

Summary
Photodynamic therapy (PDT) has been known for over a hundred years, but is only now becoming widely used. Originally developed as cancer therapy, some of its most successful applications are for non-malignant disease. The majority of mechanistic research into PDT, however, is still directed towards anti-cancer applications. In the final part of series of three reviews, we will cover the possible reasons for the well-known tumor localizing properties of photosensitizers (PS). When PS are injected into the bloodstream they bind to various serum proteins and this can affect their phamacokinetics and biodistribution. Different PS can have very different pharmacokinetics and this can directly affect the illumination parameters. Intravenously injected PS undergo a transition from being bound to serum proteins, then bound to endothelial cells, then bound to the adventitia of the vessels, then bound either to the extracellular matrix or to the cells within the tumor, and finally to being cleared from the tumor by lymphatics or blood vessels, and excreted either by the kidneys or the liver. The effect of PDT on the tumor largely depends at which stage of this continuous process light is delivered.
The anti-tumor effects of PDT are divided into three main mechanisms. Powerful anti-vascular effects can lead to thrombosis and hemorrhage in tumor blood vessels that subsequently lead to tumor death via deprivation of oxygen and nutrients. Direct tumor cell death by apoptosis or necrosis can occur if the PS has been allowed to be taken up by tumor cells. Finally the acute inflammation and release of cytokines and stress response proteins induced in the tumor by PDT can lead to an influx of leukocytes that can both contribute to tumor destruction as well as to stimulate the immune system to recognize and destroy tumor cells even at distant locations.
Keywords: Photodynamic therapy, Cancer, Pharmacokinetics, Biodistribution, Cellular destruction, Vascular shutdown, Anti-tumor immunity
Introduction
The history of the development of photodynamic therapy (PDT) for cancer can be broadly divided into two areas. One area concerns studies on the ability of certain porphyrins and related tetrapyr-role compounds to localize in cancers after injection into the bloodstream. The second area is concerned with the tumoricidal effects when cancers were illuminated with visible light after the patient had received prior treatment with various photosensitizers (PS). This is the third part of a series of three reviews. In Part 1 [1] we covered the chemical structure, photochemistry, photophysics and sub-cellular localization of PS. In Part 2 [2] we discussed the changes in cellular metabolism and intracellular signalling and modes of cell death when cells treated with PS are illuminated in culture. We now come to consider the mechanisms that operate when PDT is carried out in vivo.
The vast explosion of biomedical research in recent decades has led to the ready availability of multiple animal models of cancer. Firstly the development of immunocompromised mice and rats (nude, SCID and beige) has allowed many human tumor cell lines to be easily grown as tumors in vivo in laboratory animals. Secondly there is now a large range of inbred mice and rat strains where each individual member of a strain has identical major histocompatibility complex haplotypes. This means that tumors that arise in any individual member of that strain and from which a stable cell line can be isolated, can be freely transplanted amongst members of that strain in the presence of a fully functioning immune system. These two laboratory models (human tumor xenografts and syngeneic rodent tumors) can be implanted subcutaneously, or alternatively can be implanted orthotopically in the organ of origin. Thirdly, chemical and radiation induced carcinogenesis has allowed tumors to be induced in many species of animal with more or less reliability, and can be thought to provide a more natural tumor model than transplanted tumors. Fourthly the discovery of oncogenes and tumor suppressor genes has allowed the creation of transgenic mice that either over express an oncogene or lack a tumor suppressor gene, and more or less reliably develop cancer in a manner that is probably most similar to the human situation. The choice of the most appropriate animal model of cancer to test PDT is complex and multifactorial and is often not rationally justified. However, the fact that practical light delivery to the tumor is an integral part of PDT means that the majority of reported studies have used subcutaneous (s.c.) tumors.
PS pharmacokinetics and biodistribution
When PS are injected into the bloodstream a certain series of events commences that can take very different lengths of time to reach completion for different PS. Firstly depending on the delivery solvent or vehicle that is used for the PS injection, the PS must come to equilibrium with the components of the circulating blood. This can involve the PS disaggregating from itself or its delivery vehicle and binding instead to various protein components of serum (see later). In addition the blood has many circulating cells (erythrocytes and leukocytes) that would be available to bind injected PS [3]. Secondly the circulating PS must bind to the walls of the blood vessels, and it is thought that the nature of the various sizes and characteristics of blood vessels in the tumor and normal tissues, and different physiological types of vessels in various organs governs to a great extent where the PS localizes. Thirdly the PS will penetrate through the wall of the blood vessel at a rate that probably depends on how strong the initial binding of the PS to the intimal surface was. PS that bind strongly to the blood vessel wall will take a longer time to cross the entire wall of the blood vessel, and those that have an initial weaker binding will pass through quicker. Fourthly after extravasating, the PS will diffuse throughout the parenchyma of the organ or tumor to which it has been delivered. If the organ happens to be the liver or other metabolically active organ, the PS may be subject to changes by metabolic enzymes, but this is thought to be unlikely for most tetrapyrrole PS in common clinical use. Fifthly the PS will be eliminated from the tissue. Although this has not been much studied, if the tissue containing the PS is a tumor or other non-metabolic organ, elimination will probably by lymphatic drainage and back into the circulation via the thoracic duct. Sixthly the PS will be excreted from the body. For the majority of PS in clinical use, excretion is from the liver into the bile and thence to the intestine where it is lost via fecal elimination.
Measurement of pharmacokinetics requires sequential measurements of PS concentration in tissue or other biological substance in order to construct temporal profiles that can be compared for different PS. This is frequently done for blood where samples can be readily removed in small amounts at frequent intervals and analysis then yields values of serum half-lives (first or second order) in minutes, hours or days. Alternatively, analysis of PS concentrations in urine or fecal samples can give terminal elimination half-lives. All these methods require a method of quantitatively analyzing PS concentrations in samples of biological material. The fact that the majority of PS used for PDT are also fluorescent, has led to the development of assays involving homogenization or dissolving the tissue or biological fluid and measurement of fluorescence in solution after centrifugation or extraction steps [4]. Calibration curves are prepared with mixtures of known amounts of PS and weighed amounts of biological material. Care must be taken when these experiments are carried out on small rodents such as mice and rats because these animals accumulate a red-fluorescent compound derived from chlorophyll in their skin and digestive tract amongst other organs [5]. This can be avoided by feeding the animals a chlorophyll-free diet for enough time before the experiment to allow the interfering fluorescent compound to be eliminated [6]. In some cases the PS is quantified by a traditional analytical chemistry technique such as high performance liquid chromatography [7]. This method has the advantage of detecting possible metabolism products that have been formed from the original pure PS after injection. Finally in some cases of experimental animal models, the PS has been presynthesized to include a radioisotope label (for instance carbon-14 or tritium) or a chelator for a metal radioisotope and biological samples are then quantified using a scintillation counter [8–11]. An alternative approach to carrying analysis of samples of biological material removed sequentially, is to use a continuous monitoring method to determine PS concentration in tissue based on fluorescence. Here a non-invasive or minimally invasive approach with fiber-based fluorescence detection systems is used to measure the variation over time of a fluorescence signal linked to the PS of interest [12]. In some small animal models the skin can be removed from a subcutaneous tumor so the fiber that collects fluorescence can be in contact with the underlying tumor as well as the adjacent skin [13].
There has been a wide variation in pharmacokinetics reported for various PS in clinical and pre-clinical use. Bellnier and Dougherty [14] studied pharmacokinetics of Photofrin in patients scheduled to undergo PDT for the treatment of carcinoma of the lung or the skin. They found a triexponential three-compartment pharmacokinetic model with alpha, beta, and gamma half-lives of approximately 16 h, 7.5 days, and 155.5 days. Detectable Photofrin fluorescence was shown to persist in the serum for longer than one year. In clinical practice light is usually delivered to patients 48 h after Photofrin injection. In a Japanese study [15] skin photosensitivity was found to persist for a month or more after Photofrin injection and interestingly was significantly more pronounced for female patients.
The pharmacokinetics of 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH) was studied in cancer patients [16]. A two-compartment model yielded alpha and beta half-lives of 7.77 and 596 h. Radiolabeled Foscan pharmacokinetics was studied in tumor-bearing rats yielding a tri-exponential model with alpha, beta and gamma half-lives of 0.46, 6.91 and 82.5 h, respectively [9]. Pharmacokinetics of the silicon phthalocyanine Pc4 were studied in non-tumor-bearing mice giving a two-compartment fit with alpha and beta half-lives of approximately 10 min and 20 h with some variation depending on injected dose and solvent [17,18]. The palladium bacteriopheophorbide PS known as TOOKAD has very rapid pharmacokinetics with alpha and beta half-lives of approximately 2 min and 1.3 h and in this case graphite furnace atomic absorption spectroscopy was used to quantify the palladium atom coordinated to the tetrapyrrole [17].
PS biodistribution
In experimental animals it has sometimes been possible to establish the overall pattern of organ distribution after intravenous injection of PS. A study [10] using radioisotope-labeled Photofrin in mice with subcutaneous fibrosarcoma tumors sacrificed 24 h later found highest concentrations in liver, adrenal gland, urinary bladder, greater than pancreas, kidney, spleen, greater than stomach, bone, lung, heart, greater than muscle, much greater than brain. Only skeletal muscle, brain, and skin located contralaterally to tumors had peak concentrations lower than tumor tissue; skin overlying tumors showed concentrations not significantly different from tumor. The higher molecular weight components of Photofrin were partially retained in liver and spleen at 75 days after injection.
