetic pathways.
To test whether Kglu could be detected in vivo, we generated a pan anti-Kglu antibody. This anti-Kglu antibody detected its antigen peptide bearing a fixed glutarylated lysine (Kglu), but not peptide libraries bearing a fixed unmodified lysine (K), acetyl-lysine (Kac), malonyl-lysine (Kmal), or succinyl-lysine (Ksucc) (Figure 2A). This result demonstrated high specificity of the antibody for lysine glutarylated peptides.
Figure 2. Detection and verification of lysine glutarylation.

(A) Specificity of the anti-Kglu antibody. Dot-blot assay was carried out using anti-Kglu antibody by incubation of the peptide libraries bearing a fixed unmodified K, Kac, Kmal, Ksucc, and Kglu, respectively. Each peptide library contains 10 residues CXXXXKXXXX, where X is a mixture of 19 amino acids (excluding cysteine), C is cysteine, and the 6th residue is a fixed lysine residue: unmodified lysine (K), acetyl-lysine (Kac), malonyl-lysine (Kmal), succinyl-lysine (Ksucc), glutaryl-lysine (Kglu).
(B) Western blot of the whole-cell lysates from bacteria E. coli, yeast S. cerevisiae, Drosophila S2, mouse MEF, human HeLa cells. Western blot was performed using the whole-cell lysates from different cells.
(C) High-resolution MS/MS spectra of an E. coli tryptic peptide (SK+114.0281DaATNLLYTR) from DNA starvation/stationary phase protection protein with a mass shift of +114.0281 Da at the lysine residue (bottom) and the synthetic SKgluATNLLYTR (top). Inset shows the mass-to-charge ratios (m/z) of the precursor ions.
(D) Extracted ion chromatograms (XICs) of the synthetic peptide SKgluATNLLYTR (top), the in vivo-derived peptide (middle, from E. coli), and their mixture (bottom) by reversed-phase HPLC analysis.
To examine whether Kglu is evolutionarily conserved, we performed immunoblot analysis using the anti-Kglu antibody with whole cell lysates from different species: E. coli, S. cerevisiae, Drosophila melanogaster (S2), mouse (MEFs), and human cells (HeLa). Multiple bands were detected in all species, and these signals could be efficiently competed away by the tryptic peptides of glutarylated-BSA, but not the corresponding tryptic peptides of unmodified-BSA (Figures 2B and S2A). These results suggest that Kglu is a broadly conserved PTM and is present in both eukaryotic and prokaryotic cells.
Identification of E. coli and human Kglu substrates
To identify potential Kglu substrates in E. coli and HeLa cells, we used an affinity enrichment based proteomics approach as previously described (Chen et al., 2012; Kim et al., 2006). Glutarylated peptides were first enriched using the pan anti-Kglu antibody from tryptic digest of E. coli, and then analyzed by HPLC-mass spectrometry (MS)/MS. The acquired MS/MS spectra were analyzed by Mascot software to identify potential Kglu peptides. We used strict manual verification criteria to filter out false positives, as previously described (Chen et al., 2005), to ensure bona fide identification of glutarylated lysines (Table S1). In this pilot experiment, we detected 23 and 10 Kglu peptides from E. coli and HeLa cells, respectively.
Verification of Kglu using synthetic peptides
Because lysine glutarylated peptides have not been described in the past and affinity enrichment of Kglu peptides may identify some structural isomers, it is therefore important to confirm the identified peptides. To this end, we carried MS/MS (or tandem mass spectrometry) and HPLC co-elution experiments for the in vivo derived peptides and their synthetic counterparts. Peptides with identical primary sequences and modifications at the same residues should exhibit the same HPLC retention times and MS/MS fragmentation patterns. Therefore, comparative analysis of in vivo-derived peptides bearing the PTM of interest with their corresponding synthetic standards is an ideal approach to validate a PTM. We chemically synthesized two Kglu peptides: SKgluATNLLYTR and NFSTVDIQKgluNGVK, based on Kglu peptide candidates originally identified from E. coli and HeLa cell, respectively. We performed MS/MS and HPLC co-elution experiments to verify the chemical identity of the in vivo-derived peptides. The MS/MS spectrum of a tryptic peptide from E. coli DNA protection during starvation protein (SK+114.0281DaATNLLYTR) with a mass shift of +114.0281Da at the lysine residue has the same MS/MS spectrum as that of the synthetic peptide with a glutaryl group on lysine residue (SKgluATNLLYTR) (Figure 2C). In addition, the in vivo-derived peptide co-eluted with the synthetic one by the reversed-phase HPLC/MS analysis (Figure 2D), further confirming that the detected mass shift of + 114.0281Da in the in vivo-derived peptide is caused by lysine glutarylation. Similarly, we also confirmed lysine glutarylation in a tryptic peptide of the DNA mismatch repair protein Msh2 from HeLa cells, NFSTVDIQK+114.0311DaNGVK (Figures S2B and S2C).
Validation of Kglu by isotopic D4-glutarate labeling
Isotopic labeling is a method of choice to investigate the origin of the carbon backbones of PTMs. Succinate and malonate can be used by cells to generate lysine succinylation and malonylation, respectively, presumably by generating succinyl-CoA and malonyl-CoA intermediary metabolites (Peng et al., 2011; Zhang et al., 2011). Therefore, we hypothesized that lysine glutarylation may also be labeled with isotopically labeled glutarate via the in vivo conversion of D4-glutarate to D4-glutaryl-CoA. To test this hypothesis, we first treated HeLa cells non-isotopic glutarate. Our result showed that glutarate can slightly enhance levels of global glutarylation, but have no influence on lysine acetylation and succinylation (Figure S2D). We then treated the cells with 20 mM isotopically labeled D4-glutarate for 24 hr. Proteins whole-cell lysates were isolated and digested with trypsin. Kglu peptides were enriched by immunoprecipitation using the anti-Kglu antibody and analyzed by HPLC-MS/MS for peptide identification. Isotopically (D4) labeled Kglu peptides (with an additional +4 Da mass shift) can be distinguished from the regular lysine glutarylation on the MS spectrum. This analysis revealed that the Kglu peptide identified from HeLa cells, NFSTVDIQKgluNGVK, was indeed labeled with D4-glutarate (Figure S2E). These results further confirmed the structure of this modification, and identified glutarate as one metabolite that can be a precursor to lysine glutarylation. Together, four independent approaches, Western blotting analysis, MS/MS and HPLC co-elution with synthetic peptides, and isotopic labeling, identified and validated lysine glutarylation as a PTM and its presence in species ranging from bacteria to mammals.
