

Abstract
A novel lactonase from Mycoplasma synoviae 53 (MS53_0025) and Mycoplasma agalactiae PG2 (MAG_6390) was characterized by protein structure determination, molecular docking, gene context analysis, and library screening. The crystal structure of MS53_0025 was determined to a resolution of 2.06 Å. This protein adopts a typical amidohydrolase (β/α)8-fold and contains a binuclear zinc center located at the C-terminal end of the β-barrel. A phosphate molecule was bound in the active site and hydrogen bonds to Lys217, Lys244, Tyr245, Arg275, and Tyr278. Both docking and gene context analysis were used to narrow the theoretical substrate profile of the enzyme, thus directing empirical screening to identify that MS53_0025 and MAG_6390 catalyze the hydrolysis of d-xylono-1,4-lactone-5-phosphate (2) with kcat/Km values of 4.7 × 104 and 5.7 × 104 M–1 s–1 and l-arabino-1,4-lactone-5-phosphate (7) with kcat/Km values of 1.3 × 104 and 2.2 × 104 M–1 s–1, respectively. The identification of the substrate profile of these two phospho-furanose lactonases emerged only when all methods were integrated and therefore provides a blueprint for future substrate identification of highly related amidohydrolase superfamily members.
Enzymes of the amidohydrolase superfamily (AHS) are characterized by a distorted (β/α)8-barrel fold and a shared catalytic reaction mechanism, in which an activated water or hydroxide attacks an electrophilic center.1,2 This family of enzymes has 21762 (59%) functionally unassigned sequences and a broad substrate profile, encompassing diverse substrates such as amino acids and organophosphate esters with 42 unique reactions,3 ranging from hydrolysis, deamination, decarboxylation, isomerization, hydration to retroaldol cleavage.1 This broad spectrum of substrates and mechanisms has challenged automated function assignment by sequence identity.4 It has been proposed that as many as 40% of enzymes that are recognized as members of the AHS have been mis-annotated for function by sequence similarity alone.5,6 There are even cases of mis-annotation when structural information is available.7,8 Therefore, it has been suggested that the similarity of computationally predicted active site pockets is a more accurate method for classifying enzymes into functional families,9 while docking studies can provide substrate candidates.7,10−15 However, in the amidohydrolase superfamily, remarkable cases in which enzymes with 98% sequence identity turn over different substrates, such as atrazine versus melamine, have occurred, making functional identification even more difficult.16,17
An example of the broad substrate range of the AHS is illustrated by one of its subclasses, Cog1735 (Figure 1), which is one of 24 clusters of orthologous groups (COG) within the amidohydrolase superfamily. Cog1735 itself may be divided into nine subclusters (Figure 1A), each of which is predicted to be iso-functional, as illustrated by subgroup 3. Three homologous enzymes in this cluster, from Rhodococcus erythropolis, Mycobacterium tuberculosis, and Mycobacterium avium subsp. paratuberculosis K-10, have been tested, and all three have been identified as N-acyl-homoserine lactonaes.18−20 Other functionally annotated enzymes within Cog1735 have been shown to catalyze the hydrolysis of phosphotriesters (subgroup 8),21 and γ- and δ-lactones (subgroups 9 and 7).13,22,23 Recently, we have demonstrated that enzymes from subgroup 6 of Cog1735, Lmo2620 and BH0225, belong to the phosphotriesterase-like lactonase superfamily and catalyze the hydrolysis of d-lyxono-1,4-lactone-5-phosphate and l-ribono-1,4-lactone-5-phosphate.14 However, enzymes from subgroups 1, 2, 4, and 5 have no activity assigned to them, even though representative members have had their structures determined to atomic resolution [Protein Data Bank (PDB) entries 1BF6, 3RHG, 3K2G and 3MSR/3OVG, respectively].
Figure 1.
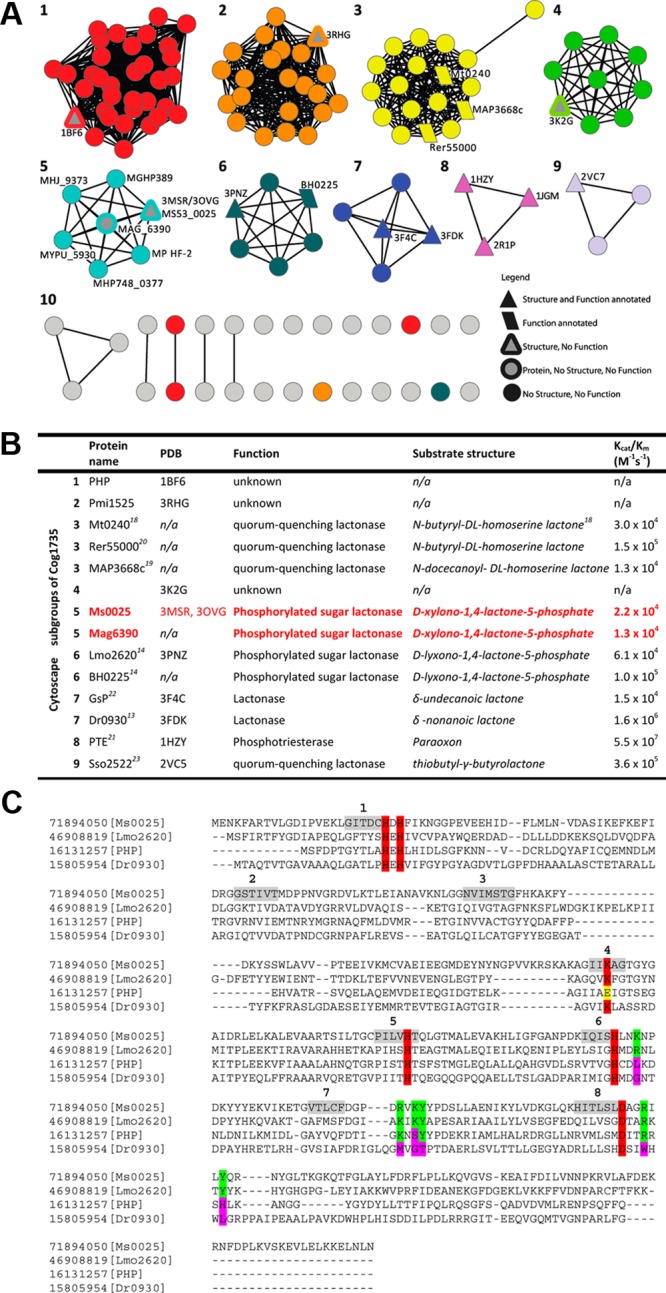
Sequence similarity network organization showing one of the 24 clusters representing the amidohydrolase superfamily sequences annotated as belonging to Cog1735. (A) Each node in the diagram represents a sequence, and each edge represents the pairwise connection between two sequences at a BLAST E value of better than 10–80. All functionally annotated proteins (trapezoids) as well as structurally characterized proteins (triangles) have been mapped onto the nodes. Ms0025 (cyan and gray triangle) and Mag6390 (cyan and gray circle) are found in subgroup 6. (B) List summarizing each of the known proteins, structures, substrate profiles, and kinetic parameters for each node. Colored red are Ms0025 and Mag6390, which are the phosphorylated sugar lactonases that are able to hydrolyze d-xylono-1,4-lactone-5-phosphate. (C) Structure-based sequence alignment of Ms0025 (gi|71894050, subgroup 6), Lmo2620 (gi|46908819, subgroup 5), phosphotriesterase homology protein (gi|16131257, subgroup 1), and Dr0930 (gi|15805954, group 7). The amino acids required for metal binding are highlighted in red. The residues implicated in phosphate binding are highlighted in green and the corresponding residues in the three other Cog1735 proteins in pink. Regions highlighted in gray represent the strands of the barrel fold.
Here we investigate the function of two unannotated proteins of the amidohydrolase superfamily from subgroup 5 of Cog1735: MAG_6390 from Mycoplasma agalactiae PG2 and MS53_0025 from Mycoplasma synoviae 53 (Table S1 of the Supporting Information). The three-dimensional structure of one of these, MS53_0025, was determined to a resolution of 2.06 Å. Guided by docking screens of metabolite10,24 and focused carbohydrate libraries, gene context, and structural similarity to other enzymes, we show that both MS53_0025 and MAG_6390 are lactonases that hydrolyze both d-xylono-1,4-lactone-5-phosphate (2) and l-arabino-1,4-lactone-5-phosphate (7) with kcat/Km values in the range of 104 M–1 s–1. These enzymes are thus a possible system of catabolism of small 1,4-lactone-5-phosphate sugars (Scheme 1), which are a potential energy source for these organisms.
Scheme 1.