In most cases where organ biodistribution data have been reported for small animals such as mice and rats, the highest concentration of PS has been found in the liver [19,20]. Chan et al. studied a set of sulfonated phthalocyanines found that the accumulation in liver was inversely proportional to the degree of sulfonation and hence to the lipophilicity of the PS molecules [21]. The liver is known to have a highly permeable blood supply with fenestrated endothelium that contains pores that allow molecules to pass easily out of the vessels. This is needed by the role of the liver in detoxifying the body of extraneous molecules (particularly organic molecules such as PS). These molecules are then excreted into the bile via the gall bladder in a similar fashion to the production of bile acids from cholesterol. The bile is routed into the duodenum where it is possible for unchanged PS to be absorbed into the circulation a second time from the small intestine (a process known as enterohepatic recycling) as well as being excreted via the feces. It is known that soon after injection, large amounts of PS can accumulate in the lungs. This has been reported [18] as the mode of dose-limiting toxicity (in the dark) when PS-injected doses were raised leading to lung hemorrhage and acute interstitial pneumonia after injection of 80 mg/kg of Pc4 into mice. The explanation is probably that large injected doses of PS can aggregate in the bloodstream and the particles so formed can easily collect in the fine capillary network of the lungs.
The amount of PS that accumulates in spleen seems to vary widely with the structure of the PS (charge and hydrophobicity). Woodburn et al. [22] studied a range of porphyrins with varying octanol/water partition coefficients and found the accumulation in the spleen varied the most compared to other organs. Egorin and co-workers [18,23] compared organ distribution of Pc4 administered by i.v. injection of the same dose (10 mg/kg) dissolved in several solvents and found that the accumulation in the spleen was the most variable.
The kidney and bladder frequently accumulate high amounts of PS despite the clearance route being almost exclusively via liver and bile [24] The digestive organs (stomach, large and small intestines) seem to take up middling amounts of PS (i.e. more than liver but less than muscle) [11]. Despite skin photosensitivity due to PS accumulating in patients’ skin being the main cause of PDT-related side-effects, skin has not been found to accumulate high amounts of PS in experimental animals. The lowest concentrations of PS are found in organs such as heart, skeletal muscle, bone, eye and brain. These are organs that are known to have relatively impermeable blood supply. The blood–brain barrier serves to exclude PS from the parenchyma of the brain, and the fact that the blood–brain barrier is frequently breached by the growth of brain tumors is probably responsible for the very large (over 100) tumor-to-normal brain ratios reported for some PS [25].
PS structure, tumour localization and delivery vehicles
Since the first observations [26] made more than 50 years ago that porphyrins and their derivatives can localize in tumors when injected into the bloodstream, the phenomenon has been intensively studied and although progress has been made, the mechanisms involved are still not completely understood [27]. A review by Boyle and Dolphin [25] collected reported tumor–normal tissue ratios for many experimental studies of PS in animal models. The complexity of comparing different PS structure, different tumor models, times after injection and doses meant that there were large variations in these ratios even with the same PS.
PS structure
As mentioned above different PS have very different pharmacokinetics and biodistribution. These considerations mean that the interval between the PS administration and light exposure is another key factor in determining PDT efficacy. With the possible exceptions of uroporphyrin and some of the larger aggregates present in Photofrin, all of the tetrapyrrole PS, which have been suggested for use as PDT drugs are more or less firmly bound to serum proteins after i.v. injection. Three classes of these compounds, which have tumor localizing properties can be delineated (although there is overlap between them).
relatively hydrophilic compounds, which are primarily bound to albumin (and possibly globulins) such as the tri and tetra-sulfonated derivatives of tetraphenylporphine (TPPS3, TPPS4) and chloroaluminum phthalocyanine (ClAlPCS3, ClAlPCS4);
amphiphilic, asymmetric compounds, which are thought to insert into the outer phospholipid and apoprotein layer of lipoprotein particles, such as the adjacent disulfonates (TPPS2a, ClAlPCS2a), benzoporphyrin derivative monoacid (BPD), lutetium texaphyrin (LuTex) and monoaspartyl chlorin(e6) (MACE) which partition between albumin and high-density lipoprotein (HDL);
hydrophobic compounds, which require a solubilization vehicle such as liposomes, cremaphor EL or Tween 80. These are thought to localize in the inner lipid core of lipoproteins particularly low-density lipoprotein (LDL) (but also HDL and very low-density lipoprotein, VLDL). Examples of these compounds are unsubstituted phthalocyanines (ZnPC, ClAlPC) naphthalocyanines (isoBOSINC), tinetiopurpurin (SnET2).
Tumor localizing PS
Since the observation of tumor localizing ability of hematoporphyrin derivative (HPD), many workers have investigated the mechanism of this tendency of PS to preferentially localize in tumors and other specific organs and anatomical sites [25,27–30]. The precise definition of tumor to normal tissue ratio has also been subject to debate. Some investigators working with s.c. tumors in experimental animals use the ratio between the tumor and peritumoral muscle or skin, while others use distant muscle and skin. In addition, there has been much effort made to determine which factors in the chemical structures of the PS are optimal for maximizing the selectivity for the tumor over normal tissue and organs. This has proved to be quite complicated because the pharmacokinetics can vary dramatically. For instance, one PS can have its best tumor to normal tissue ratio at a relatively early time point after administration such as 3 h, while with another this can be at 7 days after injection. One of the particular properties of these tetrapyr-role compounds relevant to their tumor localizing ability, is their tendency to bind strongly to serum proteins and to each other. This means that most PS when injected into the bloodstream behave as macromolecules either because they are more or less firmly bound to large protein molecules or because they have formed intermolecular aggregates of similar size. Many workers have reported [31–33] on the distribution of PS between the various classes of serum proteins when mixed with serum in vitro. These proteins are usually divided into four classes: albumin and other heavy proteins, HDL, LDL, and VLDL. However, even this study has been complicated by the fact that the most lipophilic PS are insoluble in aqueous media and need to be delivered in a solvent mixture which may alter the serum protein distribution of the PS. It has been argued that PS that preferentially bind to LDL are better tumor localizers (see below) [30], but this is by no means always the case [34].
It is useful to make a distinction between selective accumulation and selective retention. The tumor localizing ability of the PS with the faster pharmacokinetics is probably due to selective accumulation in the tumor, while the localization of PS with slower acting pharmacokinetics is more likely due to selective retention. In the selective accumulation model it is thought that the increased vascular permeability to macromolecules typical of tumor neovasculature is chiefly responsible for the preferential extravasation of the PS. These quick acting PS frequently bind to albumin which is of ideal size and Stokes radius to pass through the “pores” in the endothelium of the tumor microvessels [35]. The selective retention of PS in tumors has been the subject of much speculation. As mentioned above, a popular theory maintains that the binding of the PS to LDL is of major importance [30]. In this theory, it is proposed that cancer cells overexpress the LDL (apoB/E) receptor. Upregulation of the expression of LDL receptors is one way that rapidly growing malignant cells gain cholesterol needed for the biosynthesis of lipids needed for the rapid turnover of cellular membranes. There is experimental evidence both for and against this theory [34,36]. Other theories have been proposed to account for the selective retention of PS in tumor tissue. One is that tumors have poorly developed lymphatic drainage, and that macromolecules, which extravasate from the hyperpermeable tumor neovasculature, are retained in the extravascular space [37]. Another involves the macrophages, which infiltrate solid tumors to varying extents [27]. These tumor-associated macrophages have been shown to accumulate up to 13 times the amount of some PS compared to cancer cells [38]. The explanation for this has been proposed to be either the phagocytosis of aggregates of PS [39], or the preferential uptake by macrophages of lipoproteins, which have been altered by the binding of porphyrins [27]. Another theory proposes that the low pH commonly found in tumors has the effect of trapping some of the anionic PS, which are ionized at normal physiological pH. These PS then become neutrally charged and hence more lipophilic, when they encounter the lowered pH in the tumor environment [40]. Yet another proposal is that PS accumulation in tissues is related to the articular tissues’ propensity to accumulate lipid droplets [41].
It has been proposed that the peripheral benzodiazepine receptors located on the inner mitochondrial membrane are responsible for binding some porphyrin-based PS that accumulate in mitochondria [42,43]. Alternatively it has been suggested that mitochondrially localizing dyes such as Rhodamine 123 take advantage of differences in mitochondrial membrane potential between normal and cancer cells [44,45].
Non-covalent delivery vehicle complexes
As mentioned above, many of the most effective PS are too hydrophobic to dissolve in aqueous solvents, and this necessitates the use of a delivery vehicle to keep the molecules in a sufficiently disaggregated state to be able to travel in the blood vessels and to extravasate into tumors. It has been found that the choice of delivery vehicle can influence the tumor selectivity of the PS [46]. The castor oil derivative Cremophor EL has been used as a delivery vehicle, but Kessel and co-workers have shown that its use actually changes the lipoprotein profile [47] and can affect the biodistribution of PS and the efficacy of the tumor treatment [48]. An alternative method of PS delivery is encapsulation in liposomes. It has been suggested that when liposomal PS are administered to animals, the PS is more efficiently transferred to LDL than an aqueous formulation [49]. In vivo liposomal delivery has been shown to give advantages in either biodistribution or tumor destruction compared to non-liposomal delivery for Photofrin [50], BPD [51] and Zn-PC [52]. Other workers have used polyethylene-glycol-coated poly(lactic acid) nanoparticles [53], or albumin microspheres [54] to deliver PS.
