

Abstract
Neuroinflammation caused by microglial activation plays a key role in ischemia, neurodegeneration and many other CNS diseases. In this study, we found that Adjudin, a potential non-hormonal male contraceptive, exhibits additional function to reduce the production of proinflammatory mediators. Adjudin significantly inhibited LPS-induced IL-6 release and IL-6, IL-1β, TNF-α expression in BV2 microglial cells. Furthermore, Adjudin exhibited anti-inflammatory properties by suppression of NF-κB p65 nuclear translocation and DNA binding activity as well as ERK MAPK phosphorylation. To determine the in vivo effect of Adjudin, we used a permanent middle cerebral artery occlusion (pMCAO) mouse model and found that Adjudin could reduce ischemia-induced CD11b expression, a marker of microglial activation. Furthermore, Adjudin treatment attenuated brain edema and neurological deficits after ischemia but did not reduce infarct volume. Thus, our data suggest that Adjudin may be useful for mitigating neuroinflammation.
Keywords: Adjudin, Microglial activation, Cytokine, NF-κB, Brain edema
1. Introduction
Microglial cells are the resident macrophages of the central nervous system (CNS) and have been proposed to play a role in CNS immunity, development and repair (Streit, 2001; Town et al., 2005; Walton et al., 2006). Once activated, microglia produce multiple proinflammatory mediators, including cytokines, chemokines, reactive oxygen species (ROS), nitric oxide (NO) and prostaglandin E2 (PGE2), which are responsible for neurotoxic effect under pathological conditions (Garden and Moller, 2006). Chronic inflammation caused by microglial activation has close relationship with Alzheimer's disease, Parkinson's disease, and multiple sclerosis (Gonzalez-Scarano and Baltuch, 1999; Imamura et al., 2003; Xiang et al., 2006), while inflammatory responses by microglia exacerbate brain injury after ischemia (Yenari et al., 2010). Thus, inhibition of microglial activation would be a potential therapeutic approach to treatment of various CNS diseases.
Adjudin, 1-(2,4-dichlorobenzyl)-1H-indazole-3-carbohydrazide, formerly called AF-2364, performs potent reversible anti-spermatogenic activity by disrupting the adherens junction of germ cells to Sertoli cells (Mok et al., 2011). Previous results have shown that many indazole derivatives are non-steroidal anti-inflammatory drugs (NSAID) by inhibition of prostaglandin E2 (PGE2) synthesis, nitric oxide (NO) production, cytokine and chemokine release (Foster et al., 1990; Sironi et al., 1996; Bhatia et al., 2005; Pan et al., 2005). Recent studies indicate that the anti-inflammatory effect of indazole derivatives is associated with the inhibition of NF-κB activation (Sommers et al., 2009; Oh et al., 2010).
Since Adjudin is a small molecular derivative of indazole and has no apparent side effects in treated animals (Mok et al., 2011), in this study we aimed to investigate whether Adjudin could attenuate microglial activation and the possible mechanisms.
2. Materials and methods
2.1. Reagents and animals
LPS and DMSO were purchased from Sigma Aldrich (St. Louis, MO, USA). Adjudin was provided by The Mary M. Wohlford Laboratory, Population Council, New York. Mice were purchased from Shanghai SLAC Laboratory Animal Corporation (Shanghai, China). Animal procedures for the use of laboratory animals were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University.
2.2. Cell culture
The BV2 microglia cell line was provided by Institute of Neurology, Ruijin Hospital (Shanghai, China). Cells were maintained in Dulbecco's Modified Eagle Medium (Thermo Scientific) supplemented with 5% fetal bovine serum (GIBCO) and 1% penicillin/streptomycin (GIBCO) at 37 °C in a humidified incubator under 5% CO2. The absolute concentrations of penicillin/streptomycin were 100 units·ml−1/100 μg·ml−1.
2.3. Lactate dehydrogenase (LDH) assay
BV2 cells were lysed for 20 min in a lysing buffer containing 0.04% Triton-X, 2 mM HEPES, 0.2 mM dithiothreitol, 0.01% bovine serum albumin, and 0.1% phenol red, pH 7.5. Cell lysates (50 μl) were mixed with 150 μl 500 mM potassium phosphate buffer (pH 7.5) containing 1.5 mM NADH and 7.5 mM sodium pyruvate, and the A340 nm change was monitored over 90 s using a microplate reader (Synergy2, BioTek).
2.4. Enzyme-linked immunosorbent assay (ELISA)
The concentration of IL-6 in medium from cultured BV2 cells was measured by platinum ELISA Kit (eBioscience) according to the manufacturer's instructions. The absorbance at 450 nm was determined using a microplate reader (Synergy2, BioTek). Protein concentrations were determined by BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). The IL-6 concentration in the medium was calculated using an IL-6 standard, and normalized against the protein of the samples.