Materials and Methods
Materials
Compounds 1–10 and 12–23 (Scheme 2) were synthesized as previously described.14d-Glucono-1,5-lactone-6-phosphate (compound 11) was synthesized using yeast hexokinase to phosphorylate d-glucono-1,5-lactone with MgATP. The reaction was conducted in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.0) with 20 mM MgATP and 20 mM d-glucono-1,5-lactone for up to 3 h at 30 °C. The reaction was initiated with hexokinase from Saccharomyces cerevisiae (6 mg/mL) and followed by 13C nuclear magnetic resonance (NMR) spectroscopy with a Bruker Avance III 500 MHz NMR spectrometer equipped with an H–C–N cryoprobe. The phosphorylated sugar lactone was stable at pH 6.0 in 50 mM MES buffer for at least 3 h after completion of the reaction. The pH was increased to 7.0, the reaction mixture passed through a 30 kDa ultrafiltration membrane to remove the hexokinase, and then activated charcoal added to remove ATP and ADP. All other buffers, purification reagents, and chemicals used in this work were purchased from Sigma-Aldrich, unless otherwise stated
Scheme 2.

Cloning, Expression, and Purification of Ms0025 for Crystallography
The gene for MS53_0025 (denoted Ms0025 hereafter) from M. synoviae (gi|71894050) was codon-optimized for expression in Escherichia coli, chemically synthesized (GenScript, Inc., Piscataway, NJ), and cloned into a custom TOPO isomerase vector, pSGX3(BC), supplied by Life Technologies. The plasmid was designated NYSGXRC-9686b. The clone encodes Met-Ser-Leu followed by the polymerase chain reaction (PCR) product and finally Glu-Gly-His6. Miniprep DNA was transformed into BL21(DE3)-Codon+RIL expression cells (Stratagene), expressed, and made into a 30% glycerol stock for large scale fermentation.
Selenomethionine-labeled protein was produced for Ms0025 using High Yield SeMet medium (Orion Enterprises, Inc., Northbrook, IL) supplemented with ZnCl2. Overnight cultures of expression glycerol stocks were grown in LB medium at 30 °C with kanamycin and chloramphenicol selection. Large scale cultures (1 L volume in 2 L baffled shake flasks) were inoculated with 10 mL of saturated overnight cultures per liter. These cultures were grown for 3 h at 37 °C, and then the temperature was decreased to 22 °C for postinduction growth for 18 h. Cultures were harvested using standard centrifugation procedures and frozen prior to purification.
Cells were lysed by sonication in a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 25 mM imidazole, and 0.1% Tween 20. The cellular debris was removed by centrifugation for 30 min at 39800g. The supernatant solution was loaded onto an ÄKTAxpress purification system (GE Healthcare) in which nickel affinity and size exclusion chromatography were utilized sequentially using a 5 mL HisTrap HP column and a Superdex S200 16/60 column (GE Healthcare). Nickel binding buffer containing 50 mM Tris (pH 8.0), 500 mM NaCl, 25 mM imidazole, and 10% glycerol was used to equilibrate the HisTrap column and to wash the unbound proteins. The nickel elution buffer contained 50 mM Tris (pH 8.0), 500 mM NaCl, 500 mM imidazole, and 10% glycerol. The size exclusion chromatography buffer contained 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) (pH 7.5), 150 mM NaCl, 10% glycerol, and 5 mM dithiothreitol (DTT). Fractions were pooled on the basis of retention times and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) results. The protein concentration was 12.7 mg/mL based on the absorbance at 280 nm. The purity of the protein was >95%, and the protein identity was confirmed by electrospray mass spectrometry. The expression plasmid (NYSGXRC clone 9686b2BCt7p1) is available through the PSI Material Repository (http://psimr.asu.edu/). The DNA sequence and experimental details are available in the TargetTrack Database (http://sbkb.org/tt/) as Project Target “NYSGXRC-9686b”.
Purification of Ms0025 and Mutants for Activity Screening
The site-directed single mutants of Ms0025 (K29A, E34Q, and H100N) were constructed using the QuikChange mutagenesis kit from Stratagene. The coding regions of all mutated plasmid DNA were verified by complete sequencing, and stocks were prepared as previously noted. To express Ms0025 and variants, BL21(DE3) star cells (Novagen) were transformed with the plasmid containing the gene. A single freshly transformed colony was cultured in LB medium supplemented with 50 μg/mL kanamycin at 37 °C overnight. The overnight culture was inoculated into 1 L of LB medium and cultured at 30 °C while being vigorously shaken. When the OD600 reached 0.6, the expression of the target protein was induced with 0.5 mM isopropyl d-thiogalactopyranoside (IPTG). At this point, 1.0 mM Zn(OAc)2 was added, and the cells were further cultured at room temperature for 18 h and then harvested by centrifugation. A total of 8 g of frozen cells was resuspended in 40 mL of binding buffer [20 mM HEPES and 500 mM NaCl (pH 7.9)] and lysed by sonication at 0 °C. After centrifugation, the nucleic acids were removed by adding 2% (w/v) protamine sulfate. The supernatant solution after centrifugation was filtered with a 0.2 μm syringe filter (VWR) and loaded on a HisTrap HP column preloaded with nickel. The protein was eluted with a linear gradient of elution buffer [20 mM HEPES, 250 mM NaCl, and 500 mM imidazole (pH 7.9)]. The crude protein obtained from the HisTrap HP column was concentrated and loaded onto a HiLoad 26/60 Superdex 200 gel filtration column (Amersham Pharmacia) for removal of imidazole and further purification.
Cloning, Expression, and Purification of Mag6390
The gene encoding MAG_6390 (denoted Mag6390 hereafter) from M. agalactiae PG2 was codon optimized, synthesized, and cloned into pUC57 (GenScript). Expression constructs were generated by PCR amplification using 5′-TTAAGAAGGAGATATACCATGGCTAAAGACAAGTTCGTCCGCAC-3′ as the forward primer and 5′-GATTGGAAGTAGAGGTTCTCTGCCAGCAGTTTCAGATCCTTCTTCAG-3′ as the reverse primer. PCR was performed using KOD Hot Start DNA Polymerse (Novagen). The conditions were as follows: 2 min at 95 °C followed by 40 cycles of 30 s at 95 °C, 30 s at 66 °C, and 30 s at 72 °C. The amplified fragment was cloned into the C-terminal TEV cleavable StrepII-6x-His-tag-containing vector, CHS30, by ligation-independent cloning.25
Expression vectors were transformed into E. coli strain BL21(DE3) containing the pRIL plasmid (Stratagene) and used to inoculate 25 mL of PAG medium with 25 μg/mL kanamycin and 34 μg/mL chloramphenicol. The culture was allowed to grow overnight at 37 °C in a shaking incubator. Twenty milliliters of the overnight culture was used to inoculate 2 L of PASM-5052 autoinduction medium (Studier) containing 150 mM 2,2-bipyridyl, 1.0 mM ZnCl2, and 1.0 mM MnCl2. The culture was placed in a LEX48 airlift fermenter and incubated at 37 or 22 °C overnight. The culture was harvested and pelleted by centrifugation.
Cells were suspended in lysis buffer containing 20 mM HEPES (pH 7.5), 500 mM NaCl, 20 mM imidazole, and 10% glycerol and lysed by sonication. Lysates were clarified by centrifugation at 35000g for 30 min. Proteins were purified on an ÄKTAxpress FPLC system (GE Healthcare). Clarified lysates were loaded onto a 1 mL HisTrap HP column (GE Healthcare), washed with 10 column volumes of lysis buffer, and eluted with 20 mM HEPES (pH 7.5), 500 mM NaCl, 500 mM imidazole, and 10% glycerol. The purified sample was loaded onto a HiLoad S200 16/60 HR gel filtration column, which was equilibrated in 20 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, and 5 mM DTT. Peak fractions were collected, and protein was analyzed by SDS–PAGE. Samples were concentrated to 7.5 mg/mL using Amicon Ultra centrifugal filters (Millipore), snap-frozen in liquid nitrogen, and stored at −80 °C.
Metal Analysis
The protein concentrations were determined spectrophotometrically at 280 nm using a 1 cm path length quartz cuvette with a SPECTRAmax-340 UV–vis spectrophotometer. The extinction coefficient at 280 nm was calculated to be 26610 M–1 cm–1 for Ms0025 and 31080 M–1 cm–1 for Mag6390 (http://ca.expasy.org/tools/protparam.html). The metal content of the purified protein was determined using inductively coupled plasma emission mass spectrometry (ICP-MS). Before ICP-MS measurements, the protein samples were treated with concentrated nitric acid for 20 min at 100 °C and then diluted with distilled H2O until the final concentration of nitric acid was 1%. The average metal contents of Ms0025 and Mag6390 were 1.6 and 1.5 Zn2+ ions per subunit, respectively.
