

Abstract
Cancer is a heterogeneous set of diseases characterized by different molecular and cellular features. Over the past decades, researchers have attempted to grasp the complexity of cancer by mapping the genetic aberrations associated with it. In these efforts, the contribution of mitochondria to the pathogenesis of cancer has tended to be neglected. However, more recently, a growing body of evidence suggests that mitochondria play a key role in cancer. In fact, dysfunctional mitochondria not only contribute to the metabolic reprogramming of cancer cells but they also modulate a plethora of cellular processes involved in tumorigenesis. In this review, we describe the link between mutations to mitochondrial enzymes and tumor formation. We also discuss the hypothesis that mutations to mitochondrial and nuclear DNA could cooperate to promote the survival of cancer cells in an evolving metabolic landscape.
Keywords: Cancer, Metabolism, Mitochondria, TCA cycle, mtDNA mutations, Oncometabolites, Evolution
Review
Background
Current evidence suggests that the eukaryotic cell originates from the symbiosis between a hydrogen-dependent archaebacterium, the host cell, and a hydrogen-producing eubacterium, the ancestor of modern mitochondria, started two billion years ago [1,2]. This cooperation granted to the newly formed eukaryotic cell several evolutionary advantages, including a more efficient metabolism [1], the detoxification from the harms of the raising levels of atmospheric oxygen [1], and the ability to form multicellular organisms [3]. During evolution, the interaction between mitochondria and the host cell evolved into a more intimate relationship and mitochondria lost control of many of their functions by transferring part of their genome to the nucleus [4]. However, although subordinate to the nucleus, mitochondria maintained the capacity to communicate to the rest of the cells. Mitochondria are in fact the gatekeepers of the eukaryote's cell viability by regulating programmed cell death [5], and they control nuclear functions by the production of reactive oxygen species (ROS), by the modulation of calcium levels [6], and by the trafficking of small molecule metabolites [7]. It is therefore not surprising that the aberrant communication between mitochondria and the rest of the cell may lead to alterations of cellular homeostasis and, in multicellular organisms, to organismal dysfunction. Indeed, altered mitochondrial function has been related to diverse pathological conditions, including cardiovascular disorders, muscular degeneration, neurodegenerative disorders [8], and cancer [9]. Although the connection between mitochondria dysfunction and cancer has historically focused on metabolism [10], their contribution to cell homeostasis goes far beyond metabolism. In this review, we will describe how mitochondrial dysfunction caused by either nuclear or mitochondrial DNA mutations of key metabolic enzymes can initiate a complex cellular reprogramming that supports tumor formation and growth.
Defects in TCA cycle enzymes and cancer
Among the metabolic pathways that operate in the mitochondria, the tricarboxylic acid (TCA) cycle has recently been in the spotlight of the field of oncology. TCA cycle enzymes are encoded by nuclear DNA (nDNA) and are located in the mitochondrial matrix, with the exception of succinate dehydrogenase, which is embedded in the inner mitochondrial membrane, facing the matrix. In the last decade, several enzymes of the TCA cycle, which we will briefly describe in the following paragraphs, have been found mutated in both sporadic and hereditary forms of cancer.
Citrate synthase
Citrate synthase (CS) catalyzes the first committed step of the TCA cycle, i.e. the irreversible condensation of acetyl coenzyme A (AcCoA) and oxaloacetate into citrate. Citrate can then proceed into the TCA cycle or can be exported to the cytosol and used for protein acetylation or fatty acid biosynthesis [11] (Figure 1A). Evidence for a role of citrate synthase (CS) in cancer is sparse and controversial: CS was found to be increased in pancreatic ductal carcinoma [12] and renal oncocytoma [13] but downregulated in various cervical cancer cell lines [14]. Unfortunately, whether these changes are a simple reflection of variations in mitochondrial mass has not been determined. Furthermore, it is not clear how the deregulation of CS contributes to tumorigenesis. Two scenarios can be hypothesized. On the one hand, increased CS activity, by providing more citrate, could be an advantage for cancer cells that depend on increased fatty acid biosynthesis, such as pancreatic cancer [15]. On the other hand, the loss of CS, by inducing mitochondrial dysfunction could trigger a tumor-supporting glycolytic switch, commonly found in cancer cells. Interestingly, the loss of CS was linked to the induction of the epithelial-to-mesenchymal transition (EMT), suggesting that CS deficiency not only promotes a metabolic rewiring but also indirectly supports cancer cell invasion and metastasis [14].
Figure 1.