Some workers have investigated the pre-complexing with various serum proteins. The protein most frequently investigated has been LDL for two reasons. Firstly the hydrophobic core of the LDL particle can act as a solubilizing medium for hydrophobic PS in a similar way to liposomes or Cremophor, and secondly it was proposed to increase tumor targeting by taking advantage of receptor-mediated endocytosis of the complex by the LDL receptor that has been reported to be overexpressed on cancer cells (see above). Barel et al. [55], used HP precomplexed to lipoproteins to target a murine fibrosarcoma. An increased delivery of hematoporphyrin (HP) to the mouse tumor was reported with the HP–LDL complex compared to HP complexes of HDL, VLDL, or free HP. Similarly, precomplexing of BPD with LDL led to a greater accumulation of the PS in tumors as compared to BPD administration of an aqueous solution at 3 h [36]. Some authors have suggested that PS delivery with various macromolecular systems may lead to differing mechanisms of tumor destruction as PS are delivered to different sites. For example, although albumin and globulins are believed to deliver PS mainly to the vascular stroma of tumors [29], HDL apparently delivers PS to cells via a non-specific exchange with the plasma membrane. LDL probably delivers a large fraction of the PS via an active receptor-mediated pathway [56]. Zhou et al. [57] have suggested that aqueous solutions of HP lead to predominantly vascular damage, while LDL-mediated PDT leads predominantly to damage of neoplastic cells. However, this is not always true. In a study of PDT of ocular melanoma in a rabbit model LDL complexed to BPD was used. Despite the use of LDL as a carrier, early damage to the vasculature was demonstrated by light and electron microscopy [58]. Larroque et al. [28] showed that the insoluble Zn-PC could be formulated for i.v. administration as a complex with serum albumin, after which it redistributed primarily to HDL.
Enhanced PS accumulation in tumors as a means to deliver other molecules
The enhanced accumulation of tetrapyrrole PS in tumors has been proposed to act as a delivery mechanism for other molecules. The suggested strategy includes using PS with covalently attached radioisotopes (or radioisotope chelators) as a tumor-targeting method for both detection and therapy of cancer. Imaging modalities that can be used if gamma-particle emitting radionuclides can be specifically targeted to cancers (such as early metastases) include planar gamma cameras and single photon emission computed tomography (SPECT) [59]. Technetium-labeled Photosan-3 (an analog of Photofrin) [60] was proposed to be used in tumor imaging as demonstrated by accumulation in s.c. mouse tumors.
Radioiodine-labeled methylene blue has been used to detect melanoma [61] and 211-astatine labeled methylene blue (an alpha particle emitter) has been used to treat melanoma both in nude mouse models [62] and in a Phase I clinical trial [63]. It is thought that methylene blue shows particular affinity for melanoma tumors due to a specific molecular binding to melanin.
Porphyrins have been proposed as a means to deliver boron to tumors (particularly brain tumors) [64]. If a large accumulation of boron can be targeted to tumors then after application of a neutron beam the boron atoms capture neutrons (10iB(n,α)7Li) forming both a reactive alpha particle (helium nucleus) and also a high energy lithium atom both of which can damage tumor cells [65]. In principle the same compound (a boronated PS) could be used as a binary therapy drug and could interact with both red light and with a neutron beam to cause cell death. The boronated porphyrin (BOPP) in combination with red light has been tested in a Phase I clinical trial for high-grade gliomas [66].
Another strategy is to take advantage of the tumor localizing properties of porphyrin-based PS to deliver paramagnetic metal atoms that can be detected by magnetic resonance imaging. This has been demonstrated by the use of Mn-TPPS in rat brain tumors that gave a strong signal enhancement in T1 weighted images that lasted for three days [67,68].
It has been proposed that porphyrins and other tetrapyrrole PS can act as radiosensitizers in the absence of visible light [69]. While the mechanism underlying this effect is largely unknown, the therapy takes advantage of the tumor-localizing properties of the porphyrin to concentrate the cytotoxic effect of a beam of radiation in the tumor. In an animal model it was shown that only Photofrin has a positive therapeutic benefit, out of six different tetrapyrrole PS tested [70]. In a clinical trial the combination allowed a previously inoperable tumor to be shrunk enough to be surgically removed [71].
Gadolinium-substituted texaphyrin (Gd-Tex, motexafin gadolinium, Xcytrin) was originally synthesized as a contrast enhancement agent to carry out MRI visualization of tumors [72]. Its selective accumulation in tumors was demonstrated in normal and tumor-bearing rats and rabbits [73]. It was subsequently discovered that this compound was an effective radiation sensitizer due to its ability to carry our redox cycling and produce intracellular reactive oxygen species (ROS) without illumination [74]. Notably texaphyrins substituted with the metals lutetium, europium, yttrium, and cadmium did not demonstrate enhancement of radiation sensitivity [75]. Xcytrin has advanced to Phase III clinical trials for enhancing radiation therapy of brain metastases of lung cancer and has demonstrated survival benefits [76].
Mechanisms of tumour destruction
Three distinct (but possibly interrelated) mechanisms have been identified which contribute to the observed shrinkage (and frequent disappearance) of tumors when treated with PDT. These are graphically illustrated in Fig. 1. In the first case, the ROS that is generated by PDT can kill tumor cells directly by apoptosis and/or necrosis as discussed in Parts 1 and 2 of this review series [1,2]. PDT also damages the tumor-associated vasculature, leading to tissue deprivation of oxygen and nutrients and consequent tumor infarction. Finally, PDT can activate an immune response against tumor cells. These three mechanisms can also influence each other. The relative importance of each for the overall tumor response is yet to be defined. It is clear, however, that the combination of all these components is required for optimum long-term tumor cures, especially of tumors that may have metastasized.
Figure 1.

Pathways of PDT-mediated tumor destruction illustrating possible contributions from direct tumor cell killing, vascular damage and host immune response.
Direct tumor-cell death effects
Exposure of tumors to PDT in vivo can reduce the number of clonogenic tumor cells existing within the tumor through direct cellular damage; however, this is thought to be probably insufficient for tumor cure [77]. Studies [78] in rodent tumor systems employing curative procedures with several PS showed direct photodynamic tumor cell kill to be less than 2 logs and in most cases less than 1 log, i.e. far short of the 8-log reduction required for tumor cure, if tumor cell killing were the only mechanism operating. The in vitro illumination of tumor cells isolated from photosensitized tumors in vivo predicts that total eradication is feasible with a sufficiently high light dose with some PS [77]. But limitations appear to exist that do not allow the eradication to be realized for in vivo PDT treatment. Inhomogeneous PS distribution within the tumor might be one of these limitations. Korbelik and Krosl [79] have shown that both PS accumulation and tumor cell kill decrease with the distance of tumor cells from the vascular supply. Lee et al. [80] found that there were significant degrees of variability in PS concentration (both intra-tumor and inter-tumor) depending on tumor and PS type and delivery route. Zhou and colleagues [81] found that the PS (BPD) distribution differed significantly between the same rat prostate tumors whether grown subcutaneously or orthotopically implanted in the rat prostate gland, probably due to different degrees of vascularity. Another parameter that can limit direct tumor cell kill is the availability of oxygen within the tissue undergoing PDT treatment. Because the effects of almost all PDT drugs are oxygen dependent, PDT cell killing typically does not occur efficiently in hypoxic areas of tissue. In vivo studies showed that induction of tissue hypoxia, by clamping, abolished the PDT effects of porphyrins [82]. The rates of singlet oxygen generation and therefore tissue oxygen consumption and depletion within the tumor are significant when both tissue PS levels and the fluence rate of light are high [83]. An important parameter influencing the rate of tissue oxygen consumption is photobleaching of the PS because the reduction of PS levels also reduces the rate of photochemical oxygen consumption [84]. Since all these parameters can impose limits on the direct photodestruction of tumor cells, other mechanisms must operate to account for the demonstrable success of the treatment.
Since the reductions in oxygen that occur during PDT can limit the response, it may be possible to lower the light fluence rate to reduce oxygen consumption rate [85]. Busch et al. [86] compared Photofrin-mediated PDT of RIF1 tumors with a total dose of 135 J/cm2, delivered at a fluence rate of either 75 or 38 mW/cm2. The higher fluence rate generated significant regions of hypoxia even near to tumor blood vessels and a parallel decrease in tumor perfusion, which were not seen with the lower fluence rate. Snyder et al. [87] compared PDT regimens of two different fluences (48 and 128 J/cm2) using two fluence rates (14 and 112 mW/cm2) on Colo26 murine tumors with the object of either conserving or depleting tissue oxygen during PDT. Oxygen-conserving low fluence rate PDT of 14 mW/cm2 at a fluence of 128 J/cm2 yielded approximately 70–80% tumor cures, whereas the same fluence at the oxygen-depleting fluence rate of 112 mW/cm2 yielded approximately 10–15% tumor cures. Low fluence rate induced higher levels of apoptosis than high fluence rate PDT. There were PDT-protected tumor regions distant from vessels in the high fluence rate conditions, confirming regional tumor hypoxia. High fluence at a low fluence rate led to ablation of CD31-stained endothelium, whereas the same fluence at a high fluence rate maintained vessel endothelium. The optimally curative PDT regimen (128 J/cm2 at 14 mW/cm2) produced minimal inflammation. Depletion of neutrophils did not significantly change the high cure rates of that regimen but abolished curability in the maximally inflammatory regimen. This fluence rate effect was also seen in human basal cell carcinomas treated with PF-PDT [88]. A fluence rate of 150 mW/cm2 significantly diminished tumor oxygen levels during initial light delivery in a majority of carcinomas, but a fluence rate of 30 mW/cm2 increased tumor oxygenation in a majority of carcinomas.
Another method to overcome the inhibiting effect of oxygen consumption during PDT is to fractionate the light delivery to allow re-oxygenization of the tissue. However, this modality can be complicated as there is no consensus on either the optimum duration of the period when the light is on, or on the duration of the dark period. There have been examples of one or other of these periods being as short as 50 ms and as long as hours. One study in normal rat colon using aminolevulanic acid (ALA)-PDT produced up to three times more tissue necrosis with fractionated as compared with continuous light [89]. However, Babilas et al. [90] failed to find any increased effect of either light fractionation or low fluence rate in a hamster amelanotic melanoma treated with i.v. ALA-induced protoporphyrin IX (PPIX); in fact the best response was obtained with non-fractionated high fluence rate.
