

Abstract
Variants within the leucine-rich repeat kinase 2 gene are recognized as the most frequent genetic cause of Parkinson’s disease. Leucine-rich repeat kinase 2 variation related to susceptibility to disease displays many features that reflect the nature of complex late-onset sporadic disorders, such as Parkinson’s disease. The Genetic Epidemiology of Parkinson’s disease consortium recently performed the largest genetic association study for variants in the leucine-rich repeat kinase 2 gene across 23 different sites in 15 countries. Herein we detail the allele frequencies for the novel risk factors (p.A419V and p.M1646T) and the protective haplotype (p.N551K-R1398H-K1423K) reported in the original publication. Simple population allele frequencies can not only provide an insight into the clinical relevance of specific variants but also help genetically define patient groups. Establishing individual patient-based genomic susceptibility profiles incorporating both risk and protective factors will determine future diagnostic and treatment strategies.
Keywords: Parkinson disease, LRRK2, genetics, association study
As we enter an era of personalized medicine, defined by the individual genomic profile, it will be critical that we understand the independent and joint influences of disease-associated genetic variation1, 2. Determining appropriate genetic testing and understanding the ramifications of results will decide the utility of such approaches from a diagnostic and prognostic viewpoint. The interpretation of clinical genetic testing may be most difficult with regards to late-onset sporadic disorders such as Parkinson’s disease (PD), where a number of genetic loci have been nominated to alter disease risk, including highly penetrant mutations co-segregating with disease in families and common less penetrant risk factors3.
Recently, the Genetic Epidemiology Of Parkinson’s Disease (GEO-PD) consortium performed a large case-control study evaluating the associations of 121 different rare and common coding variants in the leucine-rich repeat kinase 2 (LRRK2) gene with susceptibility to PD4. Our study was comprised of a total of 8611 PD cases and 6929 controls representing three ethnicities (Caucasian, Asian, and Arab-Berber). The results of the study nominated new risk factors, p.M1646T in the Caucasian series and p.A419V in the Asian series, as well as a protective haplotype (p.N551K-R1398H-K1423K) across all 3 series’4, 5. In our analyses, we provided odds ratio (OR) estimates of association utilizing a number of different statistical models involving combined data from 23 GEO-PD sites. Herein, we provide population-specific frequencies which were not previously presented for p.M1646T, p.A419V, or the protective haplotype. These simple allele and haplotype frequencies can be equally helpful in the interpretation of results and in determining the clinical relevance of each variant.
As displayed in Figure 1a, the minor allele (C) for p.M1646T had a maximum frequency of 2.85% in patients with PD and 2.55% in controls for any individual country, and was more common in patients than controls for 11 of the 13 countries it was observed in (Figure 1a). The p.A419V substitution was more common in patients with PD compared to controls for the each Asian country with minor allele frequencies ranging from 0.17% to 2.94% (Figure 1b). The frequency of the protective p.N551K-R1398H-K1423K haplotype varied between countries within patients (3.01% to 10.64%) and controls (4.26% to 14.39%) (Figure 1c). The lower haplotype frequency in patients was observed for the majority of countries, with the most discrepant results occurring for the two smallest populations. Individual population frequencies and their 95% CIs are provided in Supplementary Tables 1a-1c along with the originally presented population-specific ORs1 and previously unreported population-specific p-values for association.
Figure 1.