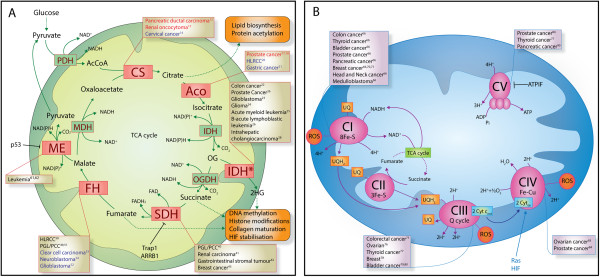
Mitochondrial dysfunctions in cancer. Schematic representation of mitochondrial enzymes involved in cancer, focusing on enzymes of the TCA cycle (A) and of the respiratory chain and ATP synthase (B). The type of cancer associated with each individual enzyme is listed in boxes. The color of the text indicates if the enzyme has been found upregulated (red), downregulated (blue), or mutated (black) in the given tumor type. CS citrate synthase, Aco aconitase, IDH isocitrate dehydrogenase, IDH* mutant IDH, OGDH oxoglutarate dehydrogenase, SDH succinate dehydrogenase, FH fumarate hydratase, ME malic enzyme, MDH malate dehydrogenase, PDH pyruvate dehydrogenase, OG 2-oxoglutarate, 2HG 2-hydroxyglutarate, HLRCC hereditary leiomyomatosis and renal cell cancer, PGL/PCC hereditary paraganglioma and pheochromocytoma, CI–CV complex I–V, Cyt c cytochrome c, UQ ubiquinone, UQH2 ubiquinol, ROS reactive oxygen species, ATPIF ATP synthase inhibitory factor. Dashed lines indicate a series of reaction in a complex pathway, whereas solid lines indicate a single step reaction.
Aconitase
Aconitate hydratase or aconitase (Aco) is a Fe-S cluster enzyme that performs the reversible isomerization of citrate to isocitrate via the intermediate cis-aconitate (Figure 1A). The role of aconitase in tumor formation has been mainly investigated in the prostate where this enzyme plays an important physiological role. In normal prostate epithelium aconitase activity is inhibited by high levels of zinc, which leads to an extraordinary accumulation of citrate [16]. In prostate cancer, however, aconitase activity is restored, re-establishing citrate oxidation [17] and decreasing fatty acid synthesis [18]. The subsequent decrease in citrate is a key metabolic feature of the transformed epithelium, making citrate a useful in vivo marker for discriminating prostate cancer from surrounding healthy regions [19]. In contrast to the tumor-promoting role of aconitase in prostate cancer, the inhibition of this enzyme has been observed in fumarate hydratase (FH)-deficient cancer cell lines. In these cells, the accumulation of the TCA cycle intermediate fumarate causes the inactivation of the iron-sulfur cluster of the enzyme, leading to a complete loss of aconitase activity (see paragraph on fumarate hydratase (FH) and [20]). Decreased expression of aconitase has also been observed in gastric cancer, and its expression is a prognostic marker of disease progression [21]. Whether mitochondrial aconitase has additional roles beyond regulating citrate availability is currently unknown.
Isocitrate dehydrogenase
Isocitrate dehydrogenase (IDH) catalyzes the reversible conversion of isocitrate into 2-oxoglutarate (OG). In eukaryotes, one nicotinamide adenine dinucleotide (NADH)-dependent (IDH3) and two nicotinamide adenine dinucleotide phosphate (NADPH)-dependent (IDH1 and IDH2) isoforms of IDH exist (Figure 1A). Mutations of both the cytoplasmic (IDH1) and the mitochondrial (IDH2) NADPH-dependent isoforms have been found in various human cancers, including colon cancer [22], glioblastoma [23], glioma [24], acute myeloid leukemia [25], prostate cancer [26], B-acute lymphoblastic leukemia [26], osteosarcoma [27], and intrahepatic cholangiocarcinoma [28]. Oncogenic mutations confer a neomorphic activity to IDHs, which instead of converting isocitrate in OG, reduce OG into the R-enantiomer of 2-hydroxyglutarate (R-2HG), which accumulates up to millimolar levels in cancer cells (See Figure 1A and [29,30]). This poorly characterized metabolite is now considered a major contributor to the oncogenic activity of mutated IDHs. Indeed, the incubation of cells with R-2HG promotes cytokine independency and blocks differentiation in hematopoietic cells, inducing leukemogenesis [31]. The tumorigenic activity of 2HG has been attributed to its inhibitory effect on various OG-dependent dioxygenases, including the hypoxia-inducible factors (HIFs) prolyl hydroxylases (PHDs), histone demethylases, and the ten-eleven translocation (TET) family of DNA demethylases [32,33]. The first evidence that 2HG acted upon DNA methylation arose in 