

Abstract
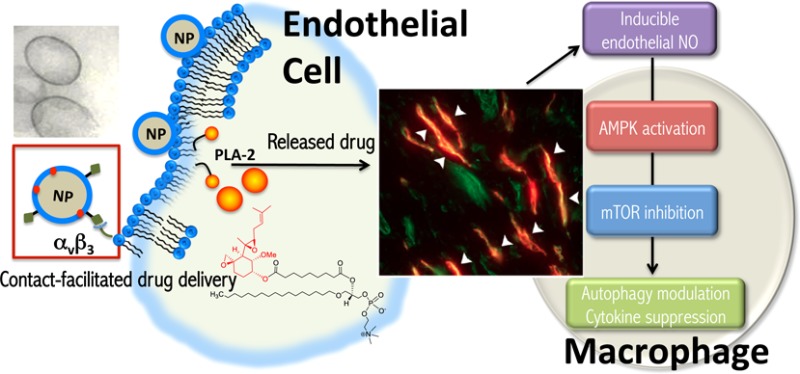
Antiangiogenesis has been extensively explored for the treatment of a variety of cancers and certain inflammatory processes. Fumagillin, a mycotoxin produced by Aspergillus fumigatus that binds methionine aminopeptidase 2 (MetAP-2), is a potent antiangiogenic agent. Native fumagillin, however, is poorly soluble and extremely unstable. We have developed a lipase-labile fumagillin prodrug (Fum-PD) that eliminated the photoinstability of the compound. Using αvβ3-integrin-targeted perfluorocarbon nanocarriers to deliver Fum-PD specifically to angiogenic vessels, we effectively suppressed clinical disease in an experimental model of rheumatoid arthritis (RA). The exact mechanism by which Fum-PD-loaded targeted nanoparticles suppressed inflammation in experimental RA, however, remained unexplained. We herein present evidence that Fum-PD nanotherapy indirectly suppresses inflammation in experimental RA through the local production of endothelial nitric oxide (NO). Fum-PD-induced NO activates AMP-activated protein kinase (AMPK), which subsequently modulates macrophage inflammatory response. In vivo, NO-induced AMPK activation inhibits mammalian target of rapamycin (mTOR) activity and enhances autophagic flux, as evidenced by p62 depletion and increased autolysosome formation. Autophagy in turn mediates the degradation of IkappaB kinase (IKK), suppressing the NF-κB p65 signaling pathway and inflammatory cytokine release. Inhibition of NO production by NG-nitro-l-arginine methyl ester (l-NAME), a nitric oxide synthase inhibitor, reverses the suppression of NF-κB-mediated inflammatory response induced by Fum-PD nanotherapy. These unexpected results uncover an activity of Fum-PD nanotherapy that may be further explored in the treatment of angiogenesis-dependent diseases.
Keywords: angiogenesis, autophagy, nitric oxide, AMP-activated protein kinase, fumagillin prodrug, nanotherapy
Angiogenesis is the new blood vessel formation that accompanies normal development as well as many disease processes. Antiangiogenic therapy has been extensively explored for the treatment of solid tumors and other inflammatory conditions. For example, antiangiogenic drugs targeting the vascular endothelial growth factor (VEGF) pathway have offered improved survival benefits in some cancer patients; however the results are often modest,1,2 and most patients treated with anti-VEGF agents eventually develop resistance, leading to cancer progression.3 Macroautophagy (hereafter referred to as autophagy) is a physiologic process that clears defective cellular organelles in the maintenance of normal cell functions but also contributes to various pathological conditions.4 As defects in autophagy predispose to cancer and other inflammatory processes,5,6 stimulating autophagy with mTOR inhibitors or other autophagy enhancers may suppress cancer and limit inflammation. In fact, the combination of autophagy modulation and chemotherapy/antiangiogenesis is currently being explored for the treatment of various cancers.5
Fumagillin, a mycotoxin produced by Aspergillus fumigatus that inhibits methionine aminopeptidase 2 (MetAP-2),7,8 is a potent inhibitor of angiogenesis.9 Native fumagillin is highly hydrophobic and extremely photounstable, thus limiting its potential clinical translation. We have successfully developed a lipase labile fumagillin prodrug (Fum-PD), which eliminated the photoinstability of native fumagillin.10 Using an αvβ3-integrin-targeted perfluorocarbon (PFC) nanocarrier system, we delivered Fum-PD specifically to the neovasculature in the inflamed joints for effective suppression of clinical disease in the KRN preclinical mouse model of rheumatoid arthritis (RA).11 Fum-PD nanotherapy led to a marked decrease in the expression of multiple inflammatory cytokines, including those released by activated macrophages such as TNF-α and IL-1β. While the antiangiogenic effect of fumagillin on endothelial cells is well described,7,8 the exact mechanism by which Fum-PD-loaded nanoparticles, which were constrained to the vasculature due to size, suppressed inflammatory cytokine release in the synovium by nonendothelial cells was unclear.
We demonstrate herein that targeted endothelial delivery of Fum-PD via nanocarriers generated a local nitrosative response that drives autophagy through the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway. Fum-PD induced the release of endothelial nitric oxide (NO), which in turn modulated macrophage inflammatory response through AMPK activation. In vivo, NO-induced phosphorylation/activation of AMPK following Fum-PD nanotherapy led to the inhibition of mTOR activity and enhanced autophagic flux, ultimately suppressing the NF-κB signaling pathway and down-regulating the subsequent inflammatory cytokine response. Thus, the effects of Fum-PD nanoparticles on autophagy could be further exploited to enhance the antiangiogenic treatment in various disease processes.
Results
αvβ3-Targeted Fum-PD Nanotherapy Suppresses Inflammatory Cytokine Response in the KRN Mouse Model of Arthritis
Fum-PD-loaded perfluorocarbon nanoparticles (NPs) were synthesized as detailed in the Methods section and characterized with multiple techniques as previously reported.10,11 The specific hydrodynamic diameter (DLS), electrophoretic potential, and polydispersity index of αvβ3-targeted PFC NPs without or with Fum-PD used in these studies are presented in Figure 1.
Figure 1.
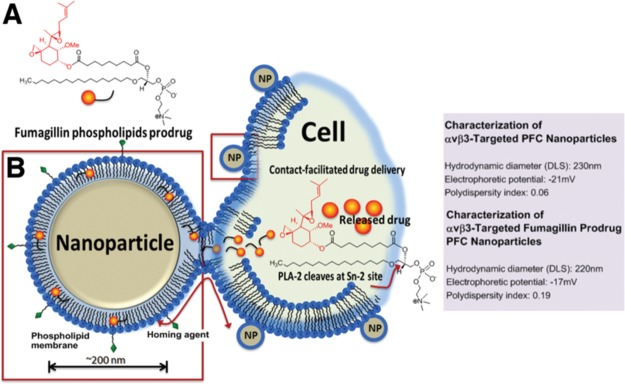
Schematic representation of Fum-PD PFC nanoparticle delivery mechanism. (A) Chemical formula of Sn-2 phospholipase labile Fum-PD. (B) Fum-PD FFC nanoparticle fuses with the targeted cell. Once inside the cell, Fum-PD is cleaved at the Sn-2 site by a phospholipase, such as PLA-2, releasing the active drug. Characteristics of PFC nanoparticles without and loaded with Fum-PD are presented on the right.
In the context of RA, we have used the KRN serum transfer model to induce symmetrical polyarticular arthritis in C57BL/6 mice that recapitulates key features of the inflammatory response observed in human RA.12 Maximal arthritis usually develops around day 6 or 7 following KRN serum injection and lasts for approximately 2 weeks. Similar to previous studies,11,13,14 we observed that ανβ3-targeted PFC NPs loaded with Fum-PD significantly suppressed clinical disease when compared to treatment with ανβ3-targeted NPs without drug (Ctrl NPs) (Figure 2A,B) or free Fum-PD (Figure S1). In parallel with improved clinical indices, we found that synovial (joint) levels of multiple macrophage-associated inflammatory cytokines (TNF-α, IL-1β, IL-6, and MCP-1) were all decreased (p < 0.01) in animals treated with αvβ3-targeted Fum-PD NPs compared to animals treated with Ctrl NPs (Figure 2C–F). The decrease in cytokine levels was significantly more profound compared to the moderate suppression of macrophage recruitment (Figure 2G). These results were unexpected, as fumagillin itself has not been shown to directly affect macrophages or macrophage-associated inflammatory cytokine release.15,16
Figure 2.