Screening of HDAC lysine deglutarylation activity in vitro
Based on the structural similarity between lysine malonylation and lysine succinylation (Figure 1A), we hypothesized lysine glutarylation could be removed by SIRT5. To test this hypothesis, we first used a fluorescence-based assay to screen all HDACs (including HDAC 1–11 and Sirtuin 1–7) for lysine deglutarylation activity, as described previously (Peng et al., 2011) and illustrated in Figure S3A. Using this assay, we found SIRT5 is the only HDAC exhibiting significant lysine deglutarylase activity in vitro (Figures 3A and S3B).
Figure 3. SIRT5 catalyzes lysine deglutarylation reactions in vitro and in vivo.
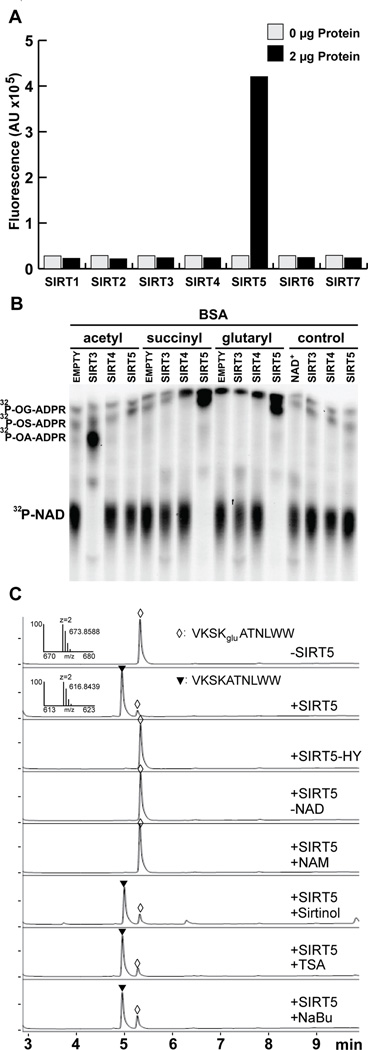
(A) Screening of HDAC lysine deglutarylation activity in vitro. Fluorometric assay to detect in vitro lysine deglutarylation activities using recombinant SIRT1-7.
(B) 32P-NAD+ consumption monitored by thin layer chromatography after an in vitro SIRT3, SIRT4, or SIRT5 enzymatic assay using chemically acylated BSA as a substrate; o-glutaryl-ADP ribose, OG-ADPR; o-succinyl-ADP ribose, OS-ADPR; o-acetyl-ADP ribose, OA-ADPR.
(C) HPLC trace of a glutarylated peptide, VKSKgluATNLWW, before and after in vitro deglutarylation reaction. The assays were carried out without hSIRT5, with hSIRT5, with enzymatically-inactive mutant of hSIRT5 (H158Y), without NAD+, with SIRT5 in the presence of nicotinamide (25 mM), or sirtinol (200 µM), or TSA (2 µM), or sodium butyrate (NaBu) (25mM). A triangle and a diamond indicate modified (HRMS, m/z, 673.8588 Da) and unmodified (HRMS, m/z, 616.8439 Da) peptides, respectively.
In a second fluorescence-independent assay, we further tested the ability of the sirtuins to deglutarylate peptide or protein substrates. We used an assay that monitors the consumption of 32P radio-labeled NAD+ by the sirtuins using two different substrates. First, acylated histone 4 (H4) peptide was generated enzymatically by incubating a peptide derived from H4 1–21 with recombinant p300 in the presence of acetyl-CoA, succinyl-CoA, or glutaryl-CoA. We observed NAD+ hydrolysis, coupled with the generation of O-acyl-ADP-ribose (OA-ADPR), indicating sirtuin catalyzed deacylation (Figure S3C). As expected, SIRT3 effectively deacetylated the acetylated peptide and SIRT5 effectively desuccinylated the succinylated peptide (Figure S3C). SIRT5 also deglutarylated the glutarylated peptide (Figure S3C). SIRT4 showed no activity against any of these modifications (Figure S3C). To test a more physiological, and more complex substrate in this assay, we chemically acylated BSA. Similar to the previous assay, we found SIRT3 deacetylated the acetyl-BSA substrate, while SIRT5 desuccinylated and deglutarylated these acylated BSA substrates (Figure 3B). These data further confirmed the deglutarylase activity for SIRT5.
The fluorescent sirtuin deacylation assay and other assays using pseudosubstrates have presented challenges to the sirtuin field. Questions about the ability of sirtuins to faithfully deacylate native peptides are often raised. Therefore, we performed a third enzymatic activity assay on known SIRT5-targeted peptides. We carried out an in vitro lysine deglutarylation reaction using the synthetic Kglu peptide, VKSKgluATNLWW, whose sequence is from DNA starvation/stationary phase protection (Dps) protein identified in our proteomic data described above. We used HPLC-MS to evaluate SIRT5-mediated lysine deglutarylation. Our data showed that SIRT5 could catalyze efficient lysine deglutarylation (Figures 3C and S3D–S3G). The reaction required NAD+ as co-factor and could be inhibited by nicotinamide (NAM) (a class III HDAC inhibitor); however, the reaction could not be inhibited using other HDAC inhibitors including trichostatin A (TSA), sodium butyrate (NaBu) (class I/II/IV HDAC inhibitors) or sirtinol (a selective SIRT1 and SIRT2 inhibitor) (Figure 3C). Additionally, the enzymatically inactive muta nt of SIRT5 (H158Y) showed no lysine deglutarylase activity (Figure 3C). Further, Sirt5 is only one of the 18 recombinant HDACs that showed significant deglutarylation activity (Figure S3G). These results demonstrate that SIRT5 can catalyze NAD+-dependent lysine deglutarylation using peptides with sites of glutarylation we identified in vivo.