2.5. Real-time PCR
Total RNA was isolated from BV2 cells by using Trizol Reagent (Invitrogen, CA, USA) and reverse-transcribed to cDNA using a PrimeScript RT reagent kit (TaKaRa). Quantitative real-time PCR was performed on ABI 7900HT by using SYBR Premix Ex Taq (TaKaRa) and the following primers: IL-6 (sense 5′-tagtccttcctaccccaatttcc-3′ and anti-sense 5′-ttggtccttagccactccttc-3′); IL-1β (sense 5′-gcaactgtt cctgaactcaact-3′ and anti-sense 5′-atcttttggggcgtcaact-3′); TNF-a (sense 5′-ccctcacactcagatcatcttct-3′ and anti-sense 5′-gctacgacgtgg gctacag-3′); GAPDH (sense 5′-aggtcggtgtgaacggatttg-3′ and anti-sense 5′-tgtagaccatgtagttgaggtca-3′). Data were analyzed by using the comparative threshold cycle (Ct) method, and results were expressed as fold difference normalized to GAPDH.
2.6. Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were prepared by using Nuclear Extract Kit (Active Motif) according to the manufacturer's instructions. DNA-protein interactions were measured by LightShift® Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL, USA) as follows: nuclear extracts (10 μg) were incubated with biotin-labeled NF-κB probe (5′-atgtg ggattttcccatgag-3′) at room temperature for 20 min. Protein–DNA complexes were resolved by 5% polyacrylamide gel, transferred to Nylon Membranes (Millipore, CA, USA), and crosslinked with 312 nm UV-light bulbs. The membranes were incubated with Streptavidin–HRP for 20 min and biotin-labeled DNA signals were visualized using ECL detection system.
2.7. Western blotting
Immunoblotting was carried out as previously described (Xia et al., 2007). Cells were lysed in RIPA buffer (Millipore, Temecula, CA, USA) containing Complete Protease Inhibitor Cocktail and 2 mM PMSF. 20 μg of total protein was separated by 10% SDS-PAGE and then transferred to 0.45 μm Nitrocellulose Membrane (Millipore, CA, USA). The membranes were incubated with primary antibodies at 4 °C overnight and then hybridized with appropriate HRP-conjugated secondary antibody (1:5000 dilution, Epitomics, Zhejiang Province, China) at room temperature for 1 h. Protein signals were visualized using ECL detection system. The primary antibodies used were as follows: ERK, phospho-ERK, p38, phospho-p38, JNK, phospho-JNK (1:1000 dilution, Cell Signaling Technology, Danvers, USA); NF-κB p65 (1:2000 dilution, Epitomics); Lamin A/C, β-actin (1:1000 dilution, Santa Cruz Biotechnology, CA, USA).
2.8. Permanent middle cerebral artery occlusion (pMCAO) model
pMCAO model in mice was performed as previously described (Yang et al., 1994). In brief, adult male ICR mice (25–30 g) were anesthetized by intraperitoneal administration of ketamine (100 mg/kg) plus xylazine (10 mg/kg). Then animal surgery was performed as follows: a 6–0 nylon suture was inserted from the left external carotid artery (ECA) into the internal carotid artery (ICA) and reached to the circle of Willis to occlude the origin of the middle cerebral artery (MCA). Successful occlusion was characterized as a reduction of cerebral blood flow (CBF) down to 10% of baseline. Sham-operated mice underwent the same procedure without suture insertion. For the animal experiments, mice were intraperitoneally administrated with 50 mg/kg Adjudin (100 mg/ml DMSO stock dissolved in corn oil at a dilution of 1:10) or vehicle (DMSO dissolved in corn oil at a dilution of 1:10) 2 h before pMCAO.
2.9. Immunohistological staining
Brain cryosections (20 μm) were fixed in 4% paraformaldehyde, blocked with 10% normal goat serum and incubated with anti-CD11b primary antibody (1:100 dilution, BD Biosciences). After washing, sections were incubated with Alexa-488-conjugated secondary antibody (1:200 dilution, Invitrogen, CA, USA) and nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (1:500 dilution, Beyotime Institute of Biotechnology, China). Confocal microscopic images were acquired using a confocal laser-scanning microscope (Leica TCS SP5 II, Germany).
2.10. Measurement of infarct volume
Brain cryosections (20 μm) were immersed in 0.1% cresyl violet for 30 min and then rinsed in distilled water for 10 min. The infarction area in each section was calculated using the ImageJ analysis software by the following formula: contralateral hemisphere area (mm2)–ipsilateral undamaged area (mm2). The infarction areas of each slice were separately summed and multiplied by the interval thickness to obtain infarct volumes.
2.11. Measurement of brain edema and brain water content
Brain edema was calculated as ipsilateral volume/contralateral volume. Brain tissue samples were weighed before and after dehydration in an oven at 100 °C for 24 h. Tissue water content (%) was calculated as [(wet weight–dry weight)/wet weight]×100.