Vascular effects
The first additional mechanism shown to operate during PDT of tumors is the vascular effect in which vascular damage, occurring after completion of the PDT treatment, contributes to long-term tumor control [91]. Microvascular collapse can be readily observed following PDT [92,93] and can lead to severe and persistent post-PDT tumor hypoxia [93]. The mechanisms underlying the vascular effects of PDT differ greatly with different PS. Photofrin-PDT leads to vessel constriction, macromolecular vessel leakage, leukocyte adhesion and thrombus formation, all apparently linked to platelet activation and release of thromboxane [94]. PDT with certain phthalocyanine derivatives causes primarily vascular leakage [95], and PDT with MACE results in blood flow stasis primarily because of platelet aggregation [96]. All of these effects may include components related to damage of the vascular endothelium. PDT may also lead to vessel constriction via inhibition of the production or release of nitric oxide by the endothelium [97]. In preclinical experiments, the microvascular PDT responses can be partially or completely inhibited by the administration of agents that affect eicosanoid generation, such as indomethacin [98] and this inhibition can markedly diminish the tumor response. Much of the above information was obtained from studies on normal microvasculature. Damage to the tumor-supplying normal vasculature may greatly affect tumor curability by PDT as demonstrated by the lack of tumor cures when the normal tissue surrounding the tumor was shielded from PDT light [98].
It is becoming more and more clear that the drug–light interval is a crucial parameter in optimizing PDT. The rate at which i.v. injected PS leave the flowing blood and bind to the blood vessel walls before leaking out into the tumor interstitium can vary markedly depending on the chemical structure (size, charge and lipophilicity). Hamblin et al. [99] carried out a study using a fluorescence scanning laser microscope to follow the pharmacokinetics of fluorescent PS and PS-conjugates in vivo in an orthotopic rat prostate cancer model obtained with MatLyLu cells injected into the prostate capsule. The tumor was surgically exposed via laparotomy to allow imaging to take place. Fig. 2A shows the image captured 10 min after injection of unconjugated ce6 into the rats. Although there has been some leakage of fluorescence out of the very tortuous vessels in the upper left of the image, the majority of the PS is still confined to the vessels but is no longer circulating but rather bound to the vessel walls. In Fig. 2B which shows the same region of the tumor captured 75 min post-injection, all of the PS has leaked out of the vessels into the tumor interstitium. This technology can be used to define the optimum times for carrying out “vascular” or “cellular” PDT using the same PS.
Figure 2.
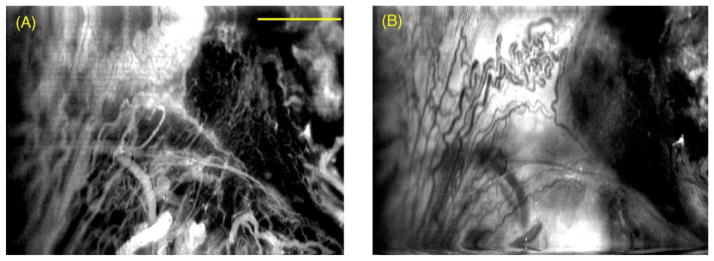
In vivo confocal fluorescence images of an orthotopic rat prostate tumor injected with the PS ce6. (A) The image captured 10 min after i.v. injection and (B) same region of the tumor captured 75 min post-injection. Scale bar is 100 μm.
Kurohane et al. [100] showed that PDT 15 min after bloodstream injection of BPD-MA caused strong suppression of tumor growth, perhaps through damaging endothelial cells in the tumor neovasculature rather than through a direct cytotoxic effect on tumor cells. Another study found that BPD-PDT 15 min post-injection gave significantly better treatment response against RIF1 tumors than a 3-h interval [101]. Chen et al. studied hypericin-PDT of RIF1 tumors comparing drug–light intervals of 0.5 and 6 h after 5 mg/kg i.v. injection [102]. Complete cures were found at the short time interval while no cures were seen at the 6 h interval. Ferrario et al. found similar results in treating a mouse mammary tumor model with mono-l-aspartyl chlorin e6 (Npe6) [103]. Maximal in vivo PDT effectiveness was achieved when light treatments were started within 2 h of drug injection. PDT effectiveness was decreased by 50% when light treatments were initiated 6 h after drug injection and was abolished with a 12-h interval between NPe6 injection and light exposure. Responsiveness to NPe6-mediated PDT was correlated with photosensitizer levels in the plasma but not in tumor tissue. No less than three separate reports have shown that tumor response to Foscan (mTHPC) does not correlate with tumor concentration of PS. Cramers et al. [104] studied nude mice bearing human mesothelioma xenografts and found there was maximal PDT response in both tumor and skin 1–3 h post-injection despite finding the highest levels of drug in the tumor at 48–120 h post-injection. Jones et al. [9] studied PDT of fibrosarcomas implanted into BDIX rats and found two times at which the anti-tumor effect of Foscan was maximal; 2 h post-injection corresponding to maximal serum concentration, and 24 h post-injection corresponding to maximal tumor concentration. Maugain et al. [3] found maximum anti-tumor activity with PDT at 6 h post-injection of Foscan into nude mice bearing Colo26 tumors and observed lower activity at shorter and longer times after injection.
Dolmans et al. carried out a novel fractionation scheme in PDT [105]. Using the murine MCaIV tumor, grown in the mammary fat pad they found the plasma half-life of the pyropheophorbide derivative MV6401 was approximately 20 min, and the drug was confined to the vascular compartment shortly after administration but accumulated in the interstitial compartment at 2–6 h after administration. Two equal MV6401 doses injected 4 h and 15 min before the light administration allowed the PS to localize in both vascular and tumor cell compartments. The fractionated drug dose PDT more effectively induced tumor growth delay than the same total dose given as a single dose either at 4 h or at 15 min before light administration.
Immune system effects
One of the key experiments to demonstrate this effect was carried out by Korbelik et al. [106] who grew the same EMT6 mammary sarcoma tumors in immunocompetent Balb/c mice and immunodeficient nude or scid mice of the same background. PF-PDT led to initial ablation of the tumors in all mice but long-term cures were obtained only in the immunocompetent mice. Adoptive transfer of bone marrow from Balb/c to SCID mice restored the ability of PDT to produce long-term cures. This established the necessity of a functioning immune system for complete tumor response to PDT. There are now thought to be two aspects to the effect of PDT on the immune response against cancer: (1) anti-tumor activity of PDT-induced inflammatory cells and (2) generation of a long-term anti-tumor immune response. These effects can be elicited by phototoxic damage that is not necessarily lethal to all tumor cells and creates an inflammatory stimulus. PDT-induced changes in the plasma membrane and membranes of cellular organelles can prompt a rapid activation of membranous phospholipases [107] leading to accelerated phospholipid degradation with a massive release of powerful inflammatory mediators [108]. Other cytokines and growth factors that are potent immunomodulators have been found to be strongly enhanced in PDT-treated mouse tumors [109,110].
The inflammatory signaling after PDT initiates a massive regulated invasion of neutrophils, mast cells, and monocytes/macrophages [111] that may outnumber resident cancer cells. Most notable is a rapid accumulation of large numbers of neutrophils, which have been shown to have a profound impact on PDT-mediated destruction of tumors, and it has been shown that depletion of neutrophils in tumor-bearing mice decreased the PDT-mediated tumor cure rate [106]. Another class of nonspecific immune effector cells whose activation substantially contributes to the anti-tumor effects of PDT is monocytes/macrophages. The tumoricidal activity of these cells was found to be increased by PDT in vivo and in vitro [112]. Adjuvant treatment with a selective macrophage-activating factor derived from Vitamin D3-binding protein was shown to potentiate the cures of PDT-treated tumors [113].
There have been substantial advances in the understanding of the PDT-induced tumor-specific immune reaction. This effect may not be relevant to the initial tumor ablation, but may be decisive in attaining long-term tumor control. Anti-cancer immunity elicited by PDT has the attributes of an inflammation primed immune development process [114] and bears similarities to the immune reaction induced by tumor inflammation caused by bacterial vaccines or some cytokines. Macrophages phagocytize large numbers of cancer cells killed or damaged by the cytotoxic effects of PDT. Directed by powerful inflammation-associated signaling, the antigen presenting cells will process tumor-specific peptides and present them on their membranes in the context of major histocompatibility class II molecules. Presentation of tumor peptide antigens, accompanied by intense accessory signals, creates conditions for the recognition of tumor antigens by CD4 helper T lymphocytes. These lymphocytes become activated and in turn sensitize cytotoxic CD8 T cells to tumor-specific epitopes. The activity of tumor-sensitized lymphocytes is not limited to the original PDT-treated site but can include disseminated and metastatic lesions of the same cancer. Thus, although the PDT treatment is localized to the tumor site, its effect can have systemic attributes due to the induction of an immune reaction. PDT-generated tumor-sensitized lymphocytes can be recovered from distant lymphoid tissues (spleen, lymph nodes) at protracted times after light treatment [115].
Korbelik and his co-workers have shown that combination therapies designed to activate constituent parts of the immune system can dramatically increase the percentage of cures of various mouse tumors. Intratumoral injection of the immunostimulant schizophyllan (SPG) (a fungal preparation) in squamous cell carcinoma (SCCVII)-bearing mice raised the relative content of Mac-1 positive host cells infiltrating the tumor and increased PF retention in these tumors. The tumor cure rate increased approximately three times when PDT was preceded by the SPG therapy. In contrast, the administration of SPG after PDT was of no benefit for tumor control [116]. In the same tumor model, local injections of killed tumor cells genetically engineered to produce granulocyte–macrophage colony-stimulating factor, administered three times in 48-h intervals, starting 2 days before the light treatment, substantially improved the curative effect of PF-mediated PDT [117]. Intratumoral injection of an extract of mycobacterium cell walls potentiated the response of EMT6 tumors to PDT mediated by PF, BPD, mTHPC, or ZnPc [118]. A study designed to investigate the role of complement activating agents to potentiate PDT looked at zymosan, an alternative complement pathway activator, that reduced the recurrence rate of PDT-treated tumors, markedly increasing the percentage of permanent cures. In contrast, a similar treatment with heat-aggregated gamma globulin (complement activator via the classical pathway) was of no significant benefit as a PDT adjuvant. Systemic complement activation with streptokinase treatment had no detectable effect on complement deposition at the tumor site without PDT, but it augmented the extent of complement activity in PDT-treated tumors [119].