Kinetic studies on SIRT5 deglutarylase activity
To define the kinetics of the deglutarylase reaction of SIRT5, we returned to the fluorescent substrates, which can be used to quantify the amount of deglutarylation over time. We first tested for its ability to process lysine side chains derivatized with dicarboxylic acids of varying lengths ranging from 3–6 carbon atoms (i.e., ε-N-malonyl-, ε-N-succinyl-, ε-N-glutaryl-, and ε-N-adipoyllysine). Two series of fluorogenic peptide substrates containing 7-amino-4-methylcoumarin (AMC) were evaluated; one series based on a simple ε-N-acetylated lysine (Figure 4A) and one containing the tripeptide sequence, Ac-Leu-Gly-Lys(acyl), which is based on H4K12 – a common substrate for deacylation (Bradner et al., 2010)(Figure S4A). Similar results were obtained for both series with high potencies of both human and murine SIRT5 recorded against succinylated and glutarylated substrates, while activities were lower against malonylated substrates (~1.3-fold relative to control wells without sirtuin), and the enzymes exhibited no measurable deacetylase or deadipoylase activity. We furthermore included an inactive histidine to tyrosine mutant of the human SIRT5, which expectedly exhibited low activity (Figure 4A). To compare the enzymatic efficiencies against different acyl groups, we then performed detailed kinetic studies using the tripeptide substrates containing malonyl-lysine (Kmal), succinyl-lysine (Ksucc), and glutaryl-lysine (Kglu) by measuring initial rate velocities as a function of substrate concentration and fitting the data to the Michaelis-Menten equation as previously described (Madsen and Olsen, 2012)(Figures S4B and Table S2A). The turnover numbers (Kcat) and catalytic efficiencies (Kcat ×Km−1) were significantly higher for desuccinylation and deglutarylation compared to the demalonylase activity in agreement with our endpoint data, and desuccinylase activity appeared ~2-fold more efficient than deglutarylase activity albeit at lower Km values for the glutarylated substrate.
Figure 4. Enzymology studies on SIRT5-catalyzed deglutarylation.
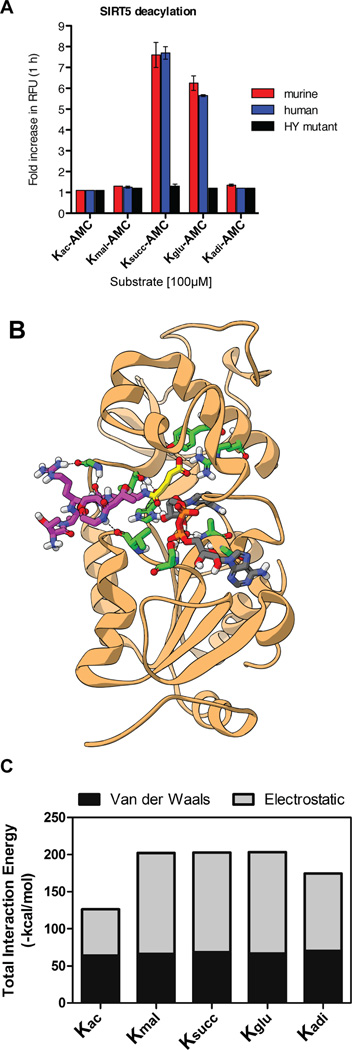
(A) Endpoint deacylation assay of mouse or human wild-type SIRT5 or human mutant SIRT5 (HY mutant) against acylated mono-lysine-AMC substrates; Kac, acetyl; Kmal, malonyl; Ksucc, succinyl; Kglu, glutaryl; Kadi, adipoyl. Error bars in this figure and subsequent ones represent standard error of the mean (SEM). (B) Crystal structure of SIRT5 with a glutarylated peptide superimposed in the catalytic pocket. (C) Summary of total interaction energy comprising the sum of Van der Waals and electrostatic interaction energies between SIRT5 and acylated histone peptides (shown in Figure S4D).
To determine if the deglutarylase activity of SIRT5 was dependent on the peptide sequence of the substrate, we measured differences in lysine deacetylation, desuccinylation and deglutarylation activities of SIRT5 using two peptide sequences we identified in our proteomics screen (Table S2B). For both of these two peptide substrates, the catalytic efficiencies for deglutarylation and desuccinylation were comparable, and significantly higher than that for deacetylation (Table S2B), which is in agreement with previous reports (Du et al., 2011; Peng et al., 2011). Together, these experiments demonstrate the major activity of SIRT5 is to remove lysine malonylation, succinylation, and glutarylation, but not acetylation.
To better understand how SIRT5 can catalyze these deacylation reactions, we performed in silico molecular modeling and simulation experiments to compare the binding interaction between SIRT5 and various acylated peptide substrates (Figures 4B, 4C, S4C–S4E, and Table S2C, detailed analyses are included in the Supplementary Experimental Procedures). The predicted energy of the interaction was essentially identical between the malonyl-, succinyl-, or glutaryl-modified histone peptides and SIRT5, but was substantially less favorable involving either the acetyl or adipoyl modifications (Figure 4C). Differences in interaction energy were almost entirely due to changes in predicted electrostatic interactions (Figure 4C). These results support our enzymatic activity data, and demonstrate how glutaryl groups can be preferably removed by SIRT5, in a similar manner as lysine succinylation and malonylation, but not acetylation or adipoylation.
SIRT5 can catalyze protein lysine deglutarylation in vivo
To validate the results of the in vitro studies, we examined lysine glutarylation and lysine succinylation in Sirt5+/+ (WT) and Sirt5−/− (KO) mouse tissues. Using mitochondrial protein lysates from tissues derived from WT and KO mice, we found that SIRT5 has a significant impact on the global lysine glutarylation state (Figures 5A and S5A–S5D). In contrast, SIRT5 has little influence on global lysine acetylation (Lombard et al., 2007). These experiments suggest that SIRT5 regulates the global mitochondrial lysine glutarylation status. Taken together, these biochemical studies reveal that SIRT5 is the only HDAC capable of acting as a lysine deglutarylase, both in vitro and in vivo. These findings also position SIRT5 to efficiently remove carboxyacyl-modifications including malonyl-lysine, succinyl-lysine, and now glutaryl-lysine.
Figure 5. Proteomic and bioinformatic analyses of Kglu substrates in WT and SIRT5KO mouse liver.

(A) Western blotting analysis of mitochondria from Sirt5+/+ (WT) and Sirt5−/− (KO) mouse livers.
(B) Distribution of Kglu sites per protein identified by proteomic analysis.
(C) Gene ontology (GO) of biological processes (BP) of proteins identified as glutarylated by proteomics.