2.12. Behavioral assessment
Modified neurologic severity scores (mNSS): mNSS is a composite of motor, reflex and balance tests. Total neurological score was calculated as the sum of scores on limb flexion (range: 0–3), walking gait (range: 0–3), beam balance (range: 0–6) and reflexes absence (range: 0–2). Therefore, neurologic function was graded on a scale of 0 to 14 (normal score 0; maximal deficit score 14) as previously described (Chen et al., 2005).
2.13. Statistical analysis
All data are presented as mean ± SEM. Data were assessed by one-way ANOVA, followed by Tukey post hoc test. P values less than 0.05 were considered statistically significant.
3. Results
3.1. Effect of Adjudin on viability of BV2 microglia
To study whether Adjudin could serve as a neuroinflammation modulator, we used BV2 microglial cell line as a model. We first evaluated its cytotoxicity to BV2 cells after 24-h incubation with different concentrations of Adjudin. As shown in Fig. 1, cell viability following treatment with 10 μM and 30 μM Adjudin was not significantly different from the control in the absence or presence of LPS. However, exposure of BV2 cells to 60 μM Adjudin led to about 40% decrease in cell survival.
Fig. 1.
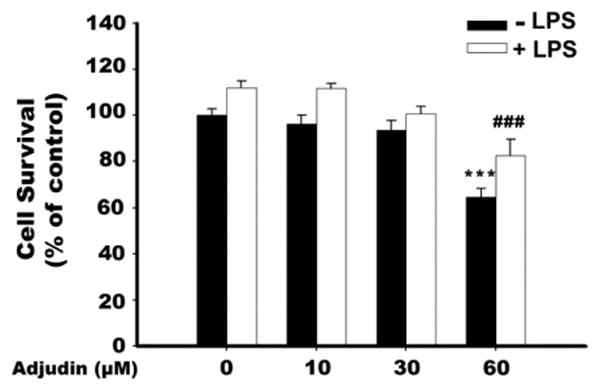
Effect of Adjudin on the cell viability of BV2 microglia. Cells were pretreated with indicated concentrations of Adjudin for 1 h and then stimulated with 200 ng/ml LPS for a 24-h incubation period. Cell viability was assessed by LDH assay. Bars represent the mean±SEM from three independent experiments. ***P<0.001 compared with control treatment; ###P<0.001 compared with the LPS alone treatment.
3.2. Adjudin reduces LPS-induced cytokine production in BV2 microglia
BV2 cells were pretreated with Adjudin (0, 10, 30, 60 μM) for 1 h and then stimulated with 200 ng/ml LPS for a 24-h incubation period. As anticipated, LPS significantly enhanced IL-6 release in the culture media, while pretreatment with Adjudin reduced IL-6 production by about 40% (Fig. 2A). We further determined whether Adjudin could regulate the expression of several cytokines important in neuroinflammation. LPS incubation for 6 h caused a remarkable increase in mRNA levels of TNF-α, IL-1β and IL-6, which could be suppressed by Adjudin in a dose-dependent manner (Fig. 2B–D).
Fig. 2.
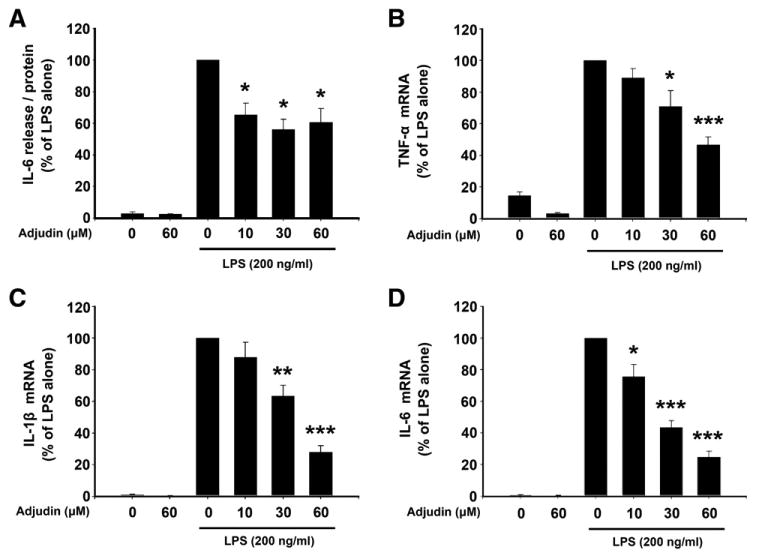
Adjudin reduces LPS-induced cytokine production in BV2 microglia. Cells were pretreated with Adjudin for 1 h and then incubated with LPS. (A) After 24 h incubation, IL-6 release in supernatants was measured by ELISA. Data were normalized against the protein of the samples. (B–D) After 6 h incubation, TNF-α (B), IL-1β (C) and IL-6 (D) mRNA expression was evaluated by qPCR. Data were normalized by GAPDH. Bars represent the mean±SEM from four independent experiments. *P<0.05; **P<0.01; ***P<0.001, compared with the LPS alone treatment.