Work from our laboratory has shown that while BPD-mediated PDT of RIF1 tumors (poorly immunogenic) in wild-type mice leads to initial tumor disappearance but no permanent cures due to local recurrence; when the tumors were genetically engineered to express the foreign protein (green fluorescent protein from jellyfish) 100% cures and long-term resistance to rechallenge were obtained [120]. The explanation for this observation is likely to be the PDT-induced immune recognition of GFP. In another study we explored the use of low-dose cyclophosphamide in combination with PDT. This treatment selectively kills CD4+, CD25+ T-suppressor cells and significantly increases the number of cures of the highly aggressive and metastatic J774 tumor in Balb/c mice. Again cured mice were resistant to rechallenge with the same tumor [121].
An interesting observation was also made by Gollnick et al., who reported that a tumor-cell lysate that was isolated following PF-PDT in vitro with could be used to vaccinate mice against the development of tumors from the same cell line [122]. PDT-induced lysates were more effective than those made from cells that were killed by ultraviolet, ionizing irradiation or freeze–thaw and only PDT-generated lysates were able to activate dendritic cells to express IL-12.
Conclusion
There may be a trend amongst second and third generation PS away from the formerly desired properties of localizing well in tumors towards fast acting vascular PS. This shift of emphasis is explained by several reasons. Firstly the achievement of high tumor-to-normal tissue ratios frequently involved long intervals between injection and illumination that were needed for the PS to clear from normal tissue, and that delay necessitated patients returning twice to the hospital for PS administration and for illumination. Secondly these long-lasting PS also tended to have more pronounced skin photosensitivity problems that could last for an inconveniently long time. Thirdly when PS are compared on the basis of moles injected, absorption spectrum and light delivered, it is likely that fast acting vascular PS are actually more effective in destroying tumors than first generation tumor-localizing dyes. Somewhat paradoxically there are also ongoing efforts to take advantage of the tumor-localizing properties of tetrapyrrole PS to deliver other imaging and therapeutically active molecules more specifically to tumors.
While the immune stimulating effects of PDT are well established in animal models these effects have not been much studied in clinical trials. There are scattered anecdotal reports of patients with terminal cancer receiving palliative PDT who survived much longer than expected. While these occurrences might be attributed to stimulation of the immune system, and while the laboratory assays necessary to confirm the generation of an anti-tumor immune response exist, they have never been applied to patients in a systematic manner. It is known that the three commonest cancer therapies; surgery, ionizing radiation and chemotherapy all tend to be immunosuppressive. It is therefore possible that PDT could emerge as the ideal cancer treatment: a local therapy capable of efficiently destroying tumors while at the same time sensitizing the immune system to seek out and destroy metatstases.
Acknowledgments
Ana P. Castano was supported by a Department of Defense CDMRP Breast Cancer Research Grant (W81XWH-04-1-0676). Tatiana N. Demidova was supported by a Wellman Center of Photomedicine graduate student fellowship. Michael R. Hamblin was supported by the US National Institutes of Health (R01-CA/AI838801). We are grateful to Tayyaba Hasan for support.
References
- 1.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one—photosensitizers, photochemistry and cellular localization. Photodiagn Photodyn Ther. 2004;1:279–93. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part two – cellular signaling, cell metabolism and modes of cell death. Photodiagn Photodyn Ther. 2005;2:1–23. doi: 10.1016/S1572-1000(05)00030-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maugain E, Sasnouski S, Zorin V, Merlin JL, Guillemin F, Bezdetnaya L. Foscan-based photodynamic treatment in vivo: correlation between efficacy and Foscan accumulation in tumor, plasma and leukocytes. Oncol Rep. 2004;12:639–45. [PubMed] [Google Scholar]
- 4.Bellnier DA, Greco WR, Parsons JC, Oseroff AR, Kuebler A, Dougherty TJ. An assay for the quantitation of Photofrin in tissues and fluids. Photochem Photobiol. 1997;66:237–44. doi: 10.1111/j.1751-1097.1997.tb08649.x. [DOI] [PubMed] [Google Scholar]
- 5.Gudgin Dickson EF, Holmes H, Jori G, et al. On the source of the oscillations observed during in vivo zinc phthalocyanine fluorescence pharmacokinetic measurements in mice. Photochem Photobiol. 1995;61:506–9. doi: 10.1111/j.1751-1097.1995.tb02353.x. [DOI] [PubMed] [Google Scholar]
- 6.Holmes H, Kennedy JC, Pottier R, Rossi R, Weagle G. A recipe for the preparation of a rodent food that eliminates chlorophyll-based tissue fluorescence. J Photochem Photobiol B. 1995;29:199. doi: 10.1016/1011-1344(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 7.Cai H, Lim CK. Comparison of HPLC, capillary electrophoretic and direct spectrofluorimetric methods for the determination of temoporfin-poly(ethylene glycol) conjugates in plasma. Analyst. 1998;123:2243–5. doi: 10.1039/a804226h. [DOI] [PubMed] [Google Scholar]
- 8.Little FM, Gomer CJ, Hyman S, Apuzzo ML. Observations in studies of quantitative kinetics of tritium labelled hematoporphyrin derivatives (HpDI and HpDII) in the normal and neoplastic rat brain model. J Neurooncol. 1984;2:361–70. doi: 10.1007/BF00178119. [DOI] [PubMed] [Google Scholar]
- 9.Jones HJ, Vernon DI, Brown SB. Photodynamic therapy effect of m-THPC (Foscan) in vivo: correlation with pharmacokinetics. Br J Cancer. 2003;89:398–404. doi: 10.1038/sj.bjc.6601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellnier DA, Ho YK, Pandey RK, Missert JR, Dougherty TJ. Distribution and elimination of Photofrin II in mice. Photochem Photobiol. 1989;50:221–8. doi: 10.1111/j.1751-1097.1989.tb04152.x. [DOI] [PubMed] [Google Scholar]
- 11.Schuitmaker JJ, Feitsma RI, Journee-De Korver JG, Dubbelman TM, Pauwels EK. Tissue distribution of bacteriochlorin a labelled with 99mTc-pertechnetate in hamster Greene melanoma. Int J Radiat Biol. 1993;64:451–8. doi: 10.1080/09553009314551641. [DOI] [PubMed] [Google Scholar]
- 12.Frisoli JK, Tudor EG, Flotte TJ, Hasan T, Deutsch TF, Schomacker KT. Pharmacokinetics of a fluorescent drug using laser-induced fluorescence. Cancer Res. 1993;53:5954–61. [PubMed] [Google Scholar]
- 13.Sheng C, Pogue BW, Wang E, Hutchins JE, Hoopes PJ. Assessment of photosensitizer dosimetry and tissue damage assay for photodynamic therapy in advanced-stage tumors. Photochem Photobiol. 2004;79:520–5. doi: 10.1562/mu-03-33.1. [DOI] [PubMed] [Google Scholar]
- 14.Bellnier DA, Dougherty TJ. A preliminary pharmacokinetic study of intravenous Photofrin in patients. J Clin Laser Med Surg. 1996;14:311–4. doi: 10.1089/clm.1996.14.311. [DOI] [PubMed] [Google Scholar]
- 15.Moriwaki SI, Misawa J, Yoshinari Y, Yamada I, Takigawa M, Tokura Y. Analysis of photosensitivity in Japanese cancer-bearing patients receiving photodynamic therapy with porfimer sodium (Photofrin) Photodermatol Photoimmunol Photomed. 2001;17:241–3. doi: 10.1034/j.1600-0781.2001.170507.x. [DOI] [PubMed] [Google Scholar]
- 16.Bellnier DA, Greco WR, Loewen GM, et al. Population pharmacokinetics of the photodynamic therapy agent 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a in cancer patients. Cancer Res. 2003;63:1806–13. [PubMed] [Google Scholar]
- 17.Brun PH, DeGroot JL, Dickson EF, Farahani M, Pottier RH. Determination of the in vivo pharmacokinetics of palladium-bacteriopheophorbide (WST09) in EMT6 tumour-bearing Balb/c mice using graphite furnace atomic absorption spectroscopy. Photochem Photobiol Sci. 2004;3:1006–10. doi: 10.1039/b403534h. [DOI] [PubMed] [Google Scholar]
- 18.Egorin MJ, Zuhowski EG, Sentz DL, Dobson JM, Callery PS, Eiseman JL. Plasma pharmacokinetics and tissue distribution in CD2F1 mice of Pc4 (NSC 676418), a silicone phthalocyanine photodynamic sensitizing agent. Cancer Chemother Pharmacol. 1999;44:283–94. doi: 10.1007/s002800050979. [DOI] [PubMed] [Google Scholar]