Proteome-wide screening of Kglu substrates in Sirt5−/− mouse liver
To characterize the landscape of lysine glutarylation, we performed proteomic screening using livers from Sirt5−/− mice, which showed marked protein hyperglutarylation (Figures 5A and S5A). Using the glutaryl-peptide enrichment and mass spectrometry strategy described above, we identified 911 Kglu sites on 229 proteins with less than 1% false discovery rate (FDR) using MaxQuant software. In addition, we further filtered out those Kglu peptides with Mascot ion score lower than 20 to increase the reliability of the results. Based on these stringent criteria, 683 lysine sites in 191 proteins were identified as glutarylated and used for further bioinformatics analyses (Table S3). While approximately half of the Kglu proteins contained only one or two Kglu sites (Figure 5B), some proteins were heavily glutarylated, including carbamoyl-phosphate synthetase 1 (CPS1, 33 Kglu sites), long-chain enoyl-CoA hydratase (HADHA, 15 Kglu sites), and aspartate aminotransferase (GOT2, 14 Kglu sites) (Table S3). We realized that some Kglu sites overlap with those Kac and Ksucc sites published previously (Chen et al., 2012; Du et al., 2011; Park et al., 2013).
Bioinformatic analyses of Kglu substrates
To identify the possible association of lysine glutarylation substrates with cellular functions, we carried out gene ontology (GO) enrichment analysis using DAVID bioinformatics resources (Huang da et al., 2009). The results of GO biological process analysis showed that Kglu was significantly enriched in many cellular metabolic processes, including oxidation-reduction (p=2.6×10−39), generation of precursor metabolites and energy (p =9.2×10−27), fatty acid metabolism (p=5.0×10−20), coenzyme metabolism (p=8.4×10−18), and aerobic respiration (p=1.2×10−13) (Figure 5C and Table S4A).The enriched GO molecular functions (GOMF) are associated with various cofactor binding processes, such as coenzyme (p=8.6×10−21), FAD (p=1.8×10−11), and vitamin (p=2.2×10−9) binding; as well as oxidation-reduction related functions, including acyl-CoA dehydrogenase activity (p=8.1×10−12), NAD+/NADP+-related aldehyde/oxo group oxidoreductase activity (p=3.1×10−9), and electron carrier activity (p=3.6×10−8) (Figure S5E and Table S4B). These data reveal that protein glutarylation could be involved in regulating diverse cellular mechanisms and enzymatic processes. GO cellular compartment (GOCC) analysis showed that Kglu proteins were highly enriched in mitochondria (Figure S5F and Table S4C). Strikingly, 148 Kglu proteins in mouse liver were mainly or partially localized to mitochondria, accounting for more than three-quarters of all identified lysine-glutarylated proteins in this study. Taken together, these lines of evidence suggest a strong link between lysine glutarylation and mitochondrial metabolism. Subsequent KEGG and Pfam enrichment analyses suggested a potential impact of protein glutarylation on metabolic pathways (Figure S5G–5L and Table S4D–4E) involving glutaryl-CoA and oxidative metabolism. In addition, we also carried out analysis of protein complexes and interaction networks, using CORUM and STRING databases, identifying protein complexes whose components are enriched with Kglu (Figure S5M–5O, and Table S4F–S4G; analyses are included in the Supplementary Experimental Procedures).
SIRT5 deglutarylates CPS1 and regulates its enzymatic activity
To begin to identify the physiological consequence of protein glutarylation, we looked to the proteins with the highest number of glutarylation sites in SIRT5KO mouse livers. We found CPS1, 33 sites; HADHA, 15; GOT2, 14; MDH2, 13; SDHA, 13; ACAA2, 11; ACAT1, 11; HADH, 11; SCP2, 11; OTC, 10 (Table S3). Because CPS1 was the protein identified with the highest level of glutarylation, and SIRT5 has been previously described to regulate CPS1 (Nakagawa et al., 2009), we sought to determine if glutarylation had a functional effect on CPS1 enzymatic activity, and was regulated by SIRT5. We found CPS1 was hyperglutarylated in SIRT5KO mice during fed, and fasted (48h) conditions, consistent with our proteomic findings above (Figure 6A). Hyperglutarylation was associated with reduced hepatic CPS1 enzymatic activity in SIRT5KO mouse liver (Figure 6B), which leads to an increase in blood ammonia levels (Nakagawa et al., 2009).
Figure 6. Carbamoyl Phosphate Synthase 1 (CPS1) is targeted for deglutarylation by SIRT5.
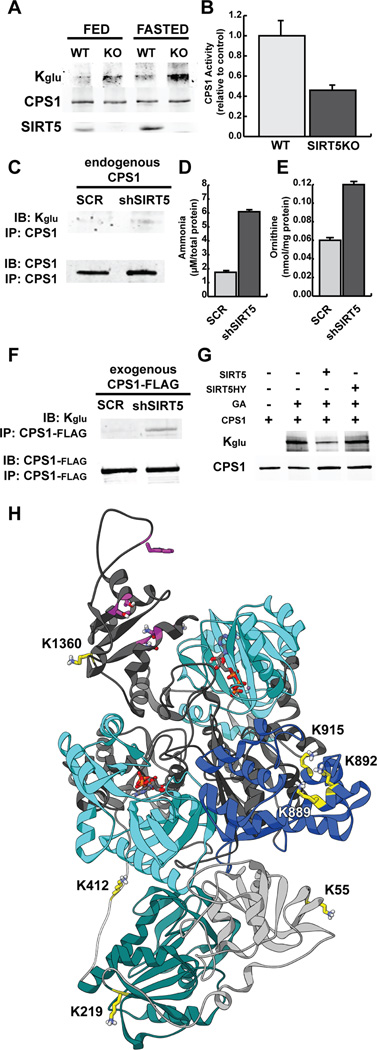
(A) CPS1 was immunoprecipitated from SIRT5 WT and KO mouse liver under fed and 48h fasted conditions and measured for glutarylation levels using an anti-Kglu antibody.
(B) CPS1 enzymatic activity was measured in hepatic lysates in SIRT5 WT and KO mice, and presented relative to WT control mice.
(C) CPS1 was immunoprecipitated from HeLa cells containing a shScramble (SCR) or shSIRT5 and measured for glutarylation levels using an anti-Kglu antibody.
(D) Ammonia levels from HeLa cells containing a shScramble (SCR) or shSIRT5 after 24h of serum starvation.
(E) Ornithine levels from HeLa cells containing a shScramble (SCR) or shSIRT5 after 24h of serum starvation.
(F) CPS1 was transfected in HEK293 cells containing a shScramble (SCR) or shSIRT5, immunoprecipitated, and measured for glutarylation levels using an anti-Kglu antibody.