Determination of the Structure of Ms0025
The purified protein was concentrated to ∼10 mg/mL and subjected to sitting-drop vapor diffusion in a 96-well plate by mixing 1 μL of the protein solution and 1 μL of the reservoir solution from the Index and Crystal screen of Hampton Research. The apoprotein with phosphate in the active site formed cubic-shaped crystals overnight from 0.2 M potassium sodium tartrate, 0.1 M sodium citrate (pH 5.6), and 2.0 M ammonium sulfate. Ms0025 was also crystallized in 2.4 M sodium malonate (pH 7.0). This protein contained two zincs and one phosphate in the active site. Crystals were cryoprotected by adding 20% (v/v) glycerol to the mother liquor and flash-frozen by being directly immersed in liquid nitrogen for data collection.
X-ray diffraction data were collected at the selenium absorption edge (λ = 0.9795 Å) at beamlines X12C and X29 at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory. Data were processed with HKL2000 or MOSFLM/SCALA.26−28 Ms0025 (PDB entry 3MSR) crystallized in hexagonal space group P6322 with one molecule per asymmetric unit and diffracted to at least 2.16 Å resolution. The structure was determined by AUTOSOL,29 followed by autobuilding in ARP/wARP.30 The second structure of Ms0025 (PDB entry 3OVG) was determined by molecular replacement using PHASER with the native (PDB entry 3MSR) structure as the search model.31 This protein crystallized in the triclinic space group with six molecules in the asymmetric unit and diffracted to 2.06 Å resolution. Atomic models for both structures were subsequently manually adjusted using COOT.32 Both structures were refined to convergence with PHENIX.33 Data collection and refinement statistics are summarized in Table 1.
Table 1. Data Collection and Refinement Statistics.
| Ms0025 | [Zn/Zn]-Ms0025 | |
|---|---|---|
| PDB entry | 3MSR | 3OVG |
| Data Collection | ||
| wavelength (Å) | 0.9795 | 0.9795 |
| resolution (Å) | 2.16 | 2.06 |
| outer shell resolution (Å) | 2.28–2.16 | 2.13–2.06 |
| space group | P6322 | P1 |
| cell dimensions | ||
| a, b, c (Å) | 111.18, 111.18, 138.23 | 89.26, 89.19, 96.06 |
| α, β, γ (deg) | 90,0, 90.0, 120 | 98.25, 92.89, 119.86 |
| no. of molecules per asymmetric unit | 1 | 6 |
| redundancy [overall (outermost shell)] | 8.0 (7.9) | 3.9 (3.9) |
| I/σ(I) [overall (outermost shell)] | 15.4 (7.1) | 11.77 (3.84) |
| Rmergea [overall (outermost shell)] | 0.132 (0.49) | 0.106 (0.347) |
| completeness (%) [overall (outermost shell)] | 99.9 (98.3) | 99.8 (96.3) |
| no. of reflections | 27521 | 152064 |
| Refinement | ||
| resolution range (Å) | 50–2.16 | 50–2.06 |
| no. of reflections | 27521 | 151931 |
| completeness (work + test) (%) | 99.4 | 99.49 |
| Rfactorb (%) | 15.2 | 17.4 |
| Rfreec (%) | 19.2 | 21 |
| no. of protein atoms | 2789 | 16647 |
| no. of water atoms | 295 | 185 |
| no. of ligand atoms | 11 | 42 |
| root-mean-square deviation for bonds (Å) | 15.38 | 23.13 |
| root-mean-square deviation for angles (deg) | 0.02 | 0.01 |
| Ramachandran plot analysis (%) | ||
| most favored region (additionally allowed) | 94.83 (5.17) | 95.43 (4.57) |
| disallowed region | 0 | 0 |
Rmerge = ∑j|Ih – ⟨Ih|⟩/∑Ih, where ⟨Ih⟩ is the average intensity over symmetry equivalents.
Rfactor = ∑|Fobs – Fcalc|/∑|Fobs|.
Rfree is the same as Rfactor but for the test set.
Kinetic Measurements and Data Analysis
The reaction rates were measured with a SPECTRAmax-340 plate reader. The hydrolysis of lactones was monitored using a pH-sensitive colorimetric assay. The net production of protons was measured using the pH indicator cresol purple.14 The reactions were conducted in 2.5 mM Bicine buffer (pH 8.3) containing 0.2 M NaCl and the compound (0.01–2 mM) to be tested as a potential substrate. The cresol purple concentration was 0.1 mM in 1% Dimethyl sulfide (DMSO), and the change in absorbance monitored at 577 nm. The effective extinction coefficient (ΔOD per mole of H+) was obtained from the titration with acetic acid at 577 nm and found to be 1.76 × 103 M–1 cm–1. The protein was stored in 50 mM HEPES (pH 7.5) and then exchanged with 10 mM Bicine buffer (pH 8.3) using a PD-10 desalting column. A background rate was observed in the absence of substrate because of the acidification by atmospheric CO2 and was subtracted (5–10 mOD/min) from the initial rates of the enzymatic reactions. The hydrolysis of compound 23 was monitored by 13C NMR using a Bruker Avance III 500 MHz NMR spectrometer equipped with an H–C–N cryoprobe at pH 7.0 using a Ms0025 concentration of 3 mg/mL. No hydrolysis product was detected after incubation with enzyme for 1 h at pH 7.0.
Kinetic parameters were obtained by fitting the initial rates directly to eq 1
| 1 |
where ν is the initial velocity, Et is the enzyme concentration, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant.
Docking Screens
The Ms0025 protein structure (PDB entry 3MSR) was prepared for initial docking using previously described protocols.7,10,13,14,24 To optimize the system, the charges on the two Zn2+ ions were reduced to +1.3 for Znα and +1.4 for Znβ, while the charges of the coordinating Nε atoms of His24, His26, and His214 and Nδ of His186 were increased by 0.25 each. To balance the system, the net charge on Asp272 was increased by 0.1 and that on the carbamylated Lys153 was increased by 0.2, as has been previously employed in docking to amidohydrolases.7,24 After the second structure of Ms0025 (PDB entry 3OVG) had been determined, chain B was used for docking. It was the chain with the lowest average B factor per atom (19.3 Å2), and it had well-defined density around the Znα and Znβ atoms and the catalytically important water (H2O-1198). The active site for docking was based on the positions of water molecules and the distal phosphate molecule.
Two libraries were used for virtual screening. First, a specially constructed high-energy intermediate (HEI) library that was based on the 5/6-membered phosphorylated sugar lactones or acid sugar lactones (Scheme S1 of the Supporting Information). This library contained 12 furanose- and pyranose-based scaffolds that expanded to 216 unique enantiomers. These molecules were used to create high-energy state compounds in which the catalytically active center has formed a bond with the hydroxide. Compounds in this library were automatically generated and can undergo either a hydrolysis or dephosphorylation reaction with different protonation profiles, creating 1728 or 112 HEI molecules, respectively. Each of these molecules was represented by up to 10 different conformations, generating a final library of 4316 high-energy state compounds for docking, termed the SUG-HEI library. The second library is our standard in-house library consisting of 57672 high-energy intermediate KEGG molecules (HEI-KEGG) that is based on unique metabolites from the Kyoto Encyclopedia of Genes and Genomes database of compounds (KEGG),34,35 which contain a reactive center annotated for the AHS mechanism.13,24
Molecular docking was performed using DOCK3.636 as with previous docking campaigns,7 using receptor and ligand bin sizes of 0.4 Å, an overlap of 0.1–0.2 Å, a bump allowance of 3, and a distance tolerance of 1.5 Å with label matching turned off. Finally, each molecule was subjected to 250 cycles of rigid-body minimization (we note that the high bump allowance is ultimately resolved by rigid-body minimization). The top 500 scoring molecules with a maximal distance of 4 Å between the reactive center of the HEI molecule and the zinc metal center were inspected visually.
Results
Determination of the Structure of Ms0025
The three-dimensional structure of the apo form of Ms0025 (PDB entry 3MSR) and its structure in complex with the binuclear metal center (PDB entry 3OVG) were determined at resolutions of 2.16 and 2.06 Å, respectively. Both structures have one phosphate ion bound at a distal part of the active site. The zinc-bound structure has two metal ions bound in the active site in all protomers. The six molecules in the asymmetric unit agree with a root-mean-square deviation (rmsd) in the range of 0.12–0.21 Å for all Cα pairs. The overall structures of the apo and zinc-bound proteins are nearly identical with an rmsd of 0.21 Å. The Ms0025 structure has an (β/α)8-barrel fold with antiparallel β-strands formed at the C-terminal end of the protein (Figure 2). There are additional antiparallel β-strands inserted between (αβ)3 and (αβ)4. The binding sites for the two zinc ions and the phosphate molecule are located at the C-terminal end of the (β/α)8-barrel.