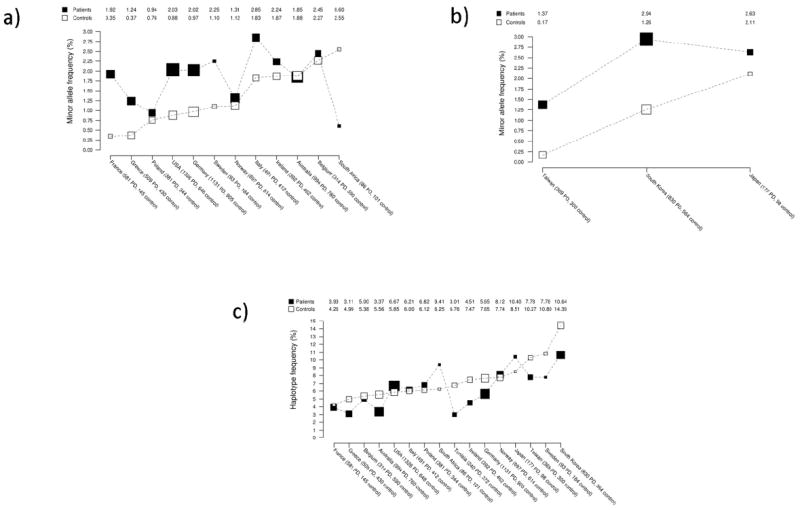
Population-specific allele frequencies.
A) Minor allele (C) frequency of p.M1646T in patients with PD and controls according to country. Larger boxes indicate larger sample sizes. Countries are ordered according to minor allele frequency (lowest to highest) in controls. Minor allele frequencies are connected by dashed lines within patients with PD and controls in order to enhance visual display.
B) Minor allele (T) frequency of p.A419V in patients with PD and controls according to country in the Asian series. Larger boxes indicate larger sample sizes. Countries are ordered according to minor allele frequency (lowest to highest) in controls. Minor allele frequencies are connected by dashed lines within patients with PD and controls in order to enhance visual display.
C) Frequency of the protective p.N551K-R1398H-K1423K (G-A-A) haplotype in patients with PD and controls according to country. Larger boxes indicate larger sample sizes. Countries are ordered according to haplotype frequency (lowest to highest) in controls. Haplotype frequencies are connected by dashed lines within patients with PD and controls in order to enhance visual display.
Of the PD genes that have been identified thus far, LRRK2 is particularly important owing to the relatively high frequency of its mutations, involvement in both familial and sporadic disease, and potential as a therapeutic target. As we have presented here, the newly identified risk-modifying susceptibility variants p.A419V, p.M1646T, and the p.N551K-R1398H-K1423K haplotype are apparent across a range of population-specific frequencies. Key next steps will involve determining the frequency of these variants in other populations and ethnicities as well as evaluating interactions with other PD susceptibility variants.
As further loci and functional variation influencing disease susceptibility are identified it will be important to assess the combined effects and determine individual genetic risk scores based on informed genetic evaluations and population ethnicity. For example, to date it appears that the genetic risk associated with the MAPT locus is not present in Asian populations; LRRK2 harbors both Asian and Caucasian ethnic-specific risk factors, while the LRRK2 protective haplotype and variants at SNCA appear to be relevant across a number of diverse populations4, 6, 7. In addition, given the late-onset sporadic nature of PD it will also be crucial to determine the joint effects of the different genetic loci identified to influence PD risk. Another recent study by the GEO-PD consortium evaluated the independent and joint effects of disease-associated genetic variation at the SNCA and MAPT loci in Caucasian populations1. The results of this study showed that individuals who harbored the risk allele at both genes had an increased risk of PD under an additive model, with no gene-gene interaction observed.
Identifying those individuals at risk of PD will require a paradigm shift in the diagnostic setting, combining clinical genetic testing and premotor symptom evaluation. Akin to the story of statins within the setting of cholesterol and vascular disease, intervention strategies for PD may need to be implemented well in advance of symptomatic presentation. Recent studies in Alzheimer’s disease have highlighted that the disease pathology may start anywhere up to 25 years before the manifestation of cognitive issues 8. Unfortunately to date however, no effective biomarker for disease progression has been identified for PD and this makes identification of early disease presentation or the effectiveness of intervention therapies difficult to interpret.
Genetic discrimination of patients will not only be crucial for preclinical diagnostics but will also play a critical role in the development of therapeutics and disease intervention strategies. The design of clinical drug trials has largely been based on the clinical manifestation of the motor symptoms and is characterized by the use of patients with PD who are in the advanced disease state. If the pathology associated with PD has initiated decades preceding the clinical manifestation of the movement disorder, these patients may not represent the group most suited to drug intervention. A two-pronged approach of symptomatic relief and neuroprotective strategies may need to be implemented well in advance of the clinical presentation, and those compounds tested to date may have been administered too late in the disease course. In addition, we may also find that the genetic discrimination of patients may also identify those at highest risk of developing therapeutic-related complications, e.g. dyskinesia and impulse control disorders, looking to pharmacogenomics to pave the way in drug administration.