(G) CPS1 was transfected in HEK293 cells and immunopurified. Glutarylation levels were measured using an anti-Kglu antibody in four samples: CPS1, glutarylated CPS1 that was chemically modified with glutaric anhydride (GA), glutarylated CPS1 after incubation with recombinant human SIRT5, glutarylated CPS1 after incubation with recombinant human SIRT5 HY mutant.
(H) A homology model of human CPS1 was built based on an E. coli CarA homologous region (light gray), encompassing an inactive glutamine amidotransferase domain (teal), and an E. coli CarB homologous region (dark gray) containing two ATP grasp domains (cyan), an oligomerization domain (blue), and a NAG-binding site (magenta). Glutarylated lysine residues targeted for SIRT5 removal are labeled and highlighted in yellow.
Because we observed basal levels of protein glutarylation in HeLa cells, we generated stable cells lines containing SIRT5 or Scramble (Scr) control shRNAs (Figure S6A). HeLa cells contain high levels of CPS1 protein (EMBL-EBI, http://www.ebi.ac.uk). Thus we attempted to immunoprecipitate endogenous CPS1 from both shSIRT5 and shScr control cell lines to determine its level of protein glutarylation. We observed CPS1 hyperglutarylation after SIRT5 knock-down compared to control (Figure 6C). Interestingly, we observed toxicity in the shSIRT5 cell culture model upon prolonged time in culture media, suggesting the shSIRT5 cells could be producing a toxic metabolite. We measured ammonia in this model after 24h of serum starvation, and observed significant increases in ammonia in the cell culture media (Figure 6D), consisted with CPS1 protein hyperglutarylation and reduced CPS1 enzymatic activity. Furthermore, we observed increased ornithine in shSIRT5 HeLa cells, supporting reduced urea/ornithine cycle flux (Figure 6E).
To further test the role of SIRT5 in deglutarylating and regulating CPS1, we generated a HEK293T containing a stable shSIRT5 or shScramble control, which do not contain endogenous CPS1. Cell lines containing the shSIRT5 had increased global protein glutarylation, but only modest increases in protein succinylation (Figure S6B). CPS1 was over-expressed in shSIRT5 and scramble control cells, and found to be hyperglutarylated (Figure 6F). Because the increases in glutarylation in these models were only associated with reduced SIRT5, we tested for the ability of SIRT5 to directly deglutarylate CPS1. To do so, we overexpressed recombinant CPS1 protein in HEK293T cells and chemically glutarylated this protein using glutaric anhydride. We then incubated glutarylated CPS1 with wild-type SIRT5, or a catalytically inactive SIRT5H158Y (SIRT5HY) mutant. We observed significant deglutarylation of CPS1 by SIRT5, but not SIRT5HY, supporting the idea that SIRT5 deglutarylates CPS1 and the absence of SIRT5 directly leads to elevated protein glutarylation (Figure 6G).
To gain insight into which Kglu sites could be targeted by SIRT5 for removal, we chemically glutarylated CPS1 and then performed an in vitro SIRT5 deglutarylation assay. We then subjected these preparations to HPLC-MS/MS analysis. We used this strategy based on the rationale that chemical glutarylation would lead to nearly complete coverage of lysine residues with the glutaryl modification, and SIRT5 would deglutarylate only specific sites, leading to a comprehensive map of deglutarylated lysines. We found 44 sites of glutarylation on chemically treated recombinant CPS1, and 31 sites of glutarylation after incubation with SIRT5 (Table S5). By comparing the change in the number of modified to unmodified CPS1 peptides after SIRT5 treatment, we identified 14 candidate lysine residues that could be efficiently deglutarylated by SIRT5 (Table S5). Finally, we compared these 14 sites to glutarylated lysines found in SIRT5KO liver, to ensure that these sites are found in a physiological setting and not an artifact of chemical acylation. In sum, we identified 8 candidate Kglu sites on CPS1 that are targeted for removal by SIRT5 (K55, K219, K412, K889, K892, K915, K1360, K1486; Table S5).
To begin to determine how glutarylation suppresses CPS1 enzymatic activity, we performed a structural analysis on CPS1 protein and identified where these eight Kglu sites were located. A human CPS1 crystal structure has not been solved, and therefore we built a homology model of CPS1 with the MODELLER protocol (Eswar et al., 2008) using the crystal structure of E. coli carbamoyl phosphate synthetase (Thoden et al., 1999) (Figure 6H).
On this homology model, we visualized the sites of glutarylation, in order to determine the possible mechanism(s) of regulation by SIRT5. K55, K219, and K412 are in or nearby the inactive glutamine amidotransferase domain in the alpha-subunit of CPS1 (Figure 6H, green). This region has been predicted to be important for CPS1 structure (Lopes-Marques et al., 2012); alternatively, these sites could be important for CPS1 interacting proteins, such as with glutaminase (Meijer, 1985). K889, K892, and K915 are all located in the CPS1 oligomerization domain, based on homology to E. coli carB (Figure 6H, blue). CPS1 is present as a homodimer, and is active in its monomeric form (Guthohrlein and Knappe, 1968). Glutarylation of these sites could disrupt the regulation of CPS1 by oligomerization. K1360 and K1486 both lie in the region of allosteric activation (Figure 6H, magenta). CPS1 requires N-acetylglutamate (NAG) as an obligate allosteric activator (Hall et al., 1958). Glutarylation of these sites could influence the activation of CPS1 by NAG, which could be reversed by SIRT5. Together, a picture emerges for SIRT5 to regulate CPS1 function by several possible mechanisms.
Protein hyperglutarylation is sensitive to dietary changes
To further explore the physiological role of glutarylation, we considered possible metabolic states that might lead to altered protein glutarylation. We monitored changes in protein glutarylation of whole cell lysate from WT and SIRT5KO mouse liver during feeding and fasting (48 hr). Similar to protein succinylation, protein glutarylation changes in response to dietary manipulation (Figures 7A and S7A). We observed, in WT mouse livers, some bands were increased with fasting, while other bands were reduced, suggesting SIRT5 could be regulating these sites (Figures 7A and S7A). Importantly, the banding pattern of protein glutarylation was different than the patterns of succinylation, suggesting that different proteins are subject to regulation by different chemical modifications. We also measured changes in acyl-CoA concentrations during a fasting time course in whole mouse liver. We found fasting induces different changes in succinyl- and glutaryl-CoA concentrations (Figure S7B), further supporting different mechanisms of regulation for different acyl-CoA species.
Figure 7. Physiology and pathophysiology of lysine glutarylation.