Fumagillin prodrug nanoparticles (Fum-PD NP) suppress KRN arthritis and inflammatory response. Mice were injected ip with 150 μL of KRN serum on day 0. Changes in ankle thickness (A) and arthritis score (B) were assessed daily. Starting on day 2, when early arthritis was clearly established, mice were randomly divided into groups and given serial daily injections of targeted NPs without drug (Ctrl NP) or Fum-PD NPs for three consecutive doses. On day 9 the animals were sacrificed; their paws were harvested and homogenized, and paw lysates analyzed for inflammatory cytokine levels (C–F). (G) Day 9 paw sections were stained with Mac-3 (macrophage marker), and the number was enumerated per high power field (HPF). Values represent mean ± SEM, n = 5 mice per treatment group. *p < 0.05, **p < 0.01, ***p < 0.001. (H) Paws were also sectioned and stained for endothelial marker CD31 (PECAM-1, red) and iNOS (green). Co-localization (yellow) is indicated with arrowheads. Note tendon autofluorescence (green, arrow). Scale bar = 50 μm.
Although the antiproliferative effect of fumagillin on endothelial cells is well established,9 the mechanism by which fumagillin nanotherapy suppressed the inflammatory response in the arthritic paws was unclear. Studies have shown that fumagillin inhibits MetAP-2,8 thereby affecting protein myristoylation and impairing the translocation of membrane proteins such as nitric oxide synthase (NOS) to the cell surface.17 As a consequence, intracellular NOS and NO could accumulate and trigger endothelial cell apoptosis.17,18 NO has complex effects, both deleterious and beneficial. Indeed, some studies have shown that NO has an anti-inflammatory effect by actively suppressing the production of inflammatory cytokines.19,20 We hypothesized that antiangiogenic therapy with Fum-PD NPs increased NO production by endothelial cells (ECs) and that NO release following EC apoptosis suppressed the inflammatory response of nearby recruited macrophages. Consistent with this hypothesis, we observed increased NOS expression in the endothelial cells of paw sections obtained from mice treated with Fum-PD NPs, but not mice treated with Ctrl NPs (Figure 2H).
Fum-PD Suppresses Macrophage Inflammatory Response through Nitric Oxide
To further confirm the hypothesis that Fum-PD enhanced NO production by ECs, we turned to an in vitro system. Mouse ECs (SVEC4-10) and day 5 thioglycollate-elicited primary peritoneal macrophages (MΦ) were individually exposed to increasing concentrations of Fum-PD (in DMSO) for 48 h, and the supernatants were assayed for NO release. Exposure of ECs to Fum-PD led to a dose-dependent release of NO (Figure 3A) and increased NOS expression (Figure 3B). In contrast, Fum-PD had no direct effects on inducible NOS (iNOS) expression and NO release by peritoneal MΦ (Figure 3A,B). Increased intracellular NO triggered EC apoptosis, as evidenced by positive terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in cells that expressed high level of iNOS (Figure 3C).
Figure 3.

Fum-PD increases NOS expression in endothelial cells. (A) Endothelial cells (EC) and day 5 thioglycollate-elicited macrophages (MΦ) were individually cultured with the indicated Fum-PD concentrations in DMSO (≤0.1%). At 48 h, supernatants were collected and assayed for nitric oxide (NO) release using Griess reagent. Values represent average ± SEM of triplicate samples derived from at least three independent experiments. **p < 0.01, ***p < 0.001. (B) ECs and peritoneal MΦ were stained for iNOS (red) 48 h after DMSO (0.1%) or Fum-PD (40 nM) exposure. (C) ECs were exposed to Fum-PD (40 nM) for 48 h, then stained for iNOS (red) and TUNEL (green) to detect apoptotic cells. Yellow (arrowheads) indicates co-localization. The staining was confirmed on at least 10 random fields. Scale bar = 50 μm.
To determine whether endothelial NO could modulate MΦ inflammatory activity, we cocultured thioglycollate-elicited peritoneal MΦ with ECs (1:1 ratio) in the presence of Fum-PD. We focused on TNF-α levels for these in vitro assays since TNF-α release was unique to peritoneal MΦ, whereas SVEC4-10 ECs released high levels of MCP-1 and IL-6 but no TNF-α (Figure S2). While Fum-PD had no direct effect on TNF-α release by MΦ (Figure 4A), exposure of EC–MΦ cocultures to Fum-PD led to a dose-dependent suppression of TNF-α release (Figure 4B). The anti-inflammatory activity of Fum-PD in EC–MΦ cocultures was recapitulated by the NO donor S-nitroso-N-acetyl-dl-penicillamine (SNAP),21 while the addition of NG-nitro-l-arginine methyl ester (l-NAME, a nonspecific NOS inhibitor) reversed the suppressive effect of Fum-PD (and SNAP) on TNF-α release. Although l-NAME is known to inhibit NO synthesis, it also acts as a scavenger of hydroxyl radicals in the millimolar range.22 However, d-NAME, an isomer of l-NAME, can scavenge hydroxyl radicals at equal rates to l-NAME.22 The fact that d-NAME did not affect Fum-PD activity (Figure 4C) implied that it was the NOS inhibitory activity of l-NAME that contributed to the observed phenotype. Taken together these results strongly suggest that, in vitro, Fum-PD indirectly suppressed MΦ inflammatory cytokine release through an NO-dependent mechanism.
Figure 4.

Fum-PD-induced NO indirectly suppresses MΦ inflammatory response in vitro. (A) Fum-PD had no direct effect on the release of TNF-α from MΦ. The experiment was repeated three times with similar results. (B, C) Fum-PD dose-dependently suppressed TNF-α release from EC–MΦ cocultures. The effect was reversed by l-NAME (20 μM), but not the inactive isomer d-NAME (20 μM). Values represent mean ± SEM of triplicate samples derived from three independent experiments. (D) EC–MΦ cocultures were exposed to Fum-PD (0–40 nM) or SNAP (25 μg/mL) without or with l-NAME (0–20 μM) and harvested at 48 h, and cell lysates were probed for phospho (P)-AMPKα, AMPKα, phospho (P)-p65, and p65. (E) EC–MΦ cocultures were exposed to Fum-PD (20 nM) without or with compound C (CC) at the indicated concentration. Supernatants were harvested and analyzed for TNF-α. The experiment is representative of two independent experiments with similar results. (F) EC–MΦ cocultures were exposed to Fum-PD without or with l-NAME (20 μM), and 48 h supernatants were assayed for IL-10. (G) RNA obtained from cocultures was analyzed by real-time PCR for expression of arginase 1, an M2 marker. *p < 0.05, **p < 0.01, ***p < 0.001 compared with no Fum-PD (0.1% DMSO).
Fum-PD-Induced NO Modulates Macrophage Activity in Vitro through AMPK Activation
NO has been shown to activate a number of signaling pathways, including AMPK.23,24 Several recent studies suggest that AMPK acts as a “central regulator” of inflammatory signaling in various cell types, macrophages in particular.25−27 In addition, AMPK activation has been shown to reduce inflammation in vivo in several preclinical models.28−30 To determine the mechanism by which Fum-PD induced NO-modulated macrophage inflammatory response, the activation status of AMPK in EC–MΦ cocultures in the presence of Fum-PD was examined. Fum-PD dose-dependently enhanced the phosphorylation of AMPK (Figure 4D). This activation of AMPK was mimicked by the addition of SNAP (Figure 4D). On the other hand, l-NAME reversed the Fum-PD-induced AMPK activation (Figure 4D). AMPK signaling has been shown to promote MΦ anti-inflammatory response by down-regulating NF-κB activation.31 Consistent with previous studies, we observed reduced NF-κB p65 phosphorylation in the presence of Fum-PD and SNAP (Figure 4D). Fum-PD suppression of NF-κB-p65 phosphorylation was reversed with the addition of l-NAME (Figure 4D).