- 19.Ismail MS, Dressler C, Koeppe P, et al. Pharmacokinetic analysis of octa-alpha-butyloxy-zinc phthalocyanine in mice bearing Lewis lung carcinoma. J Clin Laser Med Surg. 1997;15:157–61. doi: 10.1089/clm.1997.15.157. [DOI] [PubMed] [Google Scholar]
- 20.Zuk MM, Rihter BD, Kenney ME, Rodgers MA, Kreimer-Birnbaum M. Pharmacokinetic and tissue distribution studies of the photosensitizer bis(di-isobutyl octadecylsiloxy)silicon 2,3-naphthalocyanine (isoBOSINC) in normal and tumor-bearing rats. Photochem Photobiol. 1994;59:66–72. doi: 10.1111/j.1751-1097.1994.tb05002.x. [DOI] [PubMed] [Google Scholar]
- 21.Chan WS, Marshall JF, Svensen R, Bedwell J, Hart IR. Effect of sulfonation on the cell and tissue distribution of the photosensitizer aluminum phthalocyanine. Cancer Res. 1990;50:4533–8. [PubMed] [Google Scholar]
- 22.Woodburn KW, Stylli S, Hill JS, Kaye AH, Reiss JA, Phillips DR. Evaluation of tumour and tissue distribution of porphyrins for use in photodynamic therapy. Br J Cancer. 1992;65:321–8. doi: 10.1038/bjc.1992.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez MG, Moran F, Yslas EI, et al. Pharmacokinetic and tumour-photosensitizing properties of methoxyphenyl porphyrin derivative. Biomed Pharmacother. 2003;57:163–8. doi: 10.1016/s0753-3322(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 24.Richter AM, Cerruti-Sola S, Sternberg ED, Dolphin D, Levy JG. Biodistribution of tritiated benzoporphyrin derivative (3H-BPD-MA), a new potent photosensitizer, in normal and tumor-bearing mice. J Photochem Photobiol B. 1990;5:231–44. doi: 10.1016/1011-1344(90)80008-l. [DOI] [PubMed] [Google Scholar]
- 25.Boyle RW, Dolphin D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem Photobiol. 1996;64:469–85. doi: 10.1111/j.1751-1097.1996.tb03093.x. [DOI] [PubMed] [Google Scholar]
- 26.Figge FH, Weiland GS, Manganiello LO. Affinity of neoplastic, embryonic and traumatized tissues for porphyrins and metalloporphyrins. Proc Soc Exp Biol Med. 1948;68:640. doi: 10.3181/00379727-68-16580. [DOI] [PubMed] [Google Scholar]
- 27.Hamblin MR, Newman EL. On the mechanism of the tumour-localising effect in photodynamic therapy. J Photochem Photobiol B. 1994;23:3–8. doi: 10.1016/s1011-1344(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 28.Larroque C, Pelegrin A, Van Lier JE. Serum albumin as a vehicle for zinc phthalocyanine: photodynamic activities in solid tumour models. Br J Cancer. 1996;74:1886–90. doi: 10.1038/bjc.1996.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jori G. In vivo transport and pharmacokinetic behavior of tumour photosensitizers. Ciba Found Symp. 1989;146:78–86. doi: 10.1002/9780470513842.ch6. [DOI] [PubMed] [Google Scholar]
- 30.Jori G, Reddi E. The role of lipoproteins in the delivery of tumour-targeting photosensitizers. Int J Biochem. 1993;25:1369–75. doi: 10.1016/0020-711x(93)90684-7. [DOI] [PubMed] [Google Scholar]
- 31.Kessel D, Morgan A, Garbo GM. Sites and efficacy of photo-damage by tin etiopurpurin in vitro using different delivery systems. Photochem Photobiol. 1991;54:193–6. doi: 10.1111/j.1751-1097.1991.tb02006.x. [DOI] [PubMed] [Google Scholar]
- 32.Kongshaug M, Moan J, Brown SB. The distribution of porphyrins with different tumour localising ability among human plasma proteins. Br J Cancer. 1989;59:184–8. doi: 10.1038/bjc.1989.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maziere JC, Santus R, Morliere P, et al. Cellular uptake and photosensitizing properties of anticancer porphyrins in cell membranes and low and high density lipoproteins. J Photochem Photobiol B. 1990;6:61–8. doi: 10.1016/1011-1344(90)85074-7. [DOI] [PubMed] [Google Scholar]
- 34.Korbelik M. Low density lipoprotein receptor pathway in the delivery of Photofrin: how much is it relevant for selective accumulation of the photosensitizer in tumors? J Photochem Photobiol B. 1992;12:107–9. doi: 10.1016/1011-1344(92)85022-m. [DOI] [PubMed] [Google Scholar]
- 35.Yuan F, Leunig M, Berk DA, Jain RK. Microvascular permeability of albumin, vascular surface area, and vascular volume measured in human adenocarcinoma LS174T using dorsal chamber in SCID mice. Microvasc Res. 1993;45:269–89. doi: 10.1006/mvre.1993.1024. [DOI] [PubMed] [Google Scholar]
- 36.Allison BA, Pritchard PH, Levy JG. Evidence for low-density lipoprotein receptor-mediated uptake of benzoporphyrin derivative. Br J Cancer. 1994;69:833–9. doi: 10.1038/bjc.1994.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts WG, Hasan T. Role of neovasculature and vascular permeability on the tumor retention of photodynamic agents. Cancer Res. 1992;52:924–30. [PubMed] [Google Scholar]
- 38.Korbelik M, Krosl G. Photofrin accumulation in malignant and host cell populations of a murine fibrosarcoma. Photochem Photobiol. 1995;62:162–8. doi: 10.1111/j.1751-1097.1995.tb05253.x. [DOI] [PubMed] [Google Scholar]
- 39.Korbelik M, Krosl G, Chaplin DJ. Photofrin uptake by murine macrophages. Cancer Res. 1991;51:2251–5. [PubMed] [Google Scholar]
- 40.Pottier R, Kennedy JC. The possible role of ionic species in selective biodistribution of photochemotherapeutic agents toward neoplastic tissue. J Photochem Photobiol B. 1990;8:1–16. doi: 10.1016/1011-1344(90)85183-w. [DOI] [PubMed] [Google Scholar]
- 41.Freitas I. Lipid accumulation: the common feature to photosensitizer-retaining normal and malignant tissues [news] J Photochem Photobiol B. 1990;7:359–61. doi: 10.1016/1011-1344(90)85169-w. [DOI] [PubMed] [Google Scholar]
- 42.Graham A, Li G, Chen Y, et al. Structure-activity relationship of new octaethylporphyrin-based benzochlorins as photosensitizers for photodynamic therapy. Photochem Photobiol. 2003;77:561–6. doi: 10.1562/0031-8655(2003)077<0561:sronob>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 43.Wendler G, Lindemann P, Lacapere JJ, Papadopoulos V. Protoporphyrin IX binding and transport by recombinant mouse PBR. Biochem Biophys Res Commun. 2003;311:847–52. doi: 10.1016/j.bbrc.2003.10.070. [DOI] [PubMed] [Google Scholar]
- 44.Lampidis TJ, Bernal SD, Summerhayes IC, Chen LB. Selective toxicity of rhodamine 123 in carcinoma cells in vitro. Cancer Res. 1983;43:716–20. [PubMed] [Google Scholar]
- 45.Castro DJ, Saxton RE, Rodgerson DO, Fu YS, Bhuta SM, Fetterman HR, et al. Rhodamine-123 as a new laser dye: in vivo study of dye effects on murine metabolism, histology and ultrastructure. Laryngoscope. 1989;99:1057–62. doi: 10.1288/00005537-198210000-00016. [DOI] [PubMed] [Google Scholar]
- 46.Reddi E. Role of delivery vehicles for photosensitizers in the photodynamic therapy of tumours. J Photochem Photobiol B. 1997;37:189–95. doi: 10.1016/s1011-1344(96)07404-0. [DOI] [PubMed] [Google Scholar]
- 47.Woodburn K, Sykes E, Kessel D. Interactions of Solutol HS 15 and Cremophor EL with plasma lipoproteins. Int J Biochem Cell Biol. 1995;27:693–9. doi: 10.1016/1357-2725(95)00030-s. [DOI] [PubMed] [Google Scholar]
- 48.Woodburn K, Chang CK, Lee S, Henderson B, Kessel D. Biodistribution and PDT efficacy of a ketochlorin photosensitizer as a function of the delivery vehicle. Photochem Photobiol. 1994;60:154–9. doi: 10.1111/j.1751-1097.1994.tb05083.x. [DOI] [PubMed] [Google Scholar]
- 49.Ginevra F, Biffanti S, Pagnan A, Biolo R, Reddi E, Jori G. Delivery of the tumour photosensitizer zinc(II)-phthalocyanine to serum proteins by different liposomes: studies in vitro and in vivo. Cancer Lett. 1990;49:59–65. doi: 10.1016/0304-3835(90)90139-o. [DOI] [PubMed] [Google Scholar]
- 50.Jiang F, Lilge L, Logie B, Li Y, Chopp M. Photodynamic therapy of 9L gliosarcoma with liposome-delivered photofrin. Photochem Photobiol. 1997;65:701–6. doi: 10.1111/j.1751-1097.1997.tb01913.x. [DOI] [PubMed] [Google Scholar]
- 51.Richter AM, Waterfield E, Jain AK, Canaan AJ, Allison BA, Levy JG. Liposomal delivery of a photosensitizer, benzoporphyrin derivative monoacid ring A (BPD), to tumor tissue in a mouse tumor model. Photochem Photobiol. 1993;57:1000–6. doi: 10.1111/j.1751-1097.1993.tb02962.x. [DOI] [PubMed] [Google Scholar]
- 52.Polo L, Segalla A, Jori G, et al. Liposome-delivered 131I-labelled Zn(II)-phthalocyanine as a radiodiagnostic agent for tumours. Cancer Lett. 1996;109:57–61. doi: 10.1016/s0304-3835(96)04409-6. [DOI] [PubMed] [Google Scholar]