Furthermore, to date, clinical drug trials have not been fully informed, i.e. have not utilized genetically homogenous populations for specific targeted therapies. For example, it is likely that not every PD patient will benefit from LRRK2 inhibition. As highlighted in our study the protective p.N551K-R1398H-K1423K haplotype is present in some PD patients. If, as presumed, the toxic mechanism underlying LRRK2 disease is an increase in kinase activity it would support evidence that the protective haplotype lowers kinase activity5. Under these circumstances use of a LRRK2 inhibitor may be prove ineffectual and perhaps even damaging given recent insights from LRRK2 knockout model studies9, 10. Therefore the design of LRRK2 inhibitor clinical trials should use the fundamental understanding we have of individual genomics and develop inclusion/exclusion criteria based on genetic understanding of disease risk. This scenario also holds true for development of potential α-synuclein knockdown studies based on the toxic over-expression hypothesis11-13. Indeed combinatorial drug approaches combining targeted therapies may present the most effective action.
The use of next-generation sequencing technologies has exploded over the last few years. In 2011, we witnessed the first PD gene identified by using these methods14, 15. As the approaches of whole-exome and whole-genome sequencing become both more affordable and commercially available many more individuals will present at the clinic with these data. This will see an increase in the number of potential PD genes nominated and a vast quantity of rare variants in both novel and known PD loci that will need to be interpreted. Large multi-ethnic studies such as performed by the GEO-PD consortium on variants in LRRK2, SNCA and MAPT will be required to fully understand the role of each of these genes in PD and the clinical impact each will have.
Supplementary Material
Supplemental File 1.
Table of country-specific allele frequencies and association results.
Supplemental File 2.
Consortia list of GEO-PD member sites.
Supplemental File 3.
GEO-PD member site acknowledgements.
Acknowledgments
The present study and original funding for the GEO-PD Consortium was supported by grants from Michael J. Fox Foundation. Original funding for the GEO-PD was borted by a grant from The Michael J. Fox Foundation for Parkinson’s Research Edmond J. Safra Global Genetics Consortia program. Mayo Clinic is a Morris K. Udall Center of Excellence in PD Research (P50 NS072187). Finally we would like to acknowledge all the patients and control subjects who kindly donated DNA to make collaborative studies like these possible.
References
- 1.Elbaz A, Ross OA, Ioannidis JP, et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann Neurol. 2011;69(5):778–792. doi: 10.1002/ana.22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8(3):e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundal C, Fujioka S, Uitti RJ, Wszolek ZK. Autosomal dominant Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S7–10. doi: 10.1016/S1353-8020(11)70005-0. [DOI] [PubMed] [Google Scholar]
- 4.Ross OA, Soto-Ortolaza AI, Heckman MG, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol. 2011;10(10):898–908. doi: 10.1016/S1474-4422(11)70175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan EK, Peng R, Teo YY, et al. Multiple LRRK2 variants modulate risk of Parkinson disease: a Chinese multicenter study. Hum Mutat. 2010;31(5):561–568. doi: 10.1002/humu.21225. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377(9766):641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 8.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinkle KM, Yue M, Behrouz B, et al. LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor coordination behaviors. Mol Neurodegener. 2012;7(1):25. doi: 10.1186/1750-1326-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107(21):9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maraganore DM, de Andrade M, Elbaz A, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296(6):661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 12.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 13.Ross OA, Braithwaite AT, Skipper LM, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63(6):743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vilarino-Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimprich A, Benet-Pages A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1.
Table of country-specific allele frequencies and association results.
Supplemental File 2.
Consortia list of GEO-PD member sites.
Supplemental File 3.
GEO-PD member site acknowledgements.