Figure 2.
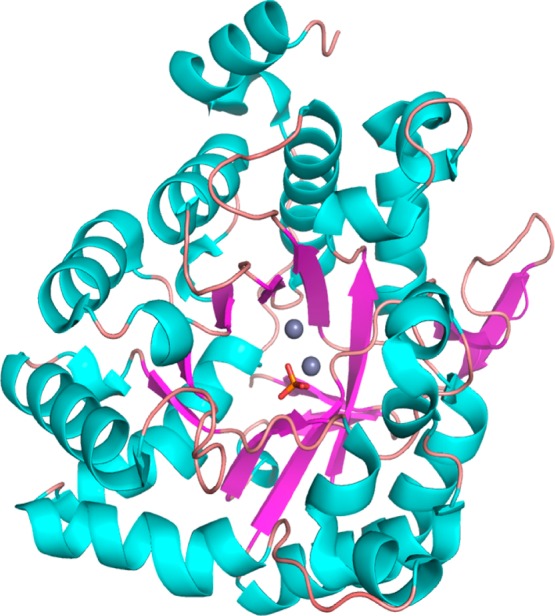
Ribbon representation of the three-dimensional structure of the (β/α)8-barrel fold of a single chain of Ms0025 (PDB entry 3MSR/3OVG). The two Zn2+ metals are colored gray, and the phosphate ligand is colored orange. The central β-barrel is colored purple, and the helices are colored cyan.
The distance between the two zinc ions is 3.6 Å. Lys153 is carboxylated (Kcx153) and coordinates with both zinc ions. The β-zinc has a tetrahedral geometry with Kcx153, His186, His214, and a water molecule, while the α-zinc is coordinated by His24, His26, Kcx153, Asp272, and two water molecules (Figure 3 and Table 2). A similar zinc coordination geometry is found in urease, phosphotriesterase, and other (β/α)8-barrel metalloproteins from the amidohydrolase superfamily.37,38 In both of the structures reported here, a phosphate molecule is bound at the C-terminal opening of the (β/α)8-barrel. The distances between the phosphorus and the two zinc ions are 8.7 and 8.8 Å. The phosphate is hydrogen bonded to Tyr245, Arg275, Lys244, Tyr278, Lys217, and two water molecules (Figure 3), and these residues are conserved in both Ms0025 and Mag6390 enzymes, and the Lmo2620 enzyme (PDB entry 3PNZ), and could indicate a conserved residue cluster for phosphate binding (Figure S2 of the Supporting Information).
Figure 3.
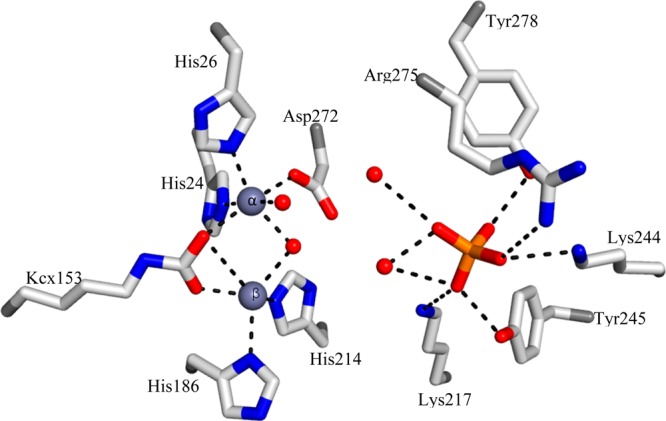
Binuclear metal center of Ms0025 (PDB entry 3OVG). Structural model for the active site showing residues that coordinate the binuclear metal center. All contacts between the two divalent zinc ions (gray spheres), water (red spheres), or phosphate (orange sticks) and active site residues (white sticks) are indicated with dashed lines. The α-zinc is coordinated by His24, His26, Kcx153, Asp272, and two water molecules, while the β-zinc exhibits tetrahedral geometry and is coordinated by Kcx153, His186, His214, and the bridging water molecule.
Table 2. Bond Lengths between Zinc or Phosphorus and Protein or Water.
| ligand–Znα | distance (Å) | ligand–Znβ | distance (Å) | ligand–PO43– | distance (Å) |
|---|---|---|---|---|---|
| His24 CE1 | 2.39 | His186 ND1 | 2.43 | Tyr245 OH | 2.66 |
| His26 NE2 | 2.29 | His214 NE2 | 2.39 | Arg275 NH1 | 3.43 |
| Asp272 OD1 | 2.22 | Kcx153 OQ1 | 2.20 | Lys244 NZ | 2.88 |
| Kcx153 OQ2 | 2.39 | H2O-1198 | 2.48 | Tyr278 OH | 2.66 |
| H2O-1198 | 2.35 | Lys217 NZ | 3.43 | ||
| H2O-1197 | 2.31 | H2O-946 | 3.22 | ||
| H2O-1206 | 2.41/3.12 |
Substrate Specificity and Kinetic Measurements
We began by interrogating the catalytic profile of Ms0025 and Mag6390, which share a sequence identity of 80% (Table S1 of the Supporting Information), by physically screening a focused library of phosphorylated and unphosphorylated lactones. The following is a list of candidate substrates that were chosen on the basis of previously observed activities for amidohydrolases in Cog1735 (Figure 1B), all of which were found to be inactive against Ms0025. For phosphotriesterase and phosphodiesterase activity: diethyl 4-nitrophenyl phosphate, ethyl 4-nitrophenyl phosphate, bis(4-nitrophenyl) phosphate, and 4-nitrophenyl phosphate. For quorum sensing activity: (S)-α-amino-γ-butyrolactone, (R)-α-amino-γ-butyrolactone, and N-butyryl-dl-homoserine lactone. For lactonase activity: β-propiolactone, β-butyrolactone, γ-butyrolactone, α-methyl-γ-butyrolactone, (±)-γ-valerolactone, (R)-γ-valerolactone, γ-caprolactone, γ-heptalactone, γ-nonanoic lactone, (S)-4-nonanolide, dl-α-hydroxy-β,β-dimethyl-γ-butyrolactone, d-gulonic acid γ-lactone, d-(+)-glucuronic acid γ-lactone, l-gulonic acid γ-lactone, δ-valerolactone, 5,6-dihydro-2H-pyran-2-one, 2H-pyran-2-one, δ-hexalactone, δ-nonalactone, (±)-5-decanolide, d-(+)-gluconic acid δ-lactone, and ε-caprolactone (Figure S1 of the Supporting Information). Unexpectedly, Ms0025 is not an archetypal AHS phosphotriesterase or lactonase, as it is unable to hydrolyze simple 1,4- or 1,5-lactones, nor was it able to hydrolyze N-acyl homoserine lactones. This prompted us to seek other clues about enzyme activity.
The Gene Context of Ms0025 Links It to Sugar Pathway Enzymes
The Ms0025 gene is located immediately upstream of a sugar phosphotransferase system (PTS), which is annotated as an ascorbate-specific transporter. All three genes, ulaABC, which comprise a system used to phosphorylate and translocate ascorbate across the cytoplasmic membrane, are adjacent to Ms002539 (Figure 4). All but one gene associated with the ascorbate PTS from E. coli K12 st., MG1655, is conserved in M. synoviae 53, including the genes ulaR, ulaD, ulaE, and ulaF(40) (Figure 4). This part of the PTS system is used to convert the 3-keto-l-gulonate-6-phosphate to d-xylulose-5-phosphate for use as a carbon source in the E. coli K12 pentose phosphate pathway.41 The only substitution in this cluster is ulaG, which hydrolyzes intracellular ascorbate-6-phosphate (10) to 3-keto-l-gulonate-6-phosphate.42,43 The UlaG enzyme is replaced in M. synoviae 53 by Ms0025. This led us to believe that we were looking for a phosphorylated substrate for Ms0025. Accordingly, ascorbate-6-phosphate (10) was assayed as a substrate of Ms0025 but demonstrated no turnover. Interestingly, even though the overall folds of UlaG and Ms0025 differ, both have a binuclear metal cluster and exhibit the same 3-fold symmetry (Figure 5). Because UlaG hydrolyzes a furanose ring of a phosphorylated 1,4-lactone, this observation nucleated our interest in a computational docking screen of metabolites and helped us to interpret the results.
Figure 4.