To further confirm that Fum-PD-mediated suppression of TNF-α release is AMPK-dependent, we cocultured EC–MΦ in the presence of Fum-PD and compound C (CC), a specific inhibitor of AMPK.32 CC dose-dependently reversed the Fum-PD-mediated inhibition of TNF-α release (Figure 4E). Although mechanistic details regarding AMPK modulation of NF-κB signaling and cytokine production in macrophages remain undefined, some studies suggest that AMPK activation polarizes macrophages toward an anti-inflammatory phenotype.
In an oversimplification, macrophages can be loosely divided into two populations, the classically and alternatively activated macrophages.33 Classically activated macrophages (M1) are generated through IFN-γ and TNF-α stimulation and secrete high levels of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-23), while alternatively activated macrophages (M2) are induced by IL-4/IL-13 and promote matrix reorganization, wound healing, fibrosis, allergy, and immunomodulation.33 Alternatively activated M2 macrophages secrete IL-10 and TGF-β, among other mediators, and up-regulate the expression of arginase 1.34 We examined the release of IL-10 and mRNA expression of arginase 1 (M2 markers) in EC–MΦ cocultures exposed to Fum-PD and observed no difference in the level of IL-10 and a dose-dependent decrease in arginase 1 expression (Figure 4F,G). While Fum-PD did not definitively polarize macrophages toward an alternatively activated M2 phenotype in vitro, a reversal of the TNF-α/IL-10 ratio suggested that Fum-PD induced a shift in macrophage activity toward an anti-inflammatory phenotype.
Fum-PD Nanotherapy Suppresses KRN Arthritis through an NO-Dependent Mechanism
Next, the KRN model was used to examine the effect of NO on arthritis progression in the context of antiangiogenic nanotherapy with Fum-PD. Arthritis was induced as above, and the animals received serial iv injections on days 2, 3, and 4 of Ctrl NPs, Fum-PD NPs, or Fum-PD NPs with concomitant administration of l-NAME or d-NAME. Systemic administration of l-NAME reversed the arthritis suppression achieved with Fum-PD NPs, while the inactive isomer d-NAME did not (Figure 5A,B). Joint-associated inflammatory cytokine levels (TNF-α, IL-1β, IL-6, and MCP-1) corroborated the clinical findings (Figure 5C–F). These results strongly suggest that in vivo Fum-PD nanotherapy suppressed inflammation and arthritis progression through an NO-dependent pathway.
Figure 5.

Fum-PD nanotherapy suppresses KRN arthritis through an NO-dependent mechanism. Mice were injected ip with 150 μL of KRN serum on day 0. Changes in ankle thickness (A) and arthritis score (B) were assessed daily. On day 2, when early arthritis was clearly established, mice were randomly divided into groups and given serial daily injections of targeted NPs without drug (Ctrl NP) or Fum-PD NP for three consecutive doses. In some groups, mice were given l-NAME or d-NAME (100 mg/kg) iv 30 min prior to the NP injection. On day 9 paws were harvested and homogenized, and paw lysates analyzed for cytokine levels (C–G) by cytometric bead array (CBA) and ELISA. Values represent mean ± SEM, n = 4 or 5 mice per treatment group. *p < 0.05, **p < 0.01, ***p < 0.001. (H, I) Lysates were also fractionated on SDS-PAGE gels and immunoblotted for phospho (P)-AMPKα, AMPKα, and phospho (P)-mTOR. Actin served as protein loading control.
Fum-PD Nanotherapy Suppresses Inflammatory Arthritis through Autophagy Modulation
To determine whether NO modulated AMPK activation in vivo, KRN arthritic mice were again treated with Fum-PD NP. Increased AMPK phosphorylation was observed (Figure 5H), and this enhanced activity was reversed by l-NAME but not d-NAME (Figure 5I). Fum-PD NP treatment also inhibited the phosphorylation of mTOR (Figure 5H), which is known to be regulated by AMPK.35 Consistent with the in vitro studies, enhanced AMPK activation was accompanied by suppression of inflammatory cytokine profiles. However, neither IFN-γ (not shown) nor IL-10 (an M2 marker) was detected in the paw lysates of treated animals. In addition, the expression of CD206, the macrophage mannose receptor that is known to be up-regulated on alternatively activated M2 macrophages, was examined.36 We observed a predominance of CD206+ macrophages in day-9 arthritic paws but no significant difference in the expression of this receptor in the synovial tissues of animals treated with Ctrl or Fum-PD NPs (Figure S3). As macrophage activation is plastic and reversible, the possibility that Fum-PD might have suppressed inflammation by inducing macrophages to differentiate toward an M2 phenotype in the early phases of disease (i.e., at a time when inflammatory M1 macrophages might predominate) could not be precluded.
We next considered whether AMPK-induced mTOR inhibition might also exert an anti-inflammatory effect in KRN arthritis through the induction of autophagy.6,37,38 Autophagy is now recognized as a major mechanism for regulating the secretion of cytokines and chemokines, especially in macrophages.6 Autophagy involves a set of evolutionary conserved gene products known as Atg proteins that are required for the formation of autophagosomes. One of these critical proteins is microtubule-associated protein light chain 3 (LC3), a mammalian homologue of yeast Atg8. The cytoplasmic form of LC3 (LC3-I) is converted to LC3-II via lipidation and recruited to the autophagosomes.39 LC3-I to LC3-II conversion therefore serves as a reliable marker for autophagosome formation. We observed a significant increase in LC3 expression (Figure 6A), percentage of cells with punctated LC3 staining (Figure 6B,C), and conversion of LC3-I to LC3-II in arthritic compared to nonarthritic paws (Figure 6D). Collectively, these results suggest that autophagy was engaged in KRN arthritis.
Figure 6.

Autophagy is engaged in KRN arthritis. Normal and day-9 arthritic paws were stained for LC3 by immunohistochemistry (IHC, A) or immunofluorescence (B). Autophagosome formation can be seen as punctated LC3 staining in arthritic paws (see higher magnification insets in B). Scale bar = 100 μm. (C) The percentage (%) of cells with punctated LC3 staining was significantly increased in arthritic paws (n = 3 mice per condition), ***p < 0.001 compared with normal paws. (D) LC3-I/LC3-II expression in paw lysates by immunoblotting. Actin serves as control for protein loading.
To determine whether autophagy modulation underlies the mechanism by which Fum-PD-induced NO suppressed inflammatory arthritis, LC3 turnover in animals treated with Fum-PD NP, without or with l-NAME coadministration, was studied. The LC3 turnover assay is based on the observation that LC3-II is degraded by lysosomal enzymes when autophagosomes fuse with lysosomes to form autolysosomes.39 Indeed Fum-PD NP treatment significantly decreased LC3-II levels (Figure 7A,B). In addition, the protein p62, also known as SQSTM1/sequestome1, is selectively incorporated into autophagosomes and degraded by autophagy.40 The expression of p62 thus inversely correlates with autophagic activity. We noted a similar decrease in p62 expression (p < 0.05) with Fum-PD NP treatment (Figure 7A,B). Furthermore, increased LC3 co-localization with the lysosome marker LAMP-1 was appreciable (Figure 7C,D), further suggesting autolysosome formation and enhanced autophagic flux following Fum-PD NP treatment. Concomitant administration of l-NAME reversed the effects of Fum-PD NP on autophagic flux, confirming the critical role of NO in autophagy modulation (Figure 7).
Figure 7.

Fum-PD NP increases autophagic flux in KRN arthritis. (A) Day-9 paw lysates derived from the different treatment groups in Figure 4 were blotted for LC3 and p62. Actin served as protein loading control. (B) Quantification: total LC3 (LC3-I and LC3-II) and p62 levels were first normalized to actin and expressed relative to Ctrl NP level, which was set at 1. Values represent mean ± SEM, n = 4 or 5 mice per treatment group. (C) Increased LC3 (green) and LAMP-1 (red) co-localization (arrowheads) following Fum-PD NP treatment. The effect was reversed by l-NAME. (D) Quantification of LC3+LAMP-1+ cells. Values represent mean ± SEM, n = 4 or 5 mice per treatment group. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar = 20 μm.