(A) Protein glutarylation was measured in hepatic whole-tissue lysates by immunoblotting with an anti-Kglu antibody in two sets (Mouse 1 and Mouse 2) of SIRT5 WT and KO mice that were fed or fasted (48h), respectively.
(B) Drosophilia were fed with 0X, 1X or 2X tryptophan for 1 week and whole-cell lysates were prepared for Western blotting analysis.
(C) Hepatic mitochondrial protein glutarylation and succinylation was measured by immunoblotting with anti-acyl-lysine antibodies in WT and glutaryl-CoA dehydrogenase knock-out (GCDHKO) mice.
(D) CPS1 was immunoprecipitated from GCDHKO mouse liver and measured for glutarylation levels using an anti-Kglu antibody.
(E) CPS1 enzymatic activity was measured in hepatic lysates from wild-type and GCDHKO mice and normalized to total CPS1 protein levels.
(F) Glutaryl-CoA or glutarate was incubated with heat-inactivated hepatic mitochondrial lysates and monitored for changes in protein glutarylation using an anti-Kglu antibody.
To determine if other dietary manipulations lead to changes in protein glutarylation, we performed a tryptophan supplementation experiment. As described above, tryptophan degradation leads to glutaryl-CoA, and therefore we predicted that increased tryptophan would lead to increased protein glutarylation. Drosophila were fed with 0X, 1X, or 2X tryptophan for 1 week, and whole cell lysates were measured for protein glutarylation by Western blot. We observed increases in protein glutarylation (Figures 7B and S7C), consistent with the idea that excess tryptophan is degraded and generates increased glutaryl-CoA, which can lead to increased protein glutarylation.
Protein hyperglutarylation in a mouse model of human glutaric acidemia
We also considered pathophysiological states which might be associated with altered protein glutarylation. Glutaric Acidemia I (GA, OMIM: 231670) is an autosomal recessive metabolic disorder in humans characterized by a progressive movement disorder that usually begins during the first year of life. GA is caused by homozygous or compound heterozygous mutations in the gene encoding glutaryl-CoA dehydrogenase (GCDH). Because GCDH catalyzes the oxidative decarboxylation of glutaryl-CoA into crotonyl-CoA in the lysine and tryptophan degradation pathways, and GA patients as well as GCDHKO mice display increased levels of glutaryl-CoA (Goodman and Frerman, 2001; Koeller et al., 2002), we predicted loss of GCDH would result in increased protein glutarylation. We purified hepatic mitochondria from wild-type and Gcdh−/− (GCDHKO) mice, and measured the state of protein acylation. Remarkably, we observed increased dramatic global protein glutarylation (Figure 7C). We also tested for succinylation, and saw no protein hypersuccinylation in the GCDHKO hepatic mitochondrial protein lysates (Figure 7C), demonstrating that the elevation of protein glutarylation is specific in this mouse model. Together, these data suggest that low GCDH activity could lead to elevated protein glutarylation.
To determine if protein glutarylation has a functional consequence in this mouse model, we assessed the glutarylation state of CPS1. We immunopurified CPS1 from wild-type and GCDHKO mice and found that CPS1 was hyperglutarylated in GCDHKO mouse liver (Figure 7D). We then measured CPS1 enzymatic activity in this model, and found hyperglutarylated CPS1 in GCDHKO mice was less active compared to wild-type (Figure 7E). Remarkably, GCDHKO mice express more CPS1 protein compared to wild-type (Figure S7D), presumably to overcome the reduction in protein activity and maintain nitrogen balance (Thies, 2010). These data suggest that one alternative fate of increased glutaryl-CoA in the absence of GCDH is protein glutarylation. To test if glutaryl-CoA could directly lead to non-enzymatic protein glutarylation, we incubated heat-inactivated mitochondrial protein lysates with 50 µM – 500 µM glutaryl-CoA. We observed increases in protein glutarylation with increasing glutaryl-CoA concentrations (Figures 7F and S7E), suggesting that glutaryl-CoA can lead to this protein modification, similar to acetyl-CoA, crotonyl-CoA, and succinyl-CoA (Tan et al., 2011; Wagner and Payne, 2013; Zhang et al., 2011). Purified bovine serum albumin (BSA) can also be non-enzymatically glutarylated with glutaryl-CoA (Figure S7F). Importantly, this was not observed using glutarate as a substrate for acylation (Figure 7F), which demonstrates the requirement for reactive thioesters in this pathway.
Discussion
In this study, we identified lysine glutarylation as an evolutionally conserved PTM, which is regulated by SIRT5 in a NAD+-dependent manner. SIRT5 was originally annotated as a deacetylase (Frye, 2000) and predicted to have deacetylase activity, but appears to preferentially deacylate negatively charged modifications, including malonylation, succinylation, and glutarylation. The enzymatic activity assays, substrate docking studies, and energy minimizations, all indicate that the SIRT5 catalytic pocket can accommodate these carboxyacyl-lysines for optimal enzymatic activity. Together, these findings support the idea that SIRT5 is not a physiological deacetylase, and instead removes a class of carboxyacyl-lysine modifications.
Our proteomic analyses of Kglu in SIRT5KO mouse liver led to the identification of 683 Kglu sites on 191 proteins. Interestingly, over three-quarters of the Kglu substrates were mitochondrial. Because SIRT5 plays an important role in the mitochondria (Nakagawa et al., 2009; Park et al., 2013), lysine glutarylation appears to play a major role in this organelle. Our bioinformatic analyses showed that lysine glutarylation is highly enriched with metabolic processes, such as cellular respiration, and fatty acid and amino acid metabolism, similar to what has been observed for acetylation and succinylation (Chen et al., 2012; Hebert et al., 2013; Park et al., 2013).
As a first step towards understanding the physiological role of glutarylation, we turned to the list of proteins with the highest levels of glutarylation. Not surprisingly, CPS1 was at the top of the list, which is also heavily succinylated and acetylated. Just like these other modifications, not all Kglu sites are candidates for deacylation. On CPS1, eight Kglu sites are candidates to be regulated by SIRT5, and predicted to have influences on CPS1 enzymatic activity. Indeed, in the hyperglutarylated state, we find CPS1 has reduced enzymatic activity. Future studies will identify other proteins whose activity could be regulated by SIRT5-mediated deglutarylation, and the proteomic datasets in this study will be a rich resource for future discovery.