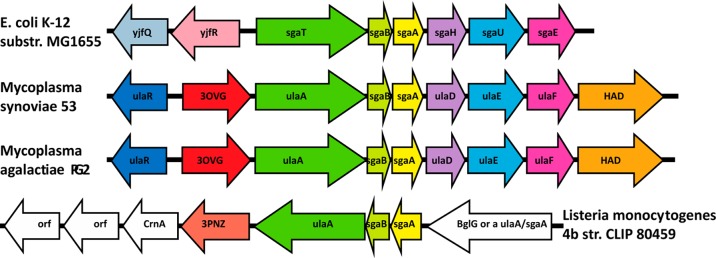
Gene organization of the operon and ulaA–G regulon involved in l-ascorbate metabolism in E. coli, which encodes a phosphotransferase system (PTS) (ulaABC genes). The putative functional analogues of the ula genes from M. synoviae 53, M. agalactiae PG2, and Listeria monocytogenes str. 4b are shown in the same colors. In red are the lactonases from M. synoviae 53, M. agalactiae PG2, and L. monocytogenes str. 4b. The gene names in order of M. synoviae 53 are ulaR/yjfq/rpiR transcriptional repressor for the l-ascorbate utilization ula divergon; Ms0025 hypothetical protein (phosphorylated sugar lactonase); ulaA/sgaT PTS system ascorbate-specific transporter subunit II, C component; sagB PTS system, IIB component, pentitol phosphotransferase enzyme II, B component; sgaA pentitol phosphotransferase enzyme II, A component; ulaD/sgaH 3-keto-l-gulonate-6-phosphate decarboxylase; ulaE/sgaUl-xylulose 5-phosphate 3-epimerase; ulaF/sagE/araDl-ribulose-5-phosphate 4-epimerase; MS53_0032, haloacid dehalogenase superfamily subclass Iib sugar phosphatase.
Figure 5.
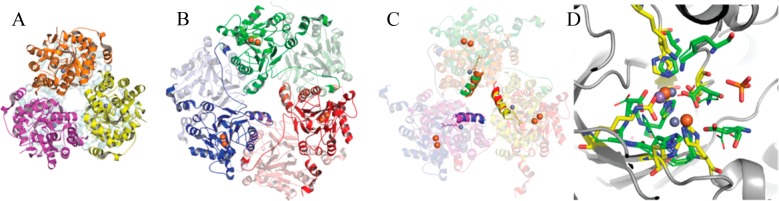
Comparison of lactonase structures of Ms0025 (PDB entry 3OVG) and UlaG (PDB entry 3BV6/2WYL). (A) Ribbon representation of the hexamer of Ms0025 that is seen in the crystal structure as a dimer (light blue) of trimers (yellow, pink, and orange). The metal ions at each active site are shown as gray spheres. The molecules are oriented with the 3-fold axis perpendicular to the plane of the page. (B) UlaG hexamer viewed down the molecular 3-fold axis, which is a trimer of dimer (red, blue, and green and dimer chain in light colors). (C) Superposition of Ms0025 and UlaG based on the superposition of the helix around the central solvent pore in UlaG (green → orange, red → yellow, blue → pink). The two zinc atoms for each protein are depicted as spheres and show a different position orientation of the active site. (D) Superposition of the holoenzyme active sites of Ms0025 and UlaG. Active site residues are represented as yellow sticks for Ms0025 and green sticks for UlaG, with Zn2+ shown as gray spheres and Mn2+ and Fe3+ shown as purple and orange spheres, respectively. In addition to metal-coordinating residues, the location of the side chain of Lys29 (Ms0025) and Lys260 (UlaG) is shown, which appears to be in a favorable position for substrate interaction, albeit coming from a different direction.
Docking to the Ms0025 Structure
We screened molecules from the KEGG library that can undergo the known amidohydrolase reactions in high-energy intermediate geometries against the apo structure, which allows for flexibility of the binding site and poses an additional challenge in identifying the natural substrate.12 In this screen, furanose sugar lactone compounds were highly ranked in catalytically competent geometries. Among these, d-galactono-1,4-lactone ranked 270 of 57672 molecules screened and was identified as a possible substrate given the gene context considerations (Figure 6A). Encouragingly, in the docked pose of this lactone, all the donor and acceptor groups interact with active site residues. The hydroxyls at positions 2 and 3 on the lactone ring interact with Lys29 and Glu34. The hydroxyl at position 5 forms a hydrogen bond with Lys217. In addition, the orientation of the lactone allows enough space for the superposition of the crystallographic phosphate that is coordinated by Lys244, Tyr245, Tyr278, and Arg275. The gap between the distal oxygen at position 6 of the lactone and the phosphate in this structural superposition is 1.5 Å, which is the length of a covalent bond, suggesting that a phosphorylated d-galactono-1,4-lactone could fit into the active site comfortably (Figure 6A).
Figure 6.
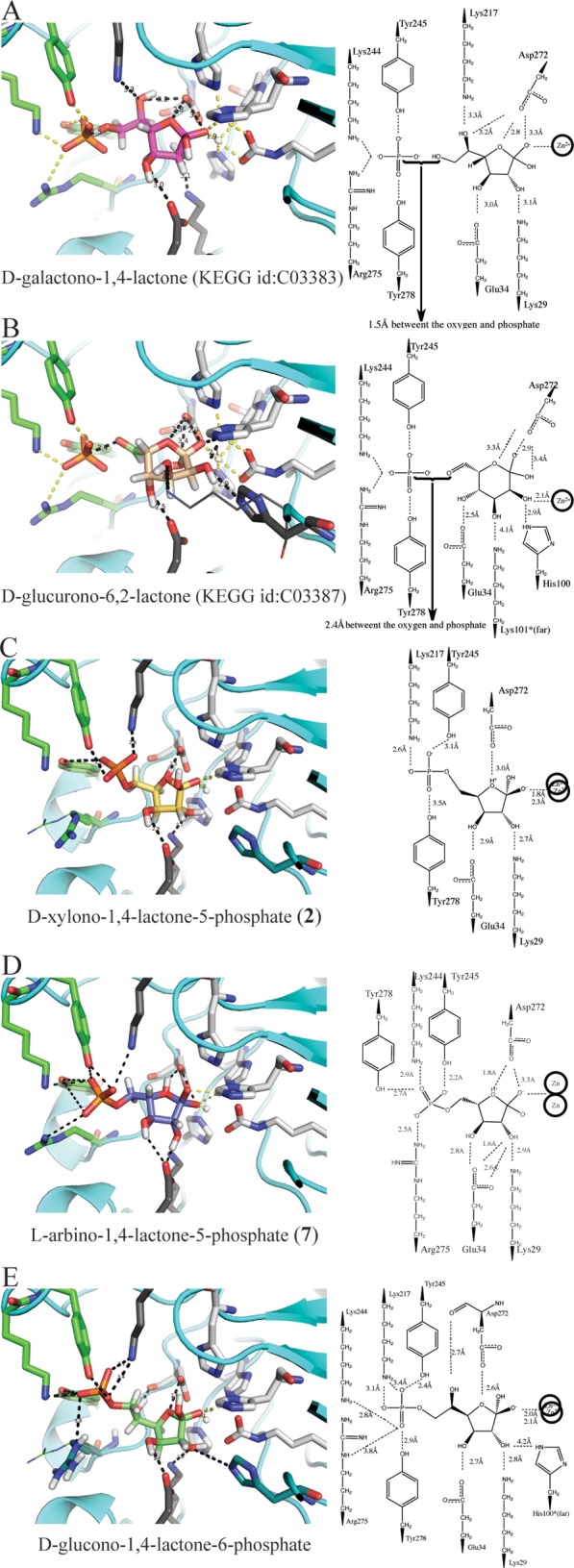
Docked compounds. (A) d-Galactono-1,4-lactone (KEGG entry C03383, pink sticks) and (B) d-glucurono-6,2-lactone (KEGG entry C03387, salmon sticks) in the active site of Ms0025 (PDB entry 3OVG), which were the initial hits from the KEGG HEI library. These are superposed with the phosphate (orange sticks) bound in the crystal structure (green sticks). On the right is a two-dimensional representation of the binding site. (C) d-Xylono-1,4-lactone-5-phosphate (2) (yellow sticks) and (D) l-arabino-1,4-lactone-5-phosphate (7) (purple sticks) show the docked pose from the dedicated SUG-HEI library (Mol-07 in Scheme S1 of the Supporting Information) that represent the best known substrates that are hydrolyzed by Ms0025 and Mag6390. (E) d-Glucono-1,4-lactone-6-phosphate (green sticks) docking pose from the dedicated SUG-HEI library (Mol-08 in Scheme S1 of the Supporting Information), in which the molecule represents the longer extension of the phosphate. For all panels, polar contacts are shown as dashed lines, white sticks represent the metal-chelating residues, dark gray sticks represent residues that were mutated, and green sticks represent residues that interact with the phosphate in the crystal structure.