Studies have shown that autophagy can modulate the transcription, processing and/or release of cytokines.41 Consequently, mice deficient in specific Atg genes produce higher levels of cytokines and have been shown to be more susceptible to certain inflammatory diseases.6 Conversely, induction of autophagy inhibits NF-κB signaling, a pathway that plays a central role in the regulation of the cytokine response in RA.42 Consistent with these studies, we found that engagement of autophagy in KRN arthritis was accompanied by an increase in NF-κB-p65 phosphorylation (Figure 8A). On the other hand, enhanced autophagic flux (Figure 7A,B) following Fum-PD NP treatment led to marked reduction in IkappaB kinase alpha (IKKα) protein level and suppression of NF-κB-p65 phosphorylation (Figure 8B and Figure S4), all of which suggest a potential explanation for the global decrease in inflammatory cytokine levels observed in Figure 2C–F. Moreover, Fum-PD NP effects on NF-κB activity were NO-dependent (Figure 8C).
Figure 8.
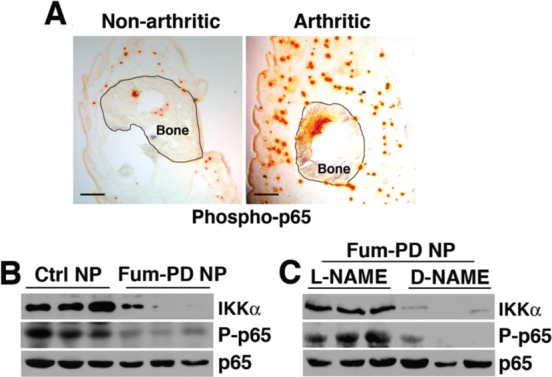
Fum-PD NP treatment suppresses NF-κB activation in KRN arthritis. (A) Paws from nonarthritic and day-9 KRN serum-induced arthritic mice were stained for phospho (P)-p65 (orange). Scale bar = 200 μm. (B) Paw lysates from day-9 arthritic paws derived from different treatment groups were blotted for total IKKα, P-p65, or total p65. (C) l-NAME, but not d-NAME, reversed Fum-PD NP-mediated IKKα degradation and suppression of p65 phosphorylation.
Discussion
NO is produced by the conversion of l-arginine to l-citrulline by different NOS isoforms:43 endothelial NOS (eNOS, Nos3) is constitutively active in endothelial cells and controls key vascular functions; neuronal NOS (nNOS) is the predominant source of NO in neurons; and inducible NOS (iNOS, Nos2) is expressed in many different cell types, including macrophages and endothelial cells. Expression and activity of iNOS were shown to be up-regulated during inflammatory responses.19 Although NO is known to play a role in inflammation, there is no consensus concerning its contribution to disease and cytokine expression in inflammatory arthritis. Elevated levels of NO activity have been reported in serum, urine, and synovial fluid of RA patients.44−46 Subsequent studies in experimental arthritis models seemed to confirm the detrimental effects of NO and the benefits of NO inhibition on disease manifestation.47,48 On the other hand, evidence also suggests that NO can have beneficial properties, and the absence of iNOS or selective inhibition of iNOS aggravated rather than attenuated experimental arthritis.49,50 We observed that Fum-PD NP-induced NO exerted anti-inflammatory effects in the studies presented herein. Perhaps the contradictory reports on the effects of NO in inflammatory joint disease may be reconciled by recognizing that NO likely has differential functions depending on the stage of the disease,51 the amount of NO generated or administered,52 and the tissues or cell types affected.53 As ανβ3-targeted Fum-PD-loaded nanoparticles homed specifically to angiogenic blood vessels, the local level of NO induced/released following Fum-PD nanoparticle exposure might be quite high but likely constrained to the neoendothelial cells or their immediate vicinity. While a small amount of NO may stimulate inflammatory cytokine/chemokine release, a large quantity of NO has been shown to suppress the inflammatory response.19
NO has also been shown to activate AMPK through different mechanisms. A recent report suggested that, in response to nitrosative stress induced by NO, ataxia-telangiectasia mutated (ATM) kinase was activated and signaled to AMPK.54 Others suggested that nitrosative stress might also activate AMPK through a mechanism involving Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) or the unfolded protein response (UPR).23,24 Regardless of the mechanism, we confirmed that Fum-PD activated AMPK in vitro and in vivo largely through a NO-dependent pathway, as l-NAME reversed the observed fumagillin-induced AMPK activation. The downstream consequences of AMPK activation are manifold and include the control of cell survival, activation, and metabolism.55
In recent years the role of AMPK in macrophage polarization has received increased attention. Macrophage responses to environmental stimuli are remarkably plastic and can give rise to different populations of cells with distinct functions. This polarization, as it is termed, may fuel disease progression or promote resolution of inflammation.33 AMPK is thought to act as a “master switch” of macrophage polarization by down-regulating NF-κB activation, although the exact mechanism by which AMPK inhibits NF-κB signaling remains unresolved.31,56−58 Our in vitro data confirmed that Fum-PD-induced AMPK activation promoted an anti-inflammatory macrophage phenotype with significantly diminished M1 cytokines (TNF-α) but without enhanced IL-10 production or arginase 1 expression (both M2 markers). That fumagillin-induced AMPK activation in vivo in KRN arthritis polarized macrophages toward an M2 phenotype was suggested but remains to be confirmed.
The activation of AMPK also leads to the inhibition of mTOR-dependent signaling activity.59 Inhibition of mTOR by AMPK activation (or by the addition of rapamycin) increases autophagy.60,61 In the KRN arthritis model, Fum-PD NPs generated a local nitrosative stress response, leading to the activation of AMPK and repression of mTOR activity, with subsequent increased autophagic flux. Although autophagy has been implicated in RA, the role of autophagy in disease progression has not been extensively explored. In a small clinical study the addition of the rapalogue everolimus to methotrexate had superior clinical efficacy compared to monotherapy with the disease-modifying drug methotrexate.62 Those results, considered with the current experimental data, shed light on the potential contribution of autophagy to RA pathogenesis. However, a more precise delineation of the role of autophagy in arthritis will require the silencing of specific Atg genes or the use of animals with selective deficiencies in specific Atg genes.
Autophagy may constitute a pathway through which signaling components are degraded, resulting in the termination or persistence of the signaling process. Several studies have shown a tight interplay between autophagy and NF-κB signaling pathway. Indeed, autophagy has been shown to mediate the selective degradation of NF-κB pathway components, including the IKK complex.63,64 Moreover, p62 is known to activate NF-κB in response to several stimuli including TNF-α and IL-1β.65 Conversely, degradation of p62 by autophagy suppresses NF-κB signaling,66,67 which is consistent with the findings that Fum-PD nanotherapy induced AMPK activation, mTOR inhibition, and enhanced autophagy. The consequent enhanced autophagic flux led to IKK degradation, which in turn suppressed p65 activation and NF-κB-driven inflammatory response in KRN arthritis. The present data also corroborated the earlier clinical report that modulation of autophagy (through mTOR inhibition) with the rapalogue everolimus62 may have additional beneficial effects in the treatment of RA.
Conclusions
In summary, the results herein suggest that antiangiogenic nanotherapy with Fum-PD NPs indirectly suppressed inflammatory cytokine production in the inflamed paws via the local production of (endothelial) NO, which likely acted on nearby recruited macrophages (Figure S5), leading to the activation of AMPK. NO-induced AMPK activation exerted a counter inflammatory response by modulating macrophage activity and enhancing autophagic flux, which in turn suppressed NF-κB-p65 activation and inflammatory cytokine release. Conversely, inhibition of NO production by l-NAME administered concomitantly with Fum-PD nanotherapy diminished AMPK activation and reversed the suppression of NF-κB-mediated inflammatory responses. The results also suggest that close examination of the effects of Fum-PD nanotherapy on autophagy may yield unanticipated secondary benefits that can be exploited to improve antiangiogenic therapies in the treatment of various inflammatory disease processes.