- 53.Allemann E, Rousseau J, Brasseur N, Kudrevich SV, Lewis K, van Lier JE. Photodynamic therapy of tumours with hexadecafluoro zinc phthalocyanine formulated in PEG-coated poly(lactic acid) nanoparticles. Int J Cancer. 1996;66:821–4. doi: 10.1002/(SICI)1097-0215(19960611)66:6<821::AID-IJC19>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 54.Margalit R, Silbiger E. Albumin microspheres as delivery systems for photodynamic drugs: physico-chemical studies and their implications for in vivo situations. J Microencapsul. 1985;2:183–96. doi: 10.3109/02652048509038524. [DOI] [PubMed] [Google Scholar]
- 55.Barel A, Jori G, Perin A, Romandini P, Pagnan A, Biffanti S. Role of high-, low- and very low-density lipoproteins in the transport and tumor-delivery of hematoporphyrin in vivo. Cancer Lett. 1986;32:145–50. doi: 10.1016/0304-3835(86)90112-6. [DOI] [PubMed] [Google Scholar]
- 56.Morliere P, Kohen E, Reyftmann JP, et al. Photosensitization by porphyrins delivered to L cell fibroblasts by human serum low density lipoproteins. A microspectrofluorometric study. Photochem Photobiol. 1987;46:183–91. doi: 10.1111/j.1751-1097.1987.tb04755.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhou CN, Milanesi C, Jori G. An ultrastructural comparative evaluation of tumors photosensitized by porphyrins administered in aqueous solution, bound to liposomes or to lipoproteins. Photochem Photobiol. 1988;48:487–92. doi: 10.1111/j.1751-1097.1988.tb02850.x. [DOI] [PubMed] [Google Scholar]
- 58.Schmidt-Erfurth U, Bauman W, Gragoudas E, et al. Photodynamic therapy of experimental choroidal melanoma using lipoprotein-delivered benzoporphyrin. Ophthalmology. 1994;101:89–99. doi: 10.1016/s0161-6420(13)31242-1. [DOI] [PubMed] [Google Scholar]
- 59.Pani R, Pellegrini R, Cinti MN, et al. New devices for imaging in nuclear medicine. Cancer Biother Radiopharm. 2004;19:121–8. doi: 10.1089/108497804773391766. [DOI] [PubMed] [Google Scholar]
- 60.Babbar AK, Singh AK, Goel HC, Chauhan UP, Sharma RK. Evaluation of (99m)Tc-labeled photosan-3, a hematoporphyrin derivative, as a potential radiopharmaceutical for tumor scintigraphy. Nucl Med Biol. 2000;27:587–92. doi: 10.1016/s0969-8051(00)00123-2. [DOI] [PubMed] [Google Scholar]
- 61.Link EM, Costa DC, Lane D, Blower PJ, Spittle MF. Radioiodinated methylene blue for diagnosing early melanoma metastases. Lancet. 1996;348:753. doi: 10.1016/s0140-6736(05)65639-5. [DOI] [PubMed] [Google Scholar]
- 62.Link EM, Carpenter RN. 211At-methylene blue for targeted radiotherapy of human melanoma xenografts: treatment of cutaneous tumors and lymph node metastases. Cancer Res. 1992;52:4385–90. [PubMed] [Google Scholar]
- 63.Link EM. Targeting melanoma with 211At/131I-methylene blue: preclinical and clinical experience. Hybridoma. 1999;18:77–82. doi: 10.1089/hyb.1999.18.77. [DOI] [PubMed] [Google Scholar]
- 64.Evstigneeva RP, Zaitsev AV, Luzgina VN, Ol’Shevskaya VA, Shtil AA. Carboranylporphyrins for boron neutron capture therapy of cancer. Curr Med Chem Anti-Cancer Agents. 2003;3:383–92. doi: 10.2174/1568011033482260. [DOI] [PubMed] [Google Scholar]
- 65.Hill JS, Kahl SB, Kaye AH, et al. Selective tumor uptake of a boronated porphyrin in an animal model of cerebral glioma. Proc Natl Acad Sci USA. 1992;89:1785–9. doi: 10.1073/pnas.89.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenthal MA, Kavar B, Hill JS, et al. Phase I and pharmacokinetic study of photodynamic therapy for high-grade gliomas using a novel boronated porphyrin. J Clin Oncol. 2001;19:519–24. doi: 10.1200/JCO.2001.19.2.519. [DOI] [PubMed] [Google Scholar]
- 67.Wilmes LJ, Hoehn-Berlage M, Els T, et al. In vivo relaxometry of three brain tumors in the rat: effect of Mn-TPPS, a tumor-selective contrast agent. J Magn Reson Imaging. 1993;3:5–12. doi: 10.1002/jmri.1880030103. [DOI] [PubMed] [Google Scholar]
- 68.Ikezaki K, Nomura T, Takahashi M, Fritz-Zieroth B, Inamura T, Fukui M. Selective and prolonged MRI enhancement by Mn-TPPS in an experimental rat brain tumour with peripheral benzodiazepine receptors. Neurol Res. 1994;16:393–7. doi: 10.1080/01616412.1994.11740260. [DOI] [PubMed] [Google Scholar]
- 69.Schaffer M, Ertl-Wagner B, Schaffer PM, et al. The application of Photofrin II as a sensitizing agent for ionizing radiation—a new approach in tumor therapy? Curr Med Chem. 2005;12:1209–15. doi: 10.2174/0929867053764653. [DOI] [PubMed] [Google Scholar]
- 70.Schaffer M, Schaffer PM, Corti L, et al. Photofrin as a specific radiosensitizing agent for tumors: studies in comparison to other porphyrins, in an experimental in vivo model. J Photochem Photobiol B. 2002;66:157–64. doi: 10.1016/s1011-1344(02)00237-3. [DOI] [PubMed] [Google Scholar]
- 71.Schaffer M, Schaffer PM, Vogesser M, et al. Application of Photofrin II as a specific radiosensitising agent in patients with bladder cancer—a report of two cases. Photochem Photobiol Sci. 2002;1:686–9. doi: 10.1039/b203732g. [DOI] [PubMed] [Google Scholar]
- 72.Young SW, Qing F, Harriman A, et al. Gadolinium(III) texaphyrin: a tumor selective radiation sensitizer that is detectable by MRI. Proc Natl Acad Sci USA. 1996;93:6610–5. doi: 10.1073/pnas.93.13.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young SW, Sidhu MK, Qing F, et al. Preclinical evaluation of gadolinium (III) texaphyrin complex. A new paramagnetic contrast agent for magnetic resonance imaging. Invest Radiol. 1994;29:330–8. doi: 10.1097/00004424-199403000-00013. [DOI] [PubMed] [Google Scholar]
- 74.Magda D, Lepp C, Gerasimchuk N, et al. Redox cycling by motexafin gadolinium enhances cellular response to ionizing radiation by forming reactive oxygen species. Int J Radiat Oncol Biol Phys. 2001;51:1025–36. doi: 10.1016/s0360-3016(01)01810-7. [DOI] [PubMed] [Google Scholar]
- 75.Miller RA, Woodburn K, Fan Q, Renschler MF, Sessler JL, Koutcher JA. In vivo animal studies with gadolinium (III) texaphyrin as a radiation enhancer. Int J Radiat Oncol Biol Phys. 1999;45:981–9. doi: 10.1016/s0360-3016(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 76.Evens AM. Motexafin gadolinium: a redox-active tumor selective agent for the treatment of cancer. Curr Opin Oncol. 2004;16:576–80. doi: 10.1097/01.cco.0000142073.29850.98. [DOI] [PubMed] [Google Scholar]
- 77.Henderson BW, Waldow SM, Mang TS, Potter WR, Malone PB, Dougherty TJ. Tumor destruction and kinetics of tumor cell death in two experimental mouse tumors following photodynamic therapy. Cancer Res. 1985;45:572–6. [PubMed] [Google Scholar]
- 78.Chan WS, Brasseur N, La Madeleine C, van Lier JE. Evidence for different mechanisms of EMT-6 tumor necrosis by photodynamic therapy with disulfonated aluminum phthalocyanine or photofrin: tumor cell survival and blood flow. Anticancer Res. 1996;16:1887–92. [PubMed] [Google Scholar]
- 79.Korbelik M, Krosl G. Cellular levels of photosensitisers in tumours: the role of proximity to the blood supply. Br J Cancer. 1994;70:604–10. doi: 10.1038/bjc.1994.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee CC, Pogue BW, O’Hara JA, et al. Spatial heterogeneity and temporal kinetics of photosensitizer (AlPcS2) concentration in murine tumors RIF-1 and MTG-B. Photochem Photobiol Sci. 2003;2:145–50. doi: 10.1039/b207598a. [DOI] [PubMed] [Google Scholar]
- 81.Zhou X, Pogue BW, Chen B, Hasan T. Analysis of effective molecular diffusion rates for verteporfin in subcutaneous versus orthotopic Dunning prostate tumors. Photochem Photobiol. 2004;79:323–31. doi: 10.1562/MU-03-31.1. [DOI] [PubMed] [Google Scholar]
- 82.Gomer CJ, Razum NJ. Acute skin response in albino mice following porphyrin photosensitization under oxic and anoxic conditions. Photochem Photobiol. 1984;40:435–9. doi: 10.1111/j.1751-1097.1984.tb04614.x. [DOI] [PubMed] [Google Scholar]
- 83.Foster TH, Murant RS, Bryant RG, Knox RS, Gibson SL, Hilf R. Oxygen consumption and diffusion effects in photodynamic therapy. Radiat Res. 1991;126:296–303. doi: 10.2307/3577919. [DOI] [PubMed] [Google Scholar]
- 84.Georgakoudi I, Nichols MG, Foster TH. The mechanism of Photofrin photobleaching and its consequences for photodynamic dosimetry. Photochem Photobiol. 1997;65:135–44. doi: 10.1111/j.1751-1097.1997.tb01889.x. [DOI] [PubMed] [Google Scholar]
- 85.Xu T, Li Y, Wu X. Application of lower fluence rate for less microvasculature damage and greater cell-killing during photodynamic therapy. Lasers Med Sci. 2004;19:150–4. doi: 10.1007/s10103-004-0310-1. [DOI] [PubMed] [Google Scholar]