3.3. Adjudin inhibits LPS-induced NF-κB activation in BV2 microglia
NF-κB has been shown to be one of the most important modulators for multiple proinflammatory cytokines, so we determined whether Adjudin could affect NF-κB signaling pathway. BV2 cells were pretreated with 30 μM Adjudin for 1 h and then stimulated with 200 ng/ml LPS for a 2-h incubation period. As shown in Fig. 3A, stimulation with LPS caused strong NF-κB DNA binding activity, which was markedly inhibited by Adjudin. The binding specificity was verified by incubating nuclear extracts from LPS-stimulated BV-2 cells with excess unlabeled specific competitor or nonspecific oligonucleotide probe. In addition, Adjudin also affected LPS-induced NF-κB p65 translocation from cytoplasm to nucleus. As shown in Fig. 3B, significant levels of NF-κB p65 were localized to the nucleus after LPS treatment, while Adjudin partially reversed this nuclear translocation.
Fig. 3.
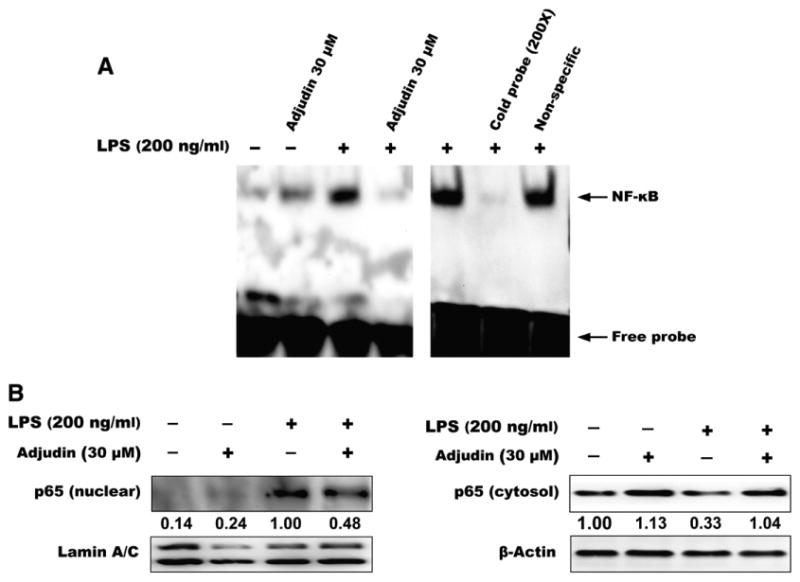
Adjudin inhibits LPS-stimulated NF-κB activation in BV2 microglia. Cells were pretreated with Adjudin for 1 h and then stimulated with LPS for 2 h. (A) Nuclei were extracted for NF-κB EMSA. (B) Nuclear and cytosol proteins were separated and analyzed using antibody against NF-κB p65. Lamin A/C was used as internal control for nuclear proteins and β-actin was used as internal control for cytosol proteins.
3.4. Adjudin decreases LPS-induced phosphorylation of ERK in BV2 microglia
We then assessed whether the suppressive effect of Adjudin on proinflammatory mediators occurred via MAPK signaling pathway. BV2 cells were pretreated for 1 h with indicated concentrations of Adjudin (0, 10, 30 μM) and then stimulated with 200 ng/ml LPS for 1 h. As anticipated, stimulation with LPS resulted in rapid activation of ERK, p38, and JNK MAPKs. However, pretreatment with 10 μM and 30 μM Adjudin only reduced ERK phosphorylation but not p38 or JNK phosphorylation (Fig. 4).
Fig. 4.
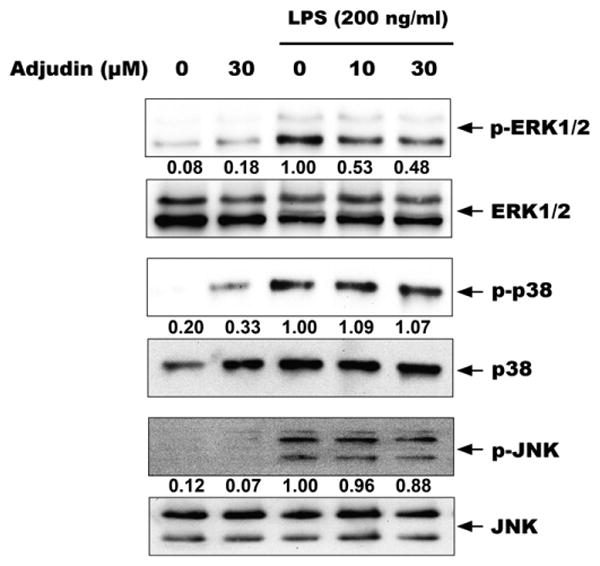
Adjudin decreases LPS-induced phosphorylation of ERK in BV2 microglia. Cells were pretreated with Adjudin for 1 h and then stimulated with LPS for 1 h. Cell lysates were analyzed by immunoblotting with antibodies specific to phospho-ERK1/2 and ERK1/2 (upper panels), phospho-p38 and p38 (middle panels), and phospho-JNK and JNK (lower panels).