In addition to the predominant mitochondrial Kglu substrates revealed by the proteomic analyses, we also identified at least 40 non-mitochondrial substrates, suggesting potential functions for lysine glutarylation outside mitochondria. Of particular interest, we identified three Kglu sites on core histone H2B (H2BK5, H2BK116, and H2BK120). Modifications on histones are critical for chromatin-mediated processes, including regulation of gene expression. Our previous studies identified new lysine acylation modifications on histones (Chen et al., 2007; Tan et al., 2011; Xie et al., 2012), including lysine crotonylation and succinylation, revealing potential cross-talk between cellular physiology and gene transcription.
Lysine malonylation, succinylation, and glutarylation likely play diverse roles in regulating aspects of cellular physiology. Metabolic pathways generating glutaryl-CoA are different from those generating succinyl-CoA and malonyl-CoA (Figure 1A). For example, glutaryl-CoA is an important intermediate in lysine and tryptophan metabolism, whereas succinyl-CoA participates in the TCA cycle and metabolism of branched-chain amino acids and odd-numbered fatty acids, and malonyl-CoA participates in fatty acid and polyketide biosynthesis. Thus, these different modifications are positioned to regulate different metabolic pathways.
Furthermore, glutarylation could be relevant in some disease states, such as glutaric acidemia. The GCDHKO mouse model of this disease has high levels of protein glutarylation, which could reduce the activity of important metabolic enzymes. Importantly, these findings also show that one alternate fate of unoxidized glutaryl-CoA in this disease is protein glutarylation, which could be either non-enzymatic, or enzymatically catalyzed. It is tempting to speculate that these findings are broadly applicable to other diseases associated with mutations in acyl-CoA dehydrogenases and could lead to subsequent protein acylation.
While increases in acetylation were previously associated with reduced sirtuin activity, we are now beginning to appreciate the complexity of identifying the molecular events leading to changes in acylation. In this study, for example, we find that reduction or ablation of the deacylase SIRT5 leads to increases in protein glutarylation; we also find that ablation of a protein that handles glutaryl-CoA (GCDH) leads to increases in protein glutarylation. Thus, future studies will need to consider the fate and/or flux of an acyl-CoA in contributing to the protein acylation balance.
Multiple chemical and biological differences exist among lysine acetylation, succinylation, and glutarylation. The negatively charged nature of lysine succinylation and glutarylation has the potential to induce significant structural changes on target proteins. Furthermore, it is possible that lysine glutarylation could have distinct regulatory role different from lysine succinylation or malonylation based on the larger size of the modification. Future studies will be focused on understanding the full regulatory scope of protein glutarylation, and interplay between these modifications in the rapidly expanding landscape of protein acylation.
Experimental Procedures
Western blotting analysis
Protein extracts were resolved by SDS-PAGE, transferred to polyvinylidene difluoride or nitrocellulose membrane, and probed with pan anti—acyl-lysine modification or protein sequence specific antibodies. See Supplemental Experimental Procedures for details.
Immunoprecipitation of Kglu peptides
The pH of the tryptic peptide solution was adjusted to pH 8.0 by adding 100mM NH4HCO3 buffer (pH 8.0) and then incubated with the anti-Kglu antibody (PTM Biolabs, Inc) immobilized with protein A agarose beads at room temperature with gentle shaking. After 4 hr of incubation, the beads were washed three times with pre-cold NETN buffer (50 mM Tris-Cl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) and twice with ETN buffer (50 mM Tris-Cl, pH 8.0, 100 mM NaCl, 1 mM EDTA), and once with cold water. The enriched peptides were eluted from the beads by washing with 0.1% TFA three times.
HPLC-MS/MS and MS data analysis
Enriched Kglu peptides were anaylzed by reversed-phase HPLC/MS/MS analysis by an Eksigent NanoLC-1D plus HPLC system (AB SCIEX) connect to an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher Scientific). MS/MS data were analyzed by Mascot software (version 2.1) and MaxQuant software (version 1.0.13.13). All the data were searched against either NCBI RefSeq E.coli k12 protein database (4123 sequences), IPI human protein database (v 3.70, 87069 sequences), or IPI mouse protein database (v 3.74, 56860 sequences). See Supplemental Experimental Procedures for details.
In vitro deglutarylation assays
Reactions were performed in a final volume of 50 µL per well in a 96-well microplate for fluorometric assays and in a final volume of 10 µL for peptides assays as previously described (Peng et al., 2011). See Supplemental Experimental Procedures for details.
32P-NAD+ consumption assay
Based on the method described by Du et al (Science, 2011), reactions were performed in a total volume of 10 µl in 50 mM Tris-HCl, pH 9.0, 4 mM MgCl2, 50 mM NaCl, 0.5 mM DTT, and 0.5 µCi 32P-NAD+ (PerkinElmer, NEG023X, 800 Ci/mmol) in the presence of either 2 µg acylated-BSA or 1 µg acylated-H4 peptide substrate. See Supplemental Experimental Procedures for details.
CPS1-FLAG immunoprecipitation
Transfected HEK293T cells were washed with PBS and scraped into NP-40 lysis buffer (25 mM Tris HCL, pH 7.5, 50 mM NaCl, 1% NP-40, 2 mM EDTA, and cocktail of protease inhibitors). After 15 min on ice, the cells were homogenized in chilled racks with a TissueLyser bead mill (Invitrogen) for 2 min at 30 Hz. Lysates were centrifuged at 14,000 × g, 20 min, and the supernatants incubated with FLAG M2 antibody resin (Sigma-Aldrich) overnight at 4 °C. The FLAG resin was washed 3X with lysis buffer, 1X with TBS, and the CPS1-FLAG protein eluted with 300 ng/µl FLAG peptide (Sigma-Aldrich).
CPS1 activity assay
CPS1 activity was assessed by measuring converted citrulline by a colorimetric method with modification (Chan et al., 2009; Pierson, 1980). See Supplemental Experimental Procedures for details.
In vitro deglutarylation of CPS1 by SIRT5
Glutarylated and unglutarylated CPS1 were incubated with SIRT5 or the catalytically inactive SIRT5HY mutant in 1x SDAC Buffer (50 mM Tris-HCl pH 9, 4 mM MgCl2, 50 mM NaCl, 0.5 mM DTT, 1 mM NAD+) for one hour at 37 °C. Reaction was stopped by adding SDS sample buffer or by freezing at −20°C.
Animal experiments
Animal studies were performed according to protocols approved by the University of Michigan or Duke University. See Supplemental Experimental Procedures for details.
See Supplementary Experimental Procedures for other experiments.
Supplementary Material
Highlights.