A subsequent docking screen was undertaken with 12 different phosphorylated and carboxylated furanose and pyranose compounds (Scheme S1 of the Supporting Information). Once each chiral center had been enumerated, 216 unique molecules were generated, which led to the creation of a library of 4316 focused SUG-HEI compounds. This library was docked, and even though we started with only two phosphorylated five-membered sugar lactones (24 ground state isomers), they populated 28% of the top 100 docking list of candidate substrates for Ms0025. Furthermore, furanose sugars (Figure 6C,E and Table S2 of the Supporting Information) were preferentially docked over pyranose sugars. Indeed, 95 of the top 100 scoring molecules contained a furanose; additionally, phosphorylated pyranose sugars were observed only 13 times in the top 500 list. Candidate substrates with a two-carbon linker between the furanose and the phosphate were accommodated well in the active site. This longer linker was able to place the phosphate groups within hydrogen bonding distance of all residues that line the distal pocket at the rim of the active site: Arg275, Tyr278, Lys277, and Tyr245. An example of a well-posed larger phosphorylated furanose compound is d-glucono-1,4-lactone-6-phosphate (Figure 6E), which could in theory be hydrolyzed to produce 6-phospho-d-gluconate. However, these molecules could not be synthesized for enzymatic analysis. Defining the preferred stereochemistry at positions 2 and 3 of the furanose ring was more difficult. Many stereoisomers had broadly similar ranks, with the possible exception of the C4 position with the R conformation, which was preferred in 85% of the furanose molecules ranked in the top 100 molecules of the SUG-HEI library.
Kinetic Measurements of Sugar Lactones
On the basis of the docking screens, the positioning of the crystallographic phosphate in the active site of Ms0025, and the structural similarity to Lmo2620, we inferred that Ms0025 and Mag6390 may recognize phosphorylated or carboxylated sugar lactones.14 Therefore, the eight stereoisomers of 1,4-pentolactone-5-phosphate (1–8) were synthesized and assayed for activity. Of these, d-xylono-1,4-lactone-5-phosphate (2) and l-arabino-1,4-lactone-5-phosphate (7) were confirmed as substrates, with kcat/Km values of 4.7 × 104 and 5.7 × 104 M–1 s–1 to kcat/Km values of 1.3 × 104 and 2.2 × 104 M–1 s–1 for Ms0025 and Mag6390, respectively (Table 3). These two compounds have the same stereochemical configuration at C2 and C3 but the opposite stereochemistry at C4. No activity could be detected with 2-deoxy-d-ribonolactone-5-phosphate (9), l-ascorbate-lactone-6-phosphate (10), or d-glucono-1,5-lactone-6-phosphate (11). The two enzymes were also screened with a series of lactones made from diacid sugars (compounds 12–19 and 20–23). Hydrolytic activity was found with compounds 12 (racemic mixture with 16), 20 (racemic mixture with 21), and 22 (Table 3 and Table S2 of the Supporting Information). In the docked poses, the furanose rings of the substrates were positioned similarly, with the reactive lactone bond positioned directly above the divalent metal center within hydrogen bonding distance of Asp227. This allows nucleophilic attack of the hydroxide from the si face of the lactone ring. To allow for the differing stereochemistry at C4, in the docking poses the furanose ring puckers in the opposite directions and twists slightly while maintaining the same contacts between the hydroxyls at C2 and C3 and the active site residues Glu34 and Lys29 (Figure 6C,D).
Table 3. Kinetic Parameters for Ms0025 and Mag6390.
| substrate | kcat (s–1) | Km (mM) | kcat/Km (M–1 s–1) |
|---|---|---|---|
| Ms0025 | |||
| 2 | 23.4 ± 0.8 | 0.50 ± 0.04 | (4.7 ± 0.4) × 104 |
| 7 | 6.7 ± 0.2 | 0.30 ± 0.02 | (2.2 ± 0.2) × 104 |
| 12 | 2.9 ± 0.2 | 1.0 ± 0.1 | (2.9 ± 0.3) × 103 |
| 20 | 7.1 ± 0.5 | 2.5 ± 0.3 | (2.8 ± 0.4) × 103 |
| 22 | 5.6 ± 0.4 | 1.7 ± 0.2 | (3.3 ± 0.4) × 103 |
| Mag6390 | |||
| 2 | 51 ± 3 | 0.9 ± 0.1 | (5.7 ± 0.7) × 104 |
| 7 | 8.7 ± 0.3 | 0.66 ± 0.06 | (1.3 ± 0.1) × 104 |
| 12 | 8.0 ± 0.5 | 3.1 ± 0.3 | (2.6 ± 0.3) × 103 |
| 20 | 4.7 ± 0.2 | 4.3 ± 0.4 | (1.1 ± 0.1) × 103 |
| 22 | 15.3 ± 0.9 | 5.0 ± 0.4 | (3.1 ± 0.3) × 103 |
Hydrolysis of the Racemic Mixture of Compounds 20 and 21
Stereospecific hydrolysis of phosphorylated 1,4-lactones (2 and 7) versus any other enantiomer (1, 3–6, and 8) suggests that Ms0025 and Mag6390 have a strict requirement for the orientation of the hydroxyl groups at C2 and C3 of the sugar ring. For the corresponding diacid sugar lactones (12–21), it was not possible to physically separate the racemic pairs of compounds. To demonstrate that a single isomer was hydrolyzed in the racemic mixture of 20 and 21, 1H NMR spectroscopy was used to monitor the rate of hydrolysis until it reached completion. In this reaction, 0.2 μM Ms0025 was mixed with compounds 20 and 21 (5.0 mM each) in 20 mM Pipes buffer (pH 6.1). The 1H NMR spectrum of the reaction was monitored and recorded every 10 min. The formation of the hydrolyzed product was monitored at 3.8 ppm, and the disappearance of the substrate was monitored at 4.1 ppm (Figure 7). Only one of the two isomeric compounds was hydrolyzed, showing a preference of C2 (R) and C3 (R).
Figure 7.

1H NMR spectral results indicating as a percentage the formation of the hydrolyzed product and the disappearance of the substrate, demonstrating a single isomer hydrolysis in the mixture of compounds 20 and 21.
Mutational Analysis
Amino acids that were not conserved between Ms0025/Mag6390 and Lmo2620 and demonstrated interactions with the docked compounds were mutated (K29A, E34Q, and H100N), with possibly the greatest impact being attributed to the K29A mutation (Figure S2 of the Supporting Information). Interestingly, these mutations did not change the substrate profile of the Ms0025 protein, though the catalytic rates were reduced (Table 4). The change in the catalytic efficiency can be mostly attributed to the decrease in the turnover rate, possibly indicating that these residues are required for the correct orientation of the substrate, for stabilization of the tetrahedral intermediate, or for the protonation of the hydrolyzed lactones and therefore do not change the substrate specificity.
Table 4. Kinetic Parameters for Ms0025 Mutants.
| substrate | protein | kcat (s–1) | Km (mM) | kcat/Km (M–1 s–1) |
|---|---|---|---|---|
| 2 | wild-type | 23.4 ± 0.8 | 0.5 ± 0.04 | (4.7 ± 0.4) × 104 |
| 2 | K29A | 1.1 ± 0.6 | 1.4 ± 0.1 | (7.9 ± 0.7) × 102 |
| 2 | E34Q | 0.9 ± 0.05 | 0.7 ± 0.1 | (1.3 ± 0.1) × 103 |
| 2 | H100N | 6.0 ± 0.3 | 2.7 ± 0.2 | (2.2 ± 0.2) × 102 |
| 7 | wild-type | 6.7 ± 0.2 | 0.30 ± 0.02 | (2.2 ± 0.2) × 104 |
| 7 | K29A | 0.42 ± 0.02 | 0.75 ± 0.07 | (5.6 ± 0.6) × 102 |
| 7 | E34Q | 0.58 ± 0.03 | 1.8 ± 0.2 | (3.2 ± 0.3) × 102 |
| 7 | H100N | 0.64 ± 0.02 | 0.83 ± 0.06 | (7.7 ± 0.6) × 102 |
| 22 | wild-type | 5.6 ± 0.4 | 1.7 ± 0.2 | (3.3 ± 0.4) × 103 |
| 22 | K29A | 0.11 ± 0.1 | 2.1 ± 0.2 | (5.2 ± 0.7) × 10 |
| 22 | E34Q | 0.40 ± 0.02 | 3.8 ± 0.3 | (1.1 ± 0.1) × 102 |
| 22 | H100N | 0.23 ± 0.01 | 1.5 ± 0.1 | (1.5 ± 0.1) × 102 |
Discussion
Ms0025 and Mag6390 are characteristic enzymes of subgroup 5 of Cog1735 of the amidohydrolase superfamily, for which no activity was previously known (Figure 1 and Table S1 of the Supporting Information), which we propose to classify as phospho-furanose lactonases. In retrospect, this may seem unsurprising, as among the characterized enzymes closest in sequence and structure to Ms0025 and Mag6390 is Lmo2620 [31% identical (Figure 1C and Figure S2 of the Supporting Information)], which hydrolyzes d-lyxono-1,4-lactone-5-phosphate (1) and l-ribono-1,4-lactone-5-phosphate (8), which closely resemble substrates d-xylono-1,4-lactone-5-phosphate (2) and l-arabino-1,4-lactone-5-phosphate (7), respectively (Table 3). However, neither Ms0025 nor Mag6390 can hydrolyze the substrates of Lmo2620, nor are these proteins active on phosphotriesters or simple lactones characteristic of other subclasses of Cog1735 or against a broader screen for lactone substrates, including β-lactones, γ-lactone, δ-lactone, ε-lactones, and cyclic monosaccharides (furanose and pyranose) (Figure S3 of the Supporting Information). Indeed, the fidelity to substrate stereochemistry is tight for these enzymes, whose substrates and nonsubstrates are separated only by the stereochemistry around one of the lactone ring hydroxyl groups. It was only when we turned to an integrated analysis of gene context, docking screens of candidate metabolites, and enzyme structure that the true substrates of these enzymes began to emerge (Figure 6C,D and Table S2 of the Supporting Information).