3.5. Adjudin attenuates ischemia-induced microglial activation
To further determine the in vivo effect of Adjudin on microglial activation, we used a mouse pMCAO model. The result showed that CD11b expression—an indicator for microglial activation—was remarkably upregulated at 24 h after pMCAO, while i.p. administration of Adjudin (50 mg/kg b.w.) 2 h before ischemia reduced CD11b expression in both cerebral cortex (Fig. 5A and C) and striatum (Fig. 5B and D). These findings suggest that Adjudin could attenuate pMCAO-induced microglial activation.
Fig. 5.
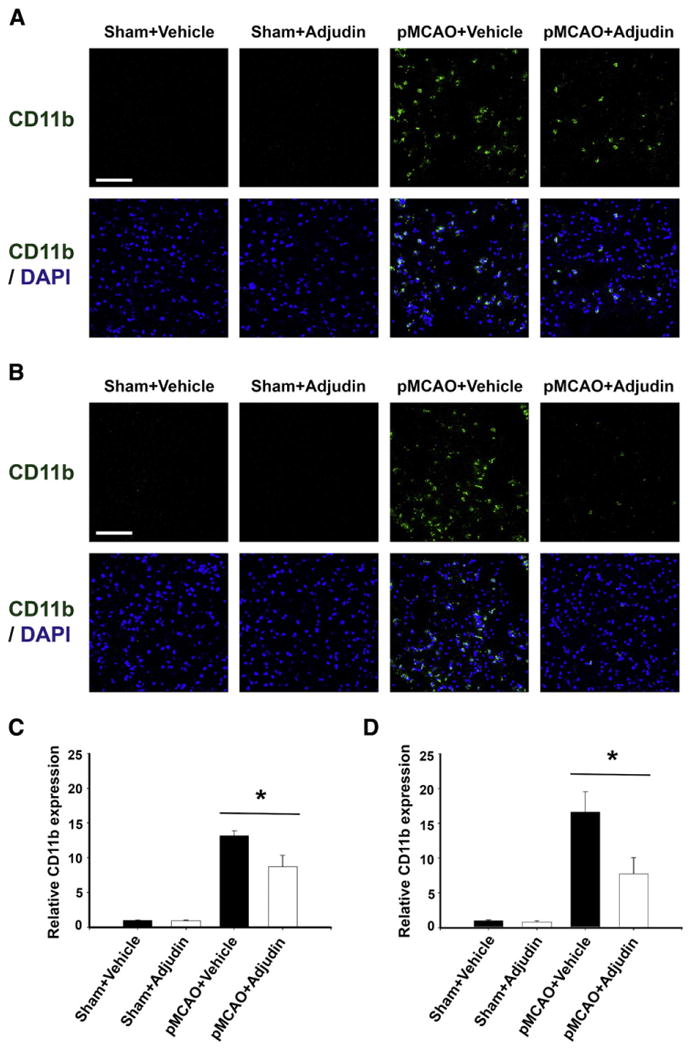
Adjudin attenuates ischemia-induced microglial activation. Immunohistological staining for CD11b in the ischemic cerebral cortex (A) and striatum (B) at 24 h after pMCAO. The blue staining represented DAPI. Scale bar=100 μm. Microglial activation in the ischemic cerebral cortex (C) and striatum (D) is quantified by the intensity of CD11b immunofluorescence. Data were mean±SEM, N=4 in each group. *P<0.05.
3.6. Adjudin reduces brain edema and neurological deficits after pMCAO
Next we investigated whether a decrease in microglial activation is related to the neuroprotective effect against ischemia. Adjudin administration did not reduce brain infarct volume (Fig. 6A and B) or the number of degenerating neurons (Supplementary Fig. 1) after ischemia. However, brain edema (Fig. 6C) and brain water content (Fig. 6D) of the ipsilateral hemisphere were smaller in the Adjudin-treated group than those in the control group at 24 h after pMCAO. Furthermore, neurological deficit improvement was observed with Adjudin treatment compared to vehicle at 72 h after pMCAO (Fig. 6E).
Fig. 6.