Lysine glutarylation is a protein post-translational modification;
SIRT5 can catalyze the enzymatic removal of lysine glutarylation;
Proteomic analyses identify a link between lysine glutarylation and metabolism;
Glutarylation suppresses CPS1 activity, which is targeted by SIRT5 for removal.
Acknowledgements
We thank Leonard Guarente for the mouse SIRT5 antibody; we are grateful to Dr Gozde Colak for critically reading this manuscript; we thank Ailan Guo and Cell Signaling Technology for helpful discussions. This work was supported by NIH grants GM105933, CA160036, and RR020839 to Y.Z., and GM101171 and CA177925 to D.B.L. Y. Z. is also supported by the Nancy and Leonard Florsheim Family Fund. M. T. are supported by the National Science and Technology Major Project of the Ministry of Science and Technology of China (2012ZX09301001-007), Natural Science Foundation of China (31370814), and Shanghai Pujiang Program (13PJ1410300). M.D.H. and his laboratory are supported by the American Heart Association grants 12SDG8840004 and 12IRG9010008, The Edward Mallinckrodt Jr. Foundation, The Ellison Medical Foundation, the National Institutes of Health (AA022146 and AG045351), and the Duke O'Brien Center for Kidney Research (5P30DK096493-02); M.F.G is supported by a post-doctoral fellowship from the NIH and NCI (CA059365-19). J.P. was supported by NIH training grant (AG000114). C.A.O and his laboratory are supported by the Danish Independent Research Council–Technology and Production Sciences (Sapere Aude Grant No. 12-132328; A.S.M.), the Danish Independent Research Council–Natural Sciences (Steno Grant No. 10-080907), the Villum Foundation, and the Carlsberg Foundation. C.A.O. is a Lundbeck Foundation Fellow. C.M. and J.S. are supported by the German Research Foundation (Deutsche Forschungsgemeinschaft grant MU1778/3-1). This work utilized the resources of the Drosophila Aging Core of the Nathan Shock Center of Excellence in the Biology of Aging (NIA: P30-AG-013283).
Abbreviations
- PTM
Protein post-translational modification
- HDAC
Histone lysine deacetylase
- KDAC
Lysine deacetylase
- FDR
False discovery rate
- Kglu
Lysine glutarylation
- Kac
Lysine acetylation
- Kbu
Lysine butyrylation
- Kmal
Lysine malonylation
- Ksucc
Lysine succinylation
- NAD+
Nicotinamide adenine dinucleotide
- CoA
Coenzyme A
- MEF
Mouse embryonic fibroblast
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- Chan CH, Ramirez-Montealegre D, Pearce DA. Altered arginine metabolism in the central nervous system (CNS) of the Cln3−/− mouse model of juvenile Batten disease. Neuropathol Appl Neurobiol. 2009;35:189–207. doi: 10.1111/j.1365-2990.2008.00984.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Kwon SW, Kim SC, Zhao Y. Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J Proteome Res. 2005;4:998–1005. doi: 10.1021/pr049754t. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao W, Yang JS, Cheng Z, Luo H, Lu Z, Tan M, Gu W, Zhao Y. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. 2012;11:1048–1062. doi: 10.1074/mcp.M112.019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- Furuya E, Uyeda K. Regulation of phosphofructokinase by a new mechanism. An activation factor binding to phosphorylated enzyme. J Biol Chem. 1980;255:11656–11659. [PubMed] [Google Scholar]
- Goodman SI, Frerman FE. Organic acidemias due to defects in lysine oxidation: 2-ketoadipic acidemia and glutaric acidemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. In The Metabolic and Molecular Bases of Inherited Disease. New York, USA: McGraw Hill; (2001). [Google Scholar]
- Guthohrlein G, Knappe J. Structure and function of carbamoylphosphate synthase. I. Transitions between two catalytically inactive forms and the active form. Eur J Biochem. 1968;7:119–127. doi: 10.1111/j.1432-1033.1968.tb19582.x. [DOI] [PubMed] [Google Scholar]
- Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230:1013–1021. [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Koeller DM, Woontner M, Crnic LS, Kleinschmidt-DeMasters B, Stephens J, Hunt EL, Goodman SI. Biochemical, pathologic and behavioral analysis of a mouse model of glutaric acidemia type I. Hum Mol Genet. 2002;11:347–357. doi: 10.1093/hmg/11.4.347. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Papandrea JD, Mitzen EJ. Fatty acid biosynthesis in Wallerian degeneration of rat sciatic nerve. Muscle Nerve. 1979;2:369–375. doi: 10.1002/mus.880020508. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Marques M, Igrejas G, Amorim A, Azevedo L. Human carbamoyl phosphate synthetase I (CPSI): insights on the structural role of the unknown function domains. Biochem Biophys Res Commun. 2012;421:409–412. doi: 10.1016/j.bbrc.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen AS, Olsen CA. Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme. J Med Chem. 2012;55:5582–5590. doi: 10.1021/jm300526r. [DOI] [PubMed] [Google Scholar]
- Meijer AJ. Channeling of ammonia from glutaminase to carbamoyl-phosphate synthetase in liver mitochondria. FEBS Lett. 1985;191:249–251. doi: 10.1016/0014-5793(85)80018-1. [DOI] [PubMed] [Google Scholar]
- Mitzen EJ, Koeppen AH. Malonate, malonyl-coenzyme A, and acetyl-coenzyme A in developing rat brain. J Neurochem. 1984;43:499–506. doi: 10.1111/j.1471-4159.1984.tb00927.x. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111 012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson DL. A rapid colorimetric assay for carbamyl phosphate synthetase I. J Biochem Biophys Methods. 1980;3:31–37. doi: 10.1016/0165-022x(80)90004-4. [DOI] [PubMed] [Google Scholar]
- Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J Biol Chem. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies N. Untersuchungen zur induzierten metabolischen Krise im Tiermodell der Glutarazidurie Typ I. Medical Faculty of the University of Hamburg; 2010. [Google Scholar]
- Thoden JB, Wesenberg G, Raushel FM, Holden HM. Carbamoyl phosphate synthetase: closure of the B-domain as a result of nucleotide binding. Biochemistry. 1999;38:2347–2357. doi: 10.1021/bi982517h. [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Widespread and Enzyme-independent N{epsilon}-Acetylation and N{epsilon}-Succinylation of Proteins in the Chemical Conditions of the Mitochondrial Matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. 2012;198:155–164. doi: 10.1083/jcb.201202056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.