- 86.Busch TM, Wileyto EP, Emanuele MJ, et al. Photodynamic therapy creates fluence rate-dependent gradients in the intratumoral spatial distribution of oxygen. Cancer Res. 2002;62:7273–9. [PubMed] [Google Scholar]
- 87.Snyder JW, Greco WR, Bellnier DA, Vaughan L, Henderson BW. Photodynamic therapy: a means to enhanced drug delivery to tumors. Cancer Res. 2003;63:8126–31. [PubMed] [Google Scholar]
- 88.Henderson BW, Busch TM, Vaughan LA, et al. Photofrin photodynamic therapy can significantly deplete or preserve oxygenation in human basal cell carcinomas during treatment, depending on fluence rate. Cancer Res. 2000;60:525–9. [PubMed] [Google Scholar]
- 89.Curnow A, Haller JC, Bown SG. Oxygen monitoring during 5-aminolaevulinic acid induced photodynamic therapy in normal rat colon. Comparison of continuous and fractionated light regimes. J Photochem Photobiol B. 2000;58:149–55. doi: 10.1016/s1011-1344(00)00120-2. [DOI] [PubMed] [Google Scholar]
- 90.Babilas P, Schacht V, Liebsch G, et al. Effects of light fractionation and different fluence rates on photodynamic therapy with 5-aminolaevulinic acid in vivo. Br J Cancer. 2003;88:1462–9. doi: 10.1038/sj.bjc.6600910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abels C. Targeting of the vascular system of solid tumours by photodynamic therapy (PDT) Photochem Photobiol Sci. 2004;3:765–71. doi: 10.1039/b314241h. [DOI] [PubMed] [Google Scholar]
- 92.Star WM, Marijnissen HP, van den Berg-Blok AE, Versteeg JA, Franken KA, Reinhold HS. Destruction of rat mammary tumor and normal tissue microcirculation by hematoporphyrin derivative photoradiation observed in vivo in sandwich observation chambers. Cancer Res. 1986;46:2532–40. [PubMed] [Google Scholar]
- 93.Henderson BW, Fingar VH. Relationship of tumor hypoxia and response to photodynamic treatment in an experimental mouse tumor. Cancer Res. 1987;47:3110–4. [PubMed] [Google Scholar]
- 94.Fingar VH, Siegel KA, Wieman TJ, Doak KW. The effects of thromboxane inhibitors on the microvascular and tumor response to photodynamic therapy. Photochem Photobiol. 1993;58:393–9. doi: 10.1111/j.1751-1097.1993.tb09580.x. [DOI] [PubMed] [Google Scholar]
- 95.Fingar VH, Wieman TJ, Karavolos PS, Doak KW, Ouellet R, van Lier JE. The effects of photodynamic therapy using differently substituted zinc phthalocyanines on vessel constriction, vessel leakage and tumor response. Photochem Photobiol. 1993;58:251–8. doi: 10.1111/j.1751-1097.1993.tb09557.x. [DOI] [PubMed] [Google Scholar]
- 96.McMahon KS, Wieman TJ, Moore PH, Fingar VH. Effects of photodynamic therapy using mono-l-aspartyl chlorin e6 on vessel constriction, vessel leakage, and tumor response. Cancer Res. 1994;54:5374–9. [PubMed] [Google Scholar]
- 97.Gilissen MJ, van de Merbelde Wit LE, Star WM, Koster JF, Sluiter W. Effect of photodynamic therapy on the endothelium-dependent relaxation of isolated rat aortas. Cancer Res. 1993;53:2548–52. [PubMed] [Google Scholar]
- 98.Fingar VH, Wieman TJ, Doak KW. Role of thromboxane and prostacyclin release on photodynamic therapy-induced tumor destruction. Cancer Res. 1990;50:2599–603. [PubMed] [Google Scholar]
- 99.Hamblin MR, Rajadhyaksha M, Momma T, Soukos NS, Hasan T. In vivo fluorescence imaging of the transport of charged chlorin e6 conjugates in a rat orthotopic prostate tumour. Br J Cancer. 1999;81:261–8. doi: 10.1038/sj.bjc.6690686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kurohane K, Tominaga A, Sato K, North JR, Namba Y, Oku N. Photodynamic therapy targeted to tumor-induced angiogenic vessels. Cancer Lett. 2001;167:49–56. doi: 10.1016/s0304-3835(01)00475-x. [DOI] [PubMed] [Google Scholar]
- 101.Chen B, Pogue BW, Goodwin IA, et al. Blood flow dynamics after photodynamic therapy with verteporfin in the RIF-1 tumor. Radiat Res. 2003;160:452–9. doi: 10.1667/RR3059. [DOI] [PubMed] [Google Scholar]
- 102.Chen B, Roskams T, de Witte PA. Antivascular tumor eradication by hypericin-mediated photodynamic therapy. Photochem Photobiol. 2002;76:509–13. doi: 10.1562/0031-8655(2002)076<0509:atebhm>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 103.Ferrario A, Kessel D, Gomer CJ. Metabolic properties and photosensitizing responsiveness of mono-l-aspartyl chlorin e6 in a mouse tumor model. Cancer Res. 1992;52:2890–3. [PubMed] [Google Scholar]
- 104.Cramers P, Ruevekamp M, Oppelaar H, Dalesio O, Baas P, Stewart FA. Foscan uptake and tissue distribution in relation to photodynamic efficacy. Br J Cancer. 2003;88:283–90. doi: 10.1038/sj.bjc.6600682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dolmans DE, Kadambi A, Hill JS, et al. Targeting tumor vasculature and cancer cells in orthotopic breast tumor by fractionated photosensitizer dosing photodynamic therapy. Cancer Res. 2002;62:4289–94. [PubMed] [Google Scholar]
- 106.Korbelik M, Krosl G, Krosl J, Dougherty GJ. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996;56:5647–52. [PubMed] [Google Scholar]
- 107.Agarwal ML, Larkin HE, Zaidi SI, Mukhtar H, Oleinick NL. Phospholipase activation triggers apoptosis in photosensitized mouse lymphoma cells. Cancer Res. 1993;53:5897–902. [PubMed] [Google Scholar]
- 108.Yamamoto N, Homma S, Sery TW, Donoso LA, Hoober JK. Photodynamic immunopotentiation: in vitro activation of macrophages by treatment of mouse peritoneal cells with haematoporphyrin derivative and light. Eur J Cancer. 1991;27:467–71. doi: 10.1016/0277-5379(91)90388-t. [DOI] [PubMed] [Google Scholar]
- 109.Herman S, Kalechman Y, Gafter U, Sredni B, Malik Z. Photofrin II induces cytokine secretion by mouse spleen cells and human peripheral mononuclear cells. Immunopharmacology. 1996;31:195–204. doi: 10.1016/0162-3109(95)00047-x. [DOI] [PubMed] [Google Scholar]
- 110.Gollnick SO, Liu X, Owczarczak B, Musser DA, Henderson BW. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997;57:3904–9. [PubMed] [Google Scholar]
- 111.Krosl G, Korbelik M, Dougherty GJ. Induction of immune cell infiltration into murine SCCVII tumour by photofrin-based photodynamic therapy. Br J Cancer. 1995;71:549–55. doi: 10.1038/bjc.1995.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamamoto N, Homma S, Nakagawa Y, et al. Activation of mouse macrophages by in vivo and in vitro treatment with a cyanine dye, lumin. J Photochem Photobiol B. 1992;13:295–306. doi: 10.1016/1011-1344(92)85069-7. [DOI] [PubMed] [Google Scholar]
- 113.Korbelik M, Naraparaju VR, Yamamoto N. Macrophage-directed immunotherapy as adjuvant to photodynamic therapy of cancer. Br J Cancer. 1997;75:202–7. doi: 10.1038/bjc.1997.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Korbelik M. Induction of tumor immunity by photodynamic therapy. J Clin Laser Med Surg. 1996;14:329–34. doi: 10.1089/clm.1996.14.329. [DOI] [PubMed] [Google Scholar]
- 115.Korbelik M, Dougherty GJ. Photodynamic therapy-mediated immune response against subcutaneous mouse tumors. Cancer Res. 1999;59:1941–6. [PubMed] [Google Scholar]
- 116.Krosl G, Korbelik M. Potentiation of photodynamic therapy by immunotherapy: the effect of schizophyllan (SPG) Cancer Lett. 1994;84:43–9. doi: 10.1016/0304-3835(94)90356-5. [DOI] [PubMed] [Google Scholar]
- 117.Krosl G, Korbelik M, Krosl J, Dougherty GJ. Potentiation of photodynamic therapy-elicited antitumor response by localized treatment with granulocyte-macrophage colony-stimulating factor. Cancer Res. 1996;56:3281–6. [PubMed] [Google Scholar]
- 118.Korbelik M, Cecic I. Enhancement of tumour response to photodynamic therapy by adjuvant mycobacterium cell-wall treatment. J Photochem Photobiol B. 1998;44:151–8. doi: 10.1016/S1011-1344(98)00138-9. [DOI] [PubMed] [Google Scholar]
- 119.Korbelik M, Sun J, Cecic I, Serrano K. Adjuvant treatment for complement activation increases the effectiveness of photodynamic therapy of solid tumors. Photochem Photobiol Sci. 2004;3:812–6. doi: 10.1039/b315663J. [DOI] [PubMed] [Google Scholar]
- 120.Castano AP, Liu Q, Hamblin MR. Photodynamic therapy cures green fluorescent protein expressing RIF1 tumors in mice. Proc SPIE. 2004;5319:50–9. [Google Scholar]
- 121.Castano AP, Hamblin MR. Combination of photodynamic therapy and low-dose cyclophosphamide as a treatment for metastatic murine tumors. Proc Am Assoc Cancer Res. 2004;45:997. [Google Scholar]
- 122.Gollnick SO, Vaughan L, Henderson BW. Generation of effective antitumor vaccines using photodynamic therapy. Cancer Res. 2002;62:1604–8. [PubMed] [Google Scholar]