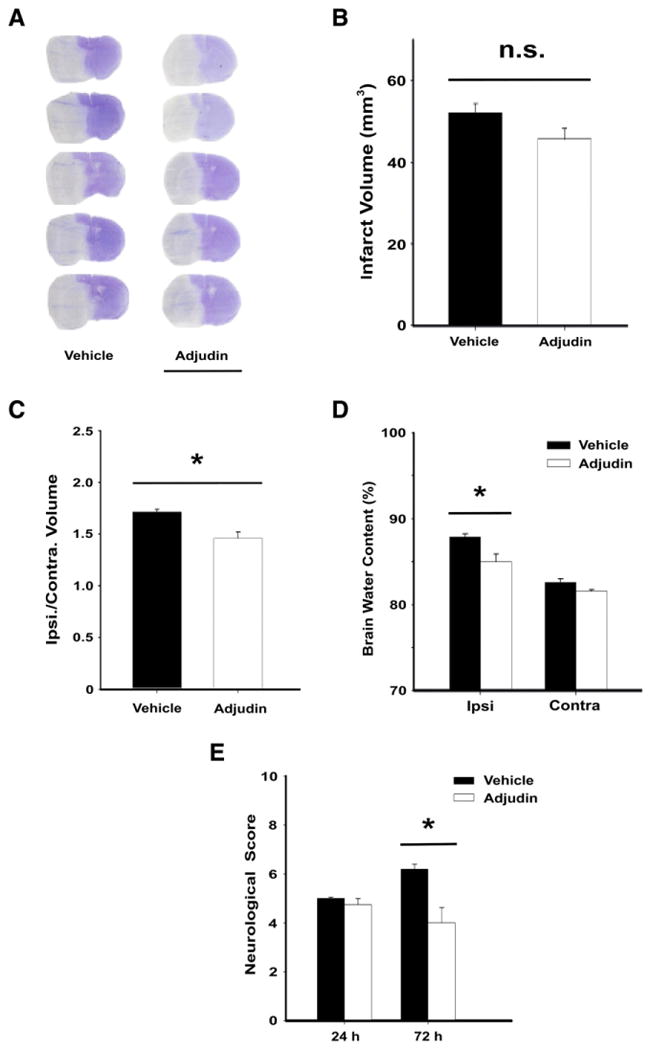
Adjudin reduces brain edema and neurological deficits after pMCAO. (A) Cresyl violet staining at 24 h after ischemia. White color indicates infarction. Scale bar=1 cm. (B) Infarct volume at 24 h after ischemia. Brain edema (C) and brain water content (D) at 24 h after ischemia. (E) Neurological deficits were assessed at 24 h and 72 h after ischemia. Data were mean±SEM, N=4–5 in each group. *P<0.05.
4. Discussion
In this study, we showed that Adjudin reduced IL-6 release and TNF-α, IL-1β, IL-6 mRNA expression in LPS-stimulated BV2 microglial cells. The anti-inflammatory property of Adjudin was associated with the inhibition of NF-κB activation and ERK MAPK phosphorylation. Using a well-recognized pMCAO model, we observed that Adjudin suppressed ischemia-induced microglial activation indicated by reduction of CD11b staining. Furthermore, Adjudin ameliorated brain edema and neurological deficits after ischemia but did not reduce infarct volume or the number of degenerating neurons.
Microglial cells are brain macrophages which serve important functions in many CNS diseases (Gonzalez-Scarano and Baltuch, 1999; Imamura et al., 2003; Xiang et al., 2006; Yenari et al., 2010). Several stimulants such as Amyloid-beta (Aβ), cytokines, LPS or IFN-γ can activate microglial cells (Hanisch and Kettenmann, 2007), and among them LPS is the most common stimulant used in in vitro studies (Qin et al., 2005). Microglial activation is complicated according to different concentrations or duration time of stimuli. Microglia after prolonged (72 h) exposure to LPS showed potentially neuroprotective effect compared with the acute (24 h) LPS stimulation (Cacci et al., 2008). Microglia activated by low level of IFN-γ seemed to promote neurogenesis despite the deleterious effect of IFN-γ reported previously (Butovsky et al., 2006). In the present study, our experimental model is stimulation with 200 ng/ml LPS for 24 h, which induced acute inflammatory responses in microglia.
NF-κB is an important upstream modulator of multiple inflammatory mediators and enzymes, including IL-1β, IL-6, TNF-α, i-NOS and COX-2 (Griscavage et al., 1996; Ghosh et al., 1998; Jobin et al., 1998). Previous studies have shown that NF-κB plays a pivotal role in inflammatory responses and neurotoxic effects by glial cells (Akama et al., 1998; Wilms et al., 2003). In the resting state, NF-κB remains in the cytoplasm as a heterotrimer complex consisting of p50, p65 and IκB sub-units. Upon activation, phosphorylation and degradation of IκB lead top50/p65 nuclear translocation, then binding to DNA and regulating gene transcription (Karin and Ben-Neriah, 2000). In the present study, we found that down-regulation of inflammatory mediators by Adjudin resulted from the inhibition of NF-κB signaling pathway at two levels: first, Adjudin prevented NF-κB p65 translocation from cytoplasm to nuclei and second, Adjudin decreased NF-κB DNA binding activity.
The MAPK family comprises ERK1/2, JNK, p38, and ERK5/BMK1 (Kyriakis and Avruch, 2001). Diverse stimuli, such as Amyloid-beta (Aβ), alpha-synuclein, LPS and IFN-γ, are able to activate MAPK signaling pathways in microglia (Koistinaho and Koistinaho, 2002; Klegeris et al., 2008). Our data showed that Adjudin could reduce ERK phosphorylation but not p38 or JNK phosphorylation. It is noteworthy that although Adjudin significantly inhibited NF-κB activation, the suppressive effect on cytokine production by Adjudin is merely 50%. There remains the possibility that Adjudin might not influence another inflammation-related transcription factor activator protein-1 (AP-1), which could be supported by our result that Adjudin did not inhibit JNK phosphorylation, since JNK is a key regulator of AP-1 by phosphorylating c-Jun subunit of AP-1 and thereby enhancing its transcriptional activity (Shaulian and Karin, 2002).