The genetic context of the two enzymes suggested a role in an ascorbic acid pathway, recognizing sugar phosphates. Whereas this was misleading in itself—the actual metabolites in this pathway tested were inactive as substrates—it did support recognition of the overall chemotype. Docking of the KEGG and then a specialized lactone library confirmed the recognition of lactones, especially pentalactones, by the enzyme. Here too, these precise molecules are not, in fact, substrates. However, juxtaposition of highly ranked docked molecules such as d-galactono-1,4-lactone (KEGG entry C03383, dock rank 270/371 of 57672 molecules docked, Figure 6A), d-fucono-1,4-lactone (KEGG entry C06158, dock rank 409/431), l-arabino-1,4-lactone (KEGG entry C01114, dock rank 408/414), and d-glucurono-6,2-lactone (KEGG entry C03387, rank 357, Figure 6B) upon the X-ray structure, with its active site phosphate, and superposition on Lmo2620 suggested 1,4-lactone-5-phosphates as candidate substrates. This provided the impetus for their synthesis and subsequent determination that they were good substrates for both Ms0025 and Mag6390. Thus, it was an integration of genetic context, library docking, structural information, and kinetic analysis—no one of which was in itself sufficient—that led to the deorphanization and functional characterization of these two enzymes. Whether such information can be systematically integrated in future function–prediction efforts is uncertain at this time; what is clear is that there will be cases in which it is needed, as certainly was the case here.
Conclusion
Ms0025 and Mag6390 are lactonases that linearize phosphorylated 1,4-lactones and, to a lesser extent, carboxy 1,4-lactones (Table 3 and Table S2 of the Supporting Information). The molecular function assigned to these enzymes is the hydrolysis of d-xylono-1,4-lactone-5-phosphate (2), with kcat/Km values of 4.7 × 104 and 5.7 × 104 M–1 s–1 and l-arabino-1,4-lactone-5-phosphate (7) with kcat/Km values of 1.3 × 104 and 2.2 × 104 M–1 s–1, for Ms0025 and Mag6390, respectively. The enzyme is able to hydrolyze diacid sugar lactones (12, 20, and 22) with the same stereochemistry as the phosphorylated compounds, however, with decreased catalytic turnover rates. This brings us closer to identifying the primary functions of the enzymes from Cog1735, in that now only subgroups 1, 2, and 4 have three-dimensional structures assigned to them yet are functionally uncharacterized. Unlike previous cases in which we have discovered activities for unannotated amidohydrolases, with Ms0025 and Mag6390, neither empirical screening,20,44,45 sequence comparisons,22,46,47 nor structural and computational screening techniques alone7,10,11,13−15 sufficed in identifying the true substrates.48 It was only the combination of these three approaches, leading to the synthesis of entirely new substrates, not present in the docking or empirical screening libraries, that ultimately illuminated the activities of these enzymes. A future challenge will be to develop methods that integrate these approaches systematically and programmatically, as other cases like these undoubtedly await.
Acknowledgments
We thank Joel Karpiak, Ryan Coleman, and Trent Balius for reading the manuscript and for technical discussions.
Supporting Information Available
Sequence similarity of enzymes predicted to have the same substrate profile as Ms0025 and Mag6390 (Table S1), sugar lactones used to create specialized libraries for docking (Scheme S1), empirically screened candidate substrates without activity against Ms0025 or Mag6390 (Figure S1), five lactones (2, 7, 12, 20, and 22) that had activity toward Ms0025, including their docking pose, docking rank, active site chemical drawing, and kinetic parameters (Table S2), and a structural alignment of Lmo2620 and Ms0025 indicating the conserved residues required for phosphate binding (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The X-ray data have been deposited in the Protein Data Bank as entries 3MSR and 3OVG.
The authors declare no competing financial interest.
Supported by National Institutes of Health Grants GM GM093342 and GM71790.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Seibert C. M.; Raushel F. M. (2005) Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44, 6383–6391. [DOI] [PubMed] [Google Scholar]
- Holm L.; Sander C. (1997) An evolutionary treasure: Unification of a broad set of amidohydrolases related to urease. Proteins 28, 72–82. [PubMed] [Google Scholar]
- Akiva E.; Brown S.; Almonacid D. E.; Barber A. E. II; Custer A. F.; Hicks M. A.; Huang C. C.; Lauck F.; Mashiyama S. T.; Meng E. C.; Mischel D.; Morris J. H.; Ojha S.; Schnoes A. M.; Stryke D.; Yunes J. M.; Ferrin T. E.; Holliday G. L.; Babbitt P. C. (2014) The Structure-Function Linkage Database. Nucleic Acids Res. 42, D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afriat-Jurnou L.; Jackson C. J.; Tawfik D. S. (2012) Reconstructing a Missing Link in the Evolution of a Recently Diverged Phosphotriesterase by Active-Site Loop Remodeling. Biochemistry 51, 6047–6055. [DOI] [PubMed] [Google Scholar]
- Richardson E. J.; Watson M. (2013) The automatic annotation of bacterial genomes. Briefings Bioinf. 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes A. M.; Brown S. D.; Dodevski I.; Babbitt P. C. (2009) Annotation Error in Public Databases: Misannotation of Molecular Function in Enzyme Superfamilies. PLoS Comput. Biol. 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornelas A.; Korczynska M.; Ragumani S.; Kumaran D.; Narindoshvili T.; Shoichet B. K.; Swaminathan S.; Raushel F. M. (2013) Functional Annotation and Three-Dimensional Structure of an Incorrectly Annotated Dihydroorotase from cog3964 in the Amidohydrolase Superfamily. Biochemistry 52, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. D.; Laskowski R. A.; Thornton J. M. (2005) Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 15, 275–284. [DOI] [PubMed] [Google Scholar]
- Volkamer A.; Kuhn D.; Rippmann F.; Rarey M. (2013) Predicting enzymatic function from global binding site descriptors. Proteins: Struct., Funct., Bioinf. 81, 479–489. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Marti-Arbona R.; Fedorov A. A.; Fedorov E.; Almo S. C.; Shoichet B. K.; Raushel F. M. (2007) Structure-based activity prediction for an enzyme of unknown function. Nature 448, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrow L.; Hiser C.; Van Voorst J. R.; Ferguson-Miller S.; Kuhn L. A. (2013) Computational Prediction and in Vitro Analysis of Potential Physiological Ligands of the Bile Acid Binding Site in Cytochrome c Oxidase. Biochemistry 52, 6995–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favia A. D.; Nobeli I.; Glaser F.; Thornton J. M. (2008) Molecular docking for substrate identification: The short-chain dehydrogenases/reductases. J. Mol. Biol. 375, 855–874. [DOI] [PubMed] [Google Scholar]
- Xiang D. F.; Kolb P.; Fedorov A. A.; Meier M. M.; Fedorov L. V.; Nguyen T. T.; Sterner R.; Almo S. C.; Shoichet B. K.; Raushel F. M. (2009) Functional Annotation and Three-Dimensional Structure of Dr0930 from Deinococcus radiodurans, a Close Relative of Phosphotriesterase in the Amidohydrolase Superfamily. Biochemistry 48, 2237–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang D. F.; Kolb P.; Fedorov A. A.; Xu C. F.; Fedorov E. V.; Narindoshivili T.; Williams H. J.; Shoichet B. K.; Almo S. C.; Raushel F. M. (2012) Structure-Based Function Discovery of an Enzyme for the Hydrolysis of Phosphorylated Sugar Lactones. Biochemistry 51, 1762–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H.; Hitchcock D. S.; Seidel R. D.; Hillerich B.; Lin H.; Almo S. C.; Sali A.; Shoichet B. K.; Raushel F. M. (2013) Assignment of Pterin Deaminase Activity to an Enzyme of Unknown Function Guided by Homology Modeling and Docking. J. Am. Chem. Soc. 135, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelarends G. J., and Whitman C. P. (2010) Mechanistic and Structural Studies of Microbial Dehalogenases: How Nature Cleaves a Carbon-Halogen Bond, Vol. 8, Elsevier, Boston. [Google Scholar]