Methods
Chemical Reagents
Unless otherwise listed, all solvents and reagents were purchased from Aldrich Chemical Co. (St. Louis, MO, USA) and used as received. Anhydrous chloroform and methanol were purchased from Aldrich Chemical Co. Perfluorooctyl bromide was acquired from Exfluor Inc. (Round Rock, TX, USA). 1-Palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine and high-purity egg yolk phospholipids were purchased from Avanti Polar Lipids, Inc. Argon and nitrogen (Ultra High Purity: UHP, 99.99%) were used for storage of materials. Fumagillin dicyclohexylamine salt was provided by the National Cancer Institute repository of natural products for investigation.
Preparation of Sn-2 Phospholipase Labile Fumagillin Prodrug
Synthesis of the Sn-2 prodrug was accomplished in two steps: (1) saponifying fumagillin dicyclohexylamine salt to fumagillol, (2) esterifying the product with 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC) as previously.10 Briefly, fumagillin dicyclohexylamine salt in 1:1 methanol/water was treated with 35% NaOH, stirred in an ice bath for 2 h, warmed to room temperature, treated with another equivalent of 35% NaOH, and then stirred in an ice bath until the starting material was not detected by TLC (∼4 h). After evaporating the methanol and resolublizing in ethyl acetate, the mixture was extracted with 5% citric acid, brine, bicarbonate, and brine, dried with MgSO4, and then concentrated in vacuo. The crude product was purified with activated charcoal in acetonitrile and then filtered through a Celite pad. Yield: colorless solid, 59 mg (70%). HRMS found: MH+ (283.3).
A solution of C16-09:0 (COOH) PC 1-hexadecyl-2-azelaoyl-sn-glycero-3-phosphocholine followed by 4-dimethylaminopyridine and N,N′-dicyclohexylcarbodiimide was added to a solution of fumagillol in dry dichloromethane. The reaction mixture was stirred overnight at ambient temperature and then passed over a short pad of silica gel using EtOAc/n-hexane. The filtered solvent was removed in vacuo, and the oil residue was purified by column chromatography on SiO2 using EtOAc/n-hexane for elution to yield the fumagillin prodrug (Fum-PD) compound as a pale yellowish solid. HR-MS found: 991.5475 (M + 2K – 2H).
Preparation of ανβ3-Targeted Fumagillin Nanoparticles
Phospholipid-encapsulated perfluorocarbon nanoparticles were prepared as a microfluidized suspension of 20% (v/v) perfluorooctyl bromide, 2.0% (w/v) surfactant comixture, and 1.7% (w/v) glycerin in pH 6.5 carbonate buffer in distilled deionized water (76.3% v/v). ανβ3-Peptidomimetic antagonist conjugated to PEG2000-phosphatidylethanolamine (Kereos, St. Louis, MO, USA) was used for homing angiogenesis. The surfactant comixture of nanoparticles included ∼97.6 mol % lecithin, 0.15 mol % αvβ3-ligand conjugated lipid, and 2.28 mol % Fum-PD (∼0.5 mM). Nontargeted nanoparticles excluded the homing ligand. The surfactant comixture was dissolved in chloroform, evaporated under reduced pressure, dried in a 40 °C vacuum oven overnight, and dispersed into water by probe sonication. The suspensions were combined with the perfluorocarbon, buffer, and glycerin with pH adjusted to 6.5 and homogenized with an S110 Microfluidics emulsifier (Microfluidics, Newton, MA, USA) at 20 000 psi for 4 min. The nanoparticles were aliquoted under a biohood into sterile vials and sealed under inert gas until use.
The ανβ3-integrin antagonist was a quinalone nonpeptide developed by Lantheus Medical Imaging (Billerica, MA, USA) and synthesized by Kereos (U.S. Patent 6,511,648 and related patents). The vitronectin antagonist was reported and characterized as the 111In-DOTA conjugate RP478 and cyan 5.5 homologue TA145.68 The homing specificity of the ligand was demonstrated and characterized with the Matrigel plug implanted in Rag1tm1Mom Tg(Tie-2-lacZ)182-Sato and C57Bl/6 mice. Nanoparticles have an IC50 of 50 pM for the Mn2+-activated ανβ3-integrin.69
The nominal hydrodynamic diameter (Dh) of the αvβ3-targeted fumagillin prodrug PFC nanoparticle and control particles based on dynamic light scattering measurements (Brookhaven ZetaPlus, Brookhaven Instruments Corporation) in aqueous solution was 252 nm, with a polydispersity of 0.14 and zeta potential of −18 mV. Incorporation of prodrug at 2.28 mol % (∼0.5 mM) within the surfactant comixture had negligible impact on particle sizes.
Arthritis Induction and Treatment
All mice were kept in a pathogen-free condition at Washington University Specialized Research Facility, and all the experiments were performed in compliance with federal laws and strictly according to protocols approved by the Division of Comparative Medicine at Washington University School of Medicine. Arthritis was induced using the KRN mouse model of inflammatory arthritis as previously described.11,13,14 Six- to eight-week-old male C57BL/6 mice (The Jackson Laboratory) were injected ip with 150 μL of KRN serum on day 0 to induce arthritis. Clinical manifestation of arthritis was assessed daily by an observer blinded to the treatment on a scale of 0–3 (0 = no swelling or erythema, 1 = slight swelling or erythema, 2 = moderate erythema and swelling in multiple digits or entire paw, 3 = pronounced erythema and swelling of entire paw, maximum score of 12 per mouse). Change from baseline in paw ankle thickness was determined daily by dial calipers, and an average change in ankle thickness was determined for each mouse from the two hind paw measurements. Mice were randomly assigned to treatment groups and injected iv at 2 μL/g (μL nanoparticles/g body weight) with αvβ3-Ctrl NPs without drug or αvβ3-Fum-PD NPs at 0.3 μg drug/g body weight or equivalent dose of free Fum-PD on days 2, 3, and 4. In the NOS inhibition experiment, mice were injected iv with 100 mg/kg of N-nitro-l-arginine methyl ester (l-NAME, Santa Cruz Biotechnology) or N-nitro-d-arginine methyl ester (d-NAME, Santa Cruz Biotechnology) in PBS 30 min before each NP injection. On day 9, the mice were sacrificed, and paws were snap frozen and kept at −80 °C for protein purification, cytokine analysis, and Western blotting. For immunohistochemistry, paws were embedded in OCT, sectioned at 9 μm, and kept at −80 °C until ready for use.
In Vitro Cell Culture and Treatment
SEVC4 endothelial cells (obtained from the American Type Culture Collection) were cultured in DMEM supplemented with 10% fetal calf serum (Gibco) and streptomycin–penicillin solution at 37 °C in a 5% CO2 atmosphere. SEVC4 cells were plated in a 96-well plate at the density of 2 × 105 cells/well and treated with different concentrations of Fum-PD (in 0.1% DMSO) or 0.1% DMSO for 48 h, and supernatants were collected. NO release was detected with Griess reagent (Sigma-Aldrich) as previously described,17 and cytokines in cell culture supernatants were detected with cytometric bead array (CBA) using the mouse inflammatory kit (BD Bioscience).
Peritonitis was induced by ip injection of sterile thioglycollate (4 wt %/vol in 1 mL of sterile saline; Sigma-Aldrich). On day 5, the mice were sacrificed by carbon dioxide exposure, and peritoneal cavities were lavaged with 8 mL of cold PBS. Cells were allowed to adhere on the surface of Petri dishes for 45 min at 37 °C and washed twice with cell culture media to remove nonadherent cells. Adherent cells (macrophages) were detached with 5 mM EDTA in DMEM supplemented with 15% horse serum for 15 min at 37 °C. Cells were washed twice with culture media and seeded with or without SEVC4 cells at a ratio of 1:1 (2 × 105 cells of each cell type/well in 96-well plates). Macrophages or cocultures were treated with different concentrations of fumagillin prodrug or SNAP (25 μg/mL). In some instances, cocultures were pretreated with l-NAME or d-NAME (20 μM) for 30 min followed by exposure to fumagillin prodrug for 48 h. Cytokine levels in cell culture supernatants were assessed with CBA. The cells in cultures were also harvested, and cell lysates prepared for Western blotting analysis.