CD11b is a β-integrin marker of microglia and the increased CD11b expression corresponds to microglial activation in various CNS diseases (Gonzalez-Scarano and Baltuch, 1999). In our study, pretreatment with 50 mg/kg Adjudin reduced CD11b positive staining in both cerebral cortex and striatum at 24 h after pMCAO, which provided evidence for suppressive effect of Adjudin on microglial activation in vivo. Next we investigated whether a decrease in microglial activation is correlated to the neuroprotective effect against ischemia. Results showed that Adjudin reduced brain edema after ischemia but did not affect infarct volume or the number of degenerating neurons. Brain swelling is correlated to neurological deficits at later time points (Manley et al., 2000), which agrees with our findings that behavioral improvement was observed by Adjudin treatment at 72 h after ischemia. Brain edema includes two major types. The cytotoxic edema represents a shift of water from the extracellular to the intracellular compartments and does not contribute to a net increase in brain volume. However, the vasogenic edema aggravates brain swelling through a shift of water from the intravascular to the extravascular compartments (Ayata and Ropper, 2002). After ischemia, blood-brain barrier (BBB) destruction leads to increased paracellular permeability, directly contributing to cerebral vasogenic edema and hemorrhagic transformation (Sandoval and Witt, 2008). Matrix metalloproteinases (MMPs) are matrix-degrading enzymes which become markedly upregulated in response to ischemic injury. Increased MMP activity results in tight junction protein degradation, causing damage to the BBB (Cunningham et al., 2005). Numerous evidence indicates that activated microglia can be a major source of secreted MMPs following stroke (del Zoppo et al., 2007), thus Adjudin might protect the brain edema partially by inhibiting microglia-mediated BBB destruction. To further assess the neuroprotective effect of Adjudin on ischemia, different regimens of Adjudin (post treatment or multiple doses), longer observation window and the choice of tMCAO model shall be considered.
In the in vitro study, 60 μM Adjudin caused about 40% reduction in the cell viability of BV2 microglial cell lines. Our previous studies have shown that Adjudin could induce apoptosis in many mitotic cells (cancer cell lines and BV2 cell line) partly by targeting mitochondrial function. We presume that Adjudin has preferential effect on proliferating cells rather than normal cells. To verify this hypothesis, we treated primary mixed glial cultures with Adjudin in the presence of LPS. As shown in Supplementary Fig. 2, cell viability of glial cultures following Adjudin treatment was not different from those in the control group at the concentration of up to 100 μM. In the in vivo study, we chose a dose of 50 mg/kg b.w. for Adjudin in animal experiments. Previous reports have shown that two doses of 50 mg/kg b.w. Adjudin (one dose per week) could induce reversible infertility in adult rats without fatality, body weight reduction or apparent side effects (Cheng et al., 2001). Histological analyses showed that cell adhesion in other organs, such as kidney, liver, brain, heart and prostate, was not affected by Adjudin (Grima et al., 2001) and toxicity studies confirmed that Adjudin has no genotoxicity or mutagenesis in acute dose even at 2000 mg/kg b.w. in rats and mice (Mruk et al., 2006). Furthermore, the serum testosterone, FSH, and LH levels of the Adjudin-treated animals had no detectable change compared with the control animals, indicating that the hypothalamic–pituitary–testicular axis is not disrupted (Cheng et al., 2001). Thus, Adjudin administration at a dose of 50 mg/kg b.w. appears to be safe in animal treatment.
In summary, this study has shown that Adjudin could inhibit microglial activation in both in vitro and in vivo models, and the anti-inflammatory property of Adjudin was due to the inhibition of NF-κB activation and ERK MAPK phosphorylation. Therefore, these data suggest potential applications of Adjudin for mitigating neuroinflammation.
Supplementary Material
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jneuroim.2012.09.012.
Acknowledgments
This study was supported by grants from NNSF China (30900756, 31270032), “Rising Star” Grant from Science and Technology, Commission of Shanghai (09QA1403400), SJTU fundings (YG2010MS19, YG2011MS44) and SJTU SMC Morning Star program (A09CX40), and partially by grants from the Ministry of Science and Technology (2013CB945604). We are also grateful to Mr. Xiang Gu for his assistance in confocal microscopy, and to Prof. Guo-yuan Yang, Mr. Falei Yuan, Mr. Xiaojie Lin, Mr. Yaohui Tang, Mr. Xiaosong He, and Miss Jun Huang for their assistance in the pMCAO model.