- Seffernick J. L.; de Souza M. L.; Sadowsky M. J.; Wackett L. P. (2001) Melamine deaminase and atrazine chlorohydrolase: 98% identical but functionally different. J. Bacteriol. 183, 2405–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afriat L.; Roodveldt C.; Manco G.; Tawfik D. S. (2006) The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry 45, 13677–13686. [DOI] [PubMed] [Google Scholar]
- Chow J. Y.; Wu L.; Yew W. S. (2009) Directed Evolution of a Quorum-Quenching Lactonase from Mycobacterium avium subsp. paratuberculosis K-10 in the Amidohydrolase Superfamily. Biochemistry 48, 4344–4353. [DOI] [PubMed] [Google Scholar]
- Uroz S.; Oger P. M.; Chapelle E.; Adeline M. T.; Faure D.; Dessaux Y. (2008) A Rhodococcus qsdA-encoded enzyme defines a novel class of large-spectrum quorum-quenching lactonases. Appl. Environ. Microbiol. 74, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. B.; Raushel F. M. (1996) Metal-substrate interactions facilitate the catalytic activity of the bacterial phosphotriesterase. Biochemistry 35, 10904–10912. [DOI] [PubMed] [Google Scholar]
- Hawwa R.; Aikens J.; Turner R. J.; Santarsiero B. D.; Mesecar A. D. (2009) Structural basis for thermostability revealed through the identification and characterization of a highly thermostable phosphotriesterase-like lactonase from Geobacillus stearothermophilus. Arch. Biochem. Biophys. 488, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias M.; Dupuy J.; Merone L.; Mandrich L.; Porzio E.; Moniot S.; Rochu D.; Lecomte C.; Rossi M.; Masson P.; Manco G.; Chabriere E. (2008) Structural basis for natural lactonase and promiscuous phosphotriesterase activities. J. Mol. Biol. 379, 1017–1028. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Ghanem E.; Li Y. C.; Raushel F. M.; Irwin J. J.; Shoichet B. K. (2006) Predicting substrates by docking high-energy intermediates to enzyme structures. J. Am. Chem. Soc. 128, 15882–15891. [DOI] [PubMed] [Google Scholar]
- Aslanidis C.; Dejong P. J. (1990) Ligation-Independent Cloning of Pcr Products (Lic-Pcr). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W. (1992) Recent Changes to the MOSFLM Package for Processing Film and Image Plate Data. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography 26, 29–35. [Google Scholar]
- Bailey S. (1994) The Ccp4 Suite: Programs for Protein Crystallography. Acta Crystallogr. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Hattne J.; Lamzin V. S. (2008) Pattern-recognition-based detection of planar objects in three-dimensional electron-density maps. Acta Crystallogr. D64, 834–842. [DOI] [PubMed] [Google Scholar]
- Langer G.; Cohen S. X.; Lamzin V. S.; Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S.; Sato Y.; Kawashima M.; Furumichi M.; Tanabe M. (2014) Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. (2010) Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- Yoshida S.; Park D. S.; Bae B.; Mackie R.; Cann I. K. O.; Nair S. K. (2011) Structural and Functional Analyses of a Glycoside Hydrolase Family 5 Enzyme with an Unexpected β-Fucosidase Activity. Biochemistry 50, 3369–3375. [DOI] [PubMed] [Google Scholar]
- Vanhooke J. L.; Benning M. M.; Raushel F. M.; Holden H. M. (1996) Three-dimensional structure of the zinc-containing phosphotriesterase with the bound substrate analog diethyl 4-methylbenzylphosphonate. Biochemistry 35, 6020–6025. [DOI] [PubMed] [Google Scholar]
- Zhang Z. G.; Aboulwafa M.; Smith M. H.; Saier M. H. (2003) The ascorbate transporter of Escherichia coli. J. Bacteriol. 185, 2243–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos A. T. R.; Ferreira H. B.; Bizarro C. V.; Bonatto S. L.; Carvalho M. O.; Pinto P. M.; Almeida D. F.; Almeida L. G. P.; Almeida R.; Alves L.; Assuncao E. N.; Azevedo V. A. C.; Bogo M. R.; Brigido M. M.; Brocchi M.; Burity H. A.; Camargo A. A.; Camargo S. S.; Carepo M. S.; Carraro D. M.; Cascardo J. C. D.; Castro L. A.; Cavalcanti G.; Chemale G.; Collevatti R. G.; Cunha C. W.; Dallagiovanna B.; Dambros B. P.; Dellagostin O. A.; Falcao C.; Fantinatti-Garboggini F.; Felipe M. S. S.; Fiorentin L.; Franco G. R.; Freitas N. S. A.; Frias D.; Grangeiro T. B.; Grisard E. C.; Guimares C. T.; Hungria M.; Jardim S. N.; Krieger M. A.; Laurino J. P.; Lima L. F. A.; Lopes M. I.; Loreto E. L. S.; Madeira H. M. F.; Manfio G. P.; Maranhao A. Q.; Martinkovics C. T.; Medeiros S. R. B.; Moreira M. A. M.; Neiva M.; Ramalho-Neto C. E.; Nicolas M. F.; Oliveira S. C.; Paixao R. F. C.; Pedrosa F. O.; Pena S. D. J.; Pereira M.; Pereira-Ferrari L.; Piffer I.; Pinto L. S.; Potrich D. P.; Salim A. C. M.; Santos F. R.; Schmitt R.; Schneider M. P. C.; Schrank A.; Schrank I. S.; Schuck A. F.; Seuanez H. N.; Silva D. W.; Silva R.; Silva S. C.; Soares C. M. A.; Souza K. R. L.; Souza R. C.; Staats C. C.; Steffens M. B. R.; Teixeira S. M. R.; Urmenyi T. P.; Vainstein M. H.; Zuccherato L. W.; Simpson A. J. G.; Zaha A. (2005) Swine and poultry pathogens: The complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. J. Bacteriol. 187, 5568–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew W. S.; Gerlt J. A. (2002) Utilization of l-ascorbate by Escherichia coli K-12: Assignments of functions to products of the yjf-sga and yia-sgb operons. J. Bacteriol. 184, 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garces F.; Fernandez F. J.; Montella C.; Penya-Soler E.; Prohens R.; Aguilar J.; Baldoma L.; Coll M.; Badia J.; Vega M. C. (2010) Molecular Architecture of the Mn2+-dependent Lactonase UlaG Reveals an RNase-like Metallo-β-lactamase Fold and a Novel Quaternary Structure. J. Mol. Biol. 398, 715–729. [DOI] [PubMed] [Google Scholar]
- Linares D.; Michaud P.; Delort A. M.; Traikia M.; Warrand J. (2011) Catabolism of l-Ascorbate by Lactobacillus rhamnosus GG. J. Agric. Food Chem. 59, 4140–4147. [DOI] [PubMed] [Google Scholar]
- Dumas D. P.; Caldwell S. R.; Wild J. R.; Raushel F. M. (1989) Purification and Properties of the Phosphotriesterase from Pseudomonas diminuta. J. Biol. Chem. 264, 19659–19665. [PubMed] [Google Scholar]
- Xiang D. F.; Xu C. F.; Kumaran D.; Brown A. C.; Sauder J. M.; Burley S. K.; Swaminathan S.; Raushel F. M. (2009) Functional Annotation of Two New Carboxypeptidases from the Amidohydrolase Superfamily of Enzymes. Biochemistry 48, 4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky M. J.; Tong Z. K.; de Souza M.; Wackett L. P. (1998) AtzC is a new member of the amidohydrolase protein superfamily and is homologous to other atrazine-metabolizing enzymes. J. Bacteriol. 180, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Arbona R.; Xu C. F.; Steele S.; Weeks A.; Kuty G. F.; Seibert C. M.; Raushel F. M. (2006) Annotating enzymes of unknown function: N-Formimino-l-glutamate deiminase is a member of the amidohydrolase superfamily. Biochemistry 45, 1997–2005. [DOI] [PubMed] [Google Scholar]
- Copley S. D. (2009) Prediction of function in protein superfamilies. F1000 Biology Reports 1, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.