Immunohistochemistry and Immunofluorescence
The frozen sections (9 μm) of harvested paws were fixed in 4% PFA for 20 min at room temperature, incubated with rat anti-mouse CD31 (1:100, cat. 102507, Biolegend Technology), rabbit anti-mouse iNOS (1:400, cat. ab3523, Abcam), rat anti-Mac-3 monoclonal antibody (1:200, cat. CL8943AP, Cedarlane Laboratories), rabbit anti-mouse LC3B (1:200, cat. L7543, Sigma-Aldrich), phospho-p65 (phosphorylation site: S536, dilution 1:100, cat. 3033, Cell Signaling Technology), and rat anti-mouse LAMP-1 (1:100, cat. SC19992, Santa Cruz) followed by FITC-conjugated anti-rabbit or TRITC-conjugated anti-rat secondary antibody (1:100, Jackson Immunoresearch Laboratory). All images were visualized on a Nikon Eclipse microscope and acquired at the same exposure with QCapture software. Merged and single-color images were loaded into ImageJ software (http://rsb.info.nih.gov/ij) for analysis. Double (LAMP-1+LC3+) positive cells or single positive (LC3+) cells were marked with different colors, each group was tallied separately, and the ratio of double to single positive cells was calculated. Data were obtained from six to eight nonoverlapping fields per section and four to six sections per treatment, 3–5 mice per treatment.
NOS immunofluorescence was performed on SVEC4 cells or primary peritoneal macrophages that were treated with fumagillin prodrug for 48 h in LAB-TEK Permanox chambered slides (Thermo Fisher Scientific). Cells were fixed in acetone at −20 °C for 5 min and incubated with rabbit anti-mouse iNOS (1:400, cat. ab3523, Abcam), followed by incubation with TRITC-conjugated anti-rabbit secondary antibody (1:100, Jackson Immunoresearch Laboratory). All images were visualized on a Nikon Eclipse microscope and acquired at the same exposure with QCapture software. All immunofluorescence grading was performed by an observer blinded to the treatment.
Apoptotic cells were detected using a DNA fragmentation (TUNEL) assay kit (Millipore) in which the addition of digoxin conjugated nucleotides to the free 3′-OH termini of the fragmented DNA strand was visualized by fluorescence microscopy with a FITC-labeled anti-digoxin antibody (1:100, Jackson Immunoresearch Laboratory).
Cytokine Analysis
Paws were homogenized in 1 mL of PBS with proteinase inhibitor cocktail and cleared by centrifugation. Cells were lysed in 100 μL of PBS with proteinase inhibitor cocktail and cleared by centrifugation. Concentrations of IFN-γ, IL-6, IL-10, IL12p70, MCP-1, and TNF-α in an equivalent volume of paw lysates, air pouch lavages, and cell culture supernatants were measured by CBA using the mouse inflammatory kit (BD Bioscience) according to the manufacturer’s instructions. Concentration of IL-1β in paw lysates was detected by using a commercially available ELISA kit (R&D System).
Western Blotting
Cell or paw lysates were prepared with 1% NP-40 lysis buffer containing proteinase inhibitor cocktail (Sigma) and 50 mM sodium fluoride (Sigma). Briefly, cells or paws were homogenized, sonicated in the lysis buffer, and cleared by centrifugation at 14 000 rpm for 10 min at 4 °C. Proteins from cell or paw lysates were quantified and equivalent amount fractionated by SDS/PAGE under reducing conditions. Membranes were blotted with anti-mouse LC3B (1:1000 cat. L7543 Sigma-Aldrich) or anti-mouse β-actin (1:500, cat. sc-1615, Santa Cruz Biotechnology) antibody; antibody anti-mouse phospho-AMPKα (Thr172, 1:1000, cat. 2535), AMPKα (1:1000, cat. 2603), phospho-mTOR (Ser2448, 1:1000, cat. 5536), IKKα (1:1000, cat. 2682), phospho-p65 (Ser536, 1:1000, cat. 3033), or p65 antibody (1:1000, cat. 8242) antibody from Cell Signaling Technology; and monoclonal mouse anti-mouse p62 antibody (1:1000, Cat. MABC32, Millipore). The blots were washed and incubated with HRP-conjugated donkey anti-goat or anti-rabbit IgG (1:2000, Jackson ImmunoResearch Laboratories) or HRP-conjugated mouse anti-mouse IgM (1:2000, μ-chain-specific; Jackson ImmunoResearch Laboratories). Bands were visualized using a SuperSignal Western blotting kit (Thermo Scientific). Briefly, Western blotting images were scanned into Carestream Molecular Imaging software. The band of interest was selected, and the average density was calculated. The intensity of the bands was normalized to that of β-actin.
RT-PCR
Cells from cocultures were harvested and placed in RNAlater stabilizing solution (Life Technologies) for at least 24 h prior to RNA processing. Total RNA was isolated using a Qiagen RNeasy mini kit (Qiagen Inc.) as recommended by the manufacturers. Two micrograms of RNA was used to synthesize cDNA by reverse transcription using the high-capacity cDNA reverse transcriptase kit (Applied Biosystems, Life Technologies) on a Multigene thermal cycler system (Labnet International Inc., Edison, NJ, USA). The reverse-transcription product served as the template for real-time PCR analysis on a 7500 RT PCR system (Applied Biosystems) using fluorogenic primers for arginase 1 (Mm00440502_m1, Applied Biosystems) according to the manufacturer’s protocol. Results from each sample were normalized to the concentration of GAPDH mRNA measured in the same samples.
Statistics
Comparisons between two groups were performed by two-tailed, unpaired t test without correction. Grading data were analyzed with the χ2 test. Comparisons between multiple groups (≥3) were performed by one-way ANOVA. Equality of variance assumption was tested, and Bonferroni’s correction for multiple comparisons was performed. Normality assumption appeared to be met, and tests for equality variance were conducted. The sample size (number of animals per treatment) chosen is based on means and variances from similar experiments with the KRN arthritis mouse model and afforded detection of 20% differences between experimental groups with a statistical power of 0.80 assuming a 2-sided alpha of <0.05. The F test was used to compare variances within each group of data, and the difference in variances was found to be not significant between groups.
Acknowledgments
The authors would like to thank Dr. P. Allen (Washington University School of Medicine) for the generous gift of KRN sera. This work was supported in part by NIH grants AI049261 and AR056468 and a bridge fund from the Dean and Department of Medicine at Washington University to C.T.N.P., and NIH grants HL112518, HL113392, CA154737, CA136398, NS073457, and DOD CA100623 to G.M.L.
The authors declare no competing financial interest.
Author Contributions
§ H.-F. Zhou and H. Yan contributed equally.
Author Present Address
⊥ Bioengineering and Beckman Institute, University of Illinois at Urbana–Champaign, Urbana, Illinois, United States.
Supporting Information Available
Supplementary Methods. Figure S1: Free Fum-PD is ineffective at suppressing KRN arthritis. Figure S2: Cytokine response in vitro following Fum-PD exposure. Figure S3: Alternatively activated M2 macrophages predominate in chronic phase of arthritis. Figure S4: Fumagillin nanotherapy-mediated NF-κB p65 suppression is NO-dependent. Figure S5: Endothelial-macrophage tight interactions in the inflamed synovium. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ebos J. M.; Kerbel R. S. Antiangiogenic Therapy: Impact on Invasion, Disease Progression, and Metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson G. C.; Hicklin D. J.; Ellis L. M. Antiangiogenic Therapy–Evolving View Based on Clinical Trial Results. Nat. Rev. Clin. Oncol. 2012, 9, 297–303. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Role of Myeloid Cells in Vascular Endothelial Growth Factor-Independent Tumor Angiogenesis. Curr. Opin. Hematol. 2010, 17, 219–224. [DOI] [PubMed] [Google Scholar]
- Levine B.; Mizushima N.; Virgin H. W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E.; DiPaola R. S. The Double-Edged Sword of Autophagy Modulation in Cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. A.; Mills K. H.; Harris J. Autophagy and Inflammatory Diseases. Immunol. Cell Biol. 2013, 91, 250–258. [DOI] [PubMed] [Google Scholar]
- Sin N.; Meng L.; Wang M. Q.; Wen J. J.; Bornmann W. G.; Crews C. M. The Anti-Angiogenic Agent Fumagillin Covalently Binds and Inhibits the Methionine Aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 6099–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Widom J.; Kemp C. W.; Crews C. M.; Clardy J. Structure of Human Methionine Aminopeptidase-2 Complexed with Fumagillin. Science 1998, 282, 1324–1327. [DOI] [PubMed] [Google Scholar]
- Ingber D.; Fujita T.; Kishimoto S.; Sudo K.; Kanamaru T.; Brem H.; Folkman J. Synthetic Analogues of Fumagillin that Inhibit Angiogenesis and Suppress Tumour Growth. Nature 1990, 348, 555–557. [DOI] [PubMed] [Google Scholar]
- Pan D.; Sanyal N.; Schmieder A. H.; Senpan A.; Kim B.; Yang X.; Hu G.; Allen J. S.; Gross R. W.; Wickline S. A.; etal. Antiangiogenic Nanotherapy with Lipase-Labile Sn-2 Fumagillin Prodrug. Nanomedicine (London) 2012, 7, 1507–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. F.; Yan H.; Senpan A.; Wickline S. A.; Pan D.; Lanza G. M.; Pham C. T.N. Suppression of Inflammation in a Mouse Model of Rheumatoid Arthritis Using Targeted Lipase-Labile Fumagillin Prodrug Nanoparticles. Biomaterials 2012, 33, 8632–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H.; Gauguier D.; Ohmura K.; Gonzalez A.; Duchatelle V.; Danoy P.; Garchon H. J.; Degott C.; Lathrop M.; Benoist C.; etal. Genetic Influences on the End-Stage Effector Phase of Arthritis. J. Exp. Med. 2001, 194, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. F.; Chan H. W.; Wickline S. A.; Lanza G. M.; Pham C. T. N. αvβ3-Targeted Nanotherapy Suppresses Inflammatory Arthritis in Mice. FASEB J. 2009, 23, 2978–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. F.; Hu G.; Wickline S. A.; Lanza G. M.; Pham C. T.N. Synergistic Effect of Antiangiogenic Nanotherapy Combined with Methotrexate in the Treatment of Experimental Inflammatory Arthritis. Nanomedicine (London) 2010, 5, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier S. G.; Lazarus D. D.; Clark E.; Doyle B.; Labenski M. T.; Thompson C. D.; Westlin W. F.; Hannig G. A Methionine Aminopeptidase-2 Inhibitor, PPI-2458, for the Treatment of Rheumatoid Arthritis. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10768–10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge J.; Madden L.; Essex D.; Binks M.; Malhotra R.; Paleolog E. M. Methionine Aminopeptidase-2 Blockade Reduces Chronic Collagen-Induced Arthritis: Potential Role for Angiogenesis Inhibition. Arthritis Res. Ther. 2007, 9, R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T.; Kaneko Y.; Tsukamoto A.; Han K.; Ichinose M.; Kimura S. Suppression of Hepatoma Growth and Angiogenesis by a Fumagillin Derivative TNP470: Possible Involvement of Nitric Oxide Synthase. Cancer Res. 1998, 58, 3751–3756. [PubMed] [Google Scholar]
- Brune B. Nitric Oxide: NO Apoptosis or Turning It ON?. Cell Death Differ. 2003, 10, 864–869. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y. The Regulatory Role of Nitric Oxide in Proinflammatory Cytokine Expression during the Induction and Resolution of Inflammation. J. Leukoc. Biol. 2010, 88, 1157–1162. [DOI] [PubMed] [Google Scholar]
- Shibata T.; Nagata K.; Kobayashi Y. Cutting edge: A Critical Role of Nitric Oxide in Preventing Inflammation upon Apoptotic Cell Clearance. J. Immunol. 2007, 179, 3407–3411. [DOI] [PubMed] [Google Scholar]
- Miller M. R.; Megson I. L. Recent Developments in Nitric Oxide Donor Drugs. Br. J. Pharmacol. 2007, 151, 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A.; Whiteman M.; Halliwell B. Scavenging of Hydroxyl Radicals but Not of Peroxynitrite by Inhibitors and Substrates of Nitric Oxide Synthases. Br. J. Pharmacol. 1997, 122, 1702–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Xie Z.; Dong Y.; Wang S.; Liu C.; Zou M. H. Identification of Nitric Oxide as an Endogenous Activator of the AMP-Activated Protein Kinase in Vascular Endothelial Cells. J. Biol. Chem. 2008, 283, 27452–27461. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Meares G. P.; Hughes K. J.; Naatz A.; Papa F. R.; Urano F.; Hansen P. A.; Benveniste E. N.; Corbett J. A. IRE1-Dependent Activation of AMPK in Response to Nitric Oxide. Mol. Cell. Biol. 2011, 31, 4286–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag D.; Carling D.; Stout R. D.; Suttles J. Adenosine 5′-Monophosphate-Activated Protein Kinase Promotes Macrophage Polarization to an Anti-Inflammatory Functional Phenotype. J. Immunol. 2008, 181, 8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Kahn B. B.; Shi H.; Xue B. Z. Macrophage α1 AMP-Activated Protein Kinase (α1AMPK) Antagonizes Fatty Acid-Induced Inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S.; Fullerton M. D.; Schertzer J. D.; Sikkema S.; Marcinko K.; Walkley C. R.; Izon D.; Honeyman J.; Chen Z. P.; van Denderen B. J.; etal. Hematopoietic AMPK β1 Reduces Mouse Adipose Tissue Macrophage Inflammation and Insulin Resistance in Obesity. J. Clin. Invest. 2011, 121, 4903–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N.; Giri S.; Prasad R.; Salem M. L.; Singh A. K.; Singh I. 5-Aminoimidazole-4-Carboxamide Ribonucleoside: a Novel Immunomodulator with Therapeutic Efficacy in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 175, 566–574. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Zmijewski J. W.; Lorne E.; Liu G.; Park Y. J.; Tsuruta Y.; Abraham E. Activation of AMPK Attenuates Neutrophil Proinflammatory Activity and Decreases the Severity of Acute Lung Injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai A.; Ma A. G.; Yong M.; Weiss C. R.; Ma Y.; Guan Q.; Bernstein C. N.; Peng Z. AMPK Agonist Downregulates Innate and Adaptive Immune Responses in TNBS-Induced Murine Acute and Relapsing Colitis. Biochem. Pharmacol. 2010, 80, 1708–1717. [DOI] [PubMed] [Google Scholar]
- Srivastava R. A.; Pinkosky S. L.; Filippov S.; Hanselman J. C.; Cramer C. T.; Newton R. S. AMP-Activated Protein Kinase: An Emerging Drug Target to Regulate Imbalances in Lipid and Carbohydrate Metabolism to Treat Cardio-Metabolic Diseases. J. Lipid Res. 2012, 53, 2490–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G.; Myers R.; Li Y.; Chen Y.; Shen X.; Fenyk-Melody J.; Wu M.; Ventre J.; Doebber T.; Fujii N.; etal. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Invest. 2001, 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. M.; Edwards J. P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F. O.; Helming L.; Gordon S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [DOI] [PubMed] [Google Scholar]
- Inoki K.; Zhu T.; Guan K. L. TSC2Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Stein M.; Keshav S.; Harris N.; Gordon S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J. Exp. Med. 1992, 176, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meley D.; Bauvy C.; Houben-Weerts J. H.; Dubbelhuis P. F.; Helmond M. T.; Codogno P.; Meijer A. J. AMP-Activated Protein Kinase and the Regulation of Autophagic Proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [DOI] [PubMed] [Google Scholar]
- Levine B.; Kroemer G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N.; Yoshimori T.; Levine B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G.; Lamark T.; Brech A.; Outzen H.; Perander M.; Overvatn A.; Stenmark H.; Johansen T. p62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. Autophagy and Cytokines. Cytokine 2011, 56, 140–144. [DOI] [PubMed] [Google Scholar]
- Simmonds R. E.; Foxwell B. M. Signalling, Inflammation and Arthritis: NF-κB and its Relevance to Arthritis and Inflammation. Rheumatology (Oxford) 2008, 47, 584–590. [DOI] [PubMed] [Google Scholar]
- Forstermann U.; Sessa W. C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33829–37837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell A. J.; Blake D. R.; Palmer R. M.; Moncada S. Increased Concentrations of Nitrite in Synovial Fluid and Serum Samples Suggest Increased Nitric Oxide Synthesis in Rheumatic Diseases. Ann. Rheum. Dis. 1992, 51, 1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski P. S.; England A. J.; Dykhuizen R.; Copland M.; Benjamin N.; Reid D. M.; Ralston S. H. Elevated Nitric Oxide Production in Rheumatoid Arthritis. Detection Using the Fasting Urinary Nitrate:Creatinine Ratio. Arthritis Rheum. 1996, 39, 643–647. [DOI] [PubMed] [Google Scholar]
- McInnes I. B.; Leung B. P.; Field M.; Wei X. Q.; Huang F. P.; Sturrock R. D.; Kinninmonth A.; Weidner J.; Mumford R.; Liew F. Y. Production of Nitric Oxide in the Synovial Membrane of Rheumatoid and Osteoarthritis Patients. J. Exp. Med. 1996, 184, 1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney-Francis N.; Allen J. B.; Mizel D. E.; Albina J. E.; Xie Q. W.; Nathan C. F.; Wahl S. M. Suppression of Arthritis by an Inhibitor of Nitric Oxide Synthase. J. Exp. Med. 1993, 178, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F.; Wu P.; Xiao N.; Qiu F.; Zeng Q. P. Nitric Oxide-Driven Hypoxia Initiates Synovial Angiogenesis, Hyperplasia and Inflammatory Lesions in Mice. PloS One 2012, 7, e34494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veihelmann A.; Landes J.; Hofbauer A.; Dorger M.; Refior H. J.; Messmer K.; Krombach F. Exacerbation of Antigen-Induced Arthritis in Inducible Nitric Oxide Synthase-Deficient Mice. Arthritis Rheum. 2001, 44, 1420–1427. [DOI] [PubMed] [Google Scholar]
- McCartney-Francis N. L.; Song X.; Mizel D. E.; Wahl S. M. Selective Inhibition of Inducible Nitric Oxide Synthase Exacerbates Erosive Joint Disease. J. Immunol. 2001, 166, 2734–2740. [DOI] [PubMed] [Google Scholar]
- Veihelmann A.; Hofbauer A.; Krombach F.; Dorger M.; Maier M.; Refior H. J.; Messmer K. Differential Function of Nitric Oxide in Murine Antigen-Induced Arthritis. Rheumatology (Oxford) 2002, 41, 509–517. [DOI] [PubMed] [Google Scholar]
- Gomaa A. A.; Elshenawy M. M.; Afifi N. A.; Mohammed E. A.; Thabit R. H. Dual Effect of Nitric Oxide Donor on Adjuvant Arthritis. Int. Immunopharmacol. 2009, 9, 439–447. [DOI] [PubMed] [Google Scholar]
- Leite A. C.; Cunha F. Q.; Dal-Secco D.; Fukada S. Y.; Girao V. C.; Rocha F. A. Effects of Nitric Oxide on Neutrophil Influx Depends on the Tissue: Role of Leukotriene B4 and Adhesion Molecules. Br. J. Pharmacol. 2009, 156, 818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi D. N.; Chowdhury R.; Trudel L. J.; Tee A. R.; Slack R. S.; Walker C. L.; Wogan G. N. Reactive Nitrogen Species Regulate Autophagy through ATM-AMPK-TSC2-Mediated Suppression of mTORC1. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E2950–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisslthaler B.; Fleming I. Activation and Signaling by the AMP-Activated Protein Kinase in Endothelial Cells. Circ. Res. 2009, 105, 114–127. [DOI] [PubMed] [Google Scholar]
- Cacicedo J. M.; Yagihashi N.; Keaney J. F. Jr.; Ruderman N. B.; Ido Y. AMPK Inhibits Fatty Acid-Induced Increases in NF-κB Transactivation in Cultured Human Umbilical Vein Endothelial Cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1299. [DOI] [PubMed] [Google Scholar]
- Okayasu T.; Tomizawa A.; Suzuki K.; Manaka K.; Hattori Y. PPARα Activators Upregulate eNOS Activity and Inhibit Cytokine-Induced NF-κB Activation through AMP-Activated Protein Kinase Activation. Life Sci. 2008, 82, 884–891. [DOI] [PubMed] [Google Scholar]
- Hattori Y.; Nakano Y.; Hattori S.; Tomizawa A.; Inukai K.; Kasai K. High Molecular Weight Adiponectin Activates AMPK and Suppresses Cytokine-Induced NF-κB Activation in Vascular Endothelial Cells. FEBS Lett. 2008, 582, 1719–1724. [DOI] [PubMed] [Google Scholar]
- Laplante M.; Sabatini D. M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T.; Klionsky D. J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codogno P.; Meijer A. J. Autophagy and Signaling: their Role in Cell Survival and Cell Death. Cell Death Differ. 2005, 12Suppl 21509–1518. [DOI] [PubMed] [Google Scholar]
- Bruyn G. A.; Tate G.; Caeiro F.; Maldonado-Cocco J.; Westhovens R.; Tannenbaum H.; Bell M.; Forre O.; Bjorneboe O.; Tak P. P.; etal. Everolimus in Patients with Rheumatoid Arthritis Receiving Concomitant Methotrexate: A 3-Month, Double-Blind, Randomised, Placebo-Controlled, Parallel-Group, Proof-of-Concept Study. Ann. Rheum. Dis. 2008, 67, 1090–1095. [DOI] [PubMed] [Google Scholar]
- Qing G.; Yan P.; Xiao G. Hsp90 Inhibition Results in Autophagy-Mediated Proteasome-Independent Degradation of IκB Kinase (IKK). Cell Res. 2006, 16, 895–901. [DOI] [PubMed] [Google Scholar]
- Salminen A.; Hyttinen J. M.; Kauppinen A.; Kaarniranta K. Context-Dependent Regulation of Autophagy by IKK-NF-κB Signaling: Impact on the Aging Process. Int. J. Cell Biol. 2012, 2012, 849541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J.; Diaz-Meco M. T. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. M.; Shin D. M.; Yuk J. M.; Shi G.; Choi D. K.; Lee S. H.; Huang S. M.; Kim J. M.; Kim C. D.; Lee J. H.; etal. Autophagy Negatively Regulates Keratinocyte Inflammatory Responses via Scaffolding Protein p62/SQSTM1. J. Immunol. 2011, 186, 1248–1258. [DOI] [PubMed] [Google Scholar]
- Duran A.; Linares J. F.; Galvez A. S.; Wikenheiser K.; Flores J. M.; Diaz-Meco M. T.; Moscat J. The Signaling Adaptor p62 is an Important NF-κB Mediator in Tumorigenesis. Cancer Cell 2008, 13, 343–354. [DOI] [PubMed] [Google Scholar]
- Meoli D. F.; Sadeghi M. M.; Krassilnikova S.; Bourke B. N.; Giordano F. J.; Dione D. P.; Su H.; Edwards D. S.; Liu S.; Harris T. D.; etal. Noninvasive Imaging of Myocardial Angiogenesis Following Experimental Myocardial Infarction. J. Clin. Invest. 2004, 113, 1684–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder A. H.; Caruthers S. D.; Zhang H.; Williams T. A.; Robertson J. D.; Wickline S. A.; Lanza G. M. Three-Dimensional MR Mapping of Angiogenesis with α5β1(αvβ3)-Targeted Theranostic Nanoparticles in the MDA-MB-435 Xenograft Mouse Model. FASEB J. 2008, 22, 4179–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.