References
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci U S A. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Ropper AH. Ischaemic brain oedema. J Clin Neurosci. 2002;9:113–124. doi: 10.1054/jocn.2001.1031. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259–G1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Cacci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L. In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia. 2008;56:412–425. doi: 10.1002/glia.20616. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, Li Y, Zhang L, Robin A, Katakowski M, Lu M, Chopp M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Silvestrini B, Grima J, Mo MY, Zhu LJ, Johansson E, Saso L, Leone MG, Palmery M, Mruk D. Two new male contraceptives exert their effects by depleting germ cells prematurely from the testis. Biol Reprod. 2001;65(2):449–461. doi: 10.1095/biolreprod65.2.449. [DOI] [PubMed] [Google Scholar]
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- Foster SJ, Bruneau P, Walker ER, McMillan RM. 2-substituted indazolinones: orally active and selective 5-lipoxygenase inhibitors with anti-inflammatory activity. Br J Pharmacol. 1990;99:113–118. doi: 10.1111/j.1476-5381.1990.tb14663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Grima J, Silvestrini B, Cheng CY. Reversible inhibition of spermatogenesis in rats using a new male contraceptive, 1-(2,4-dichlorobenzyl)-indazole-3-carbohydrazide. Biol Reprod. 2001;64(5):1500–1508. doi: 10.1095/biolreprod64.5.1500. [DOI] [PubMed] [Google Scholar]
- Griscavage JM, Wilk S, Ignarro LJ. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-kappa B. Proc. Natl Acad Sci U S A. 1996;93:3308–3312. doi: 10.1073/pnas.93.8.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003;106:518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- Jobin C, Morteau O, Han DS, Balfour Sartor R. Specific NF-kappaB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1 gene expression in HT-29 cells. Immunology. 1998;95:537–543. doi: 10.1046/j.1365-2567.1998.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 2008;29:739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Mok KW, Mruk DD, Lie PP, Lui WY, Cheng CY. Adjudin, a potential male contraceptive, exerts its effects locally in the seminiferous epithelium of mammalian testes. Reproduction. 2011;141:571–580. doi: 10.1530/REP-10-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mruk DD, Wong CH, Silvestrini B, Cheng CY. A male contraceptive targeting germ cell adhesion. Nat Med. 2006;12:1323–1328. doi: 10.1038/nm1420. [DOI] [PubMed] [Google Scholar]
- Oh JH, Park EJ, Park JW, Lee J, Lee SH, Kwon TK. A novel cyclin-dependent kinase inhibitor down-regulates tumor necrosis factor-alpha (TNF-alpha)-induced expression of cell adhesion molecules by inhibition of NF-kappaB activation in human pulmonary epithelial cells. Int Immunopharmacol. 2010;10:572–579. doi: 10.1016/j.intimp.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Pan SL, Guh JH, Peng CY, Chang YL, Cheng FC, Chang JH, Kuo SC, Lee FY, Teng CM. A potential role of YC-1 on the inhibition of cytokine release in peripheral blood mononuclear leukocytes and endotoxemic mouse models. Thromb Haemost. 2005;93:940–948. doi: 10.1160/TH04-03-0195. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, Block ML. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood–brain barrier tight junction permeability and is-chemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Sironi M, Pozzi P, Polentarutti N, Benigni F, Coletta I, Guglielmotti A, Milanese C, Ghezzi P, Vecchi A, Pinza M, Mantovani A. Inhibition of inflammatory cytokine production and protection against endotoxin toxicity by benzydamine. Cytokine. 1996;8:710–716. doi: 10.1006/cyto.1996.0094. [DOI] [PubMed] [Google Scholar]
- Sommers CD, Thompson JM, Guzova JA, Bonar SL, Rader RK, Mathialagan S, Venkatraman N, Holway VW, Kahn LE, Hu G, Garner DS, Huang HC, Chiang PC, Schindler JF, Hu Y, Meyer DM, Kishore NN. Novel tight-binding inhibitory factor-kappaB kinase (IKK-2) inhibitors demonstrate target-specific anti-inflammatory activities in cellular assays and following oral and local delivery in an in vivo model of airway inflammation. J Pharmacol Exp Ther. 2009;330:377–388. doi: 10.1124/jpet.108.147538. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and macrophages in the developing CNS. Neurotoxicology. 2001;22:619–624. doi: 10.1016/s0161-813x(01)00033-x. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall GP, II, Scheffler B, Steindler DA. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–825. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J. 2003;17:500–502. doi: 10.1096/fj.02-0314fje. [DOI] [PubMed] [Google Scholar]
- Xia W, Mruk DD, Cheng CY. C-type natriuretic peptide regulates blood–testis barrier dynamics in adult rat testes. Proc Natl Acad Sci U S A. 2007;104:3841–3846. doi: 10.1073/pnas.0610100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM. Microglia activation in the brain as inflammatory biomarker of Alzheimer's disease neuropathology and clinical dementia. Dis Markers. 2006;22:95–102. doi: 10.1155/2006/276239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7:378–391. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jneuroim.2012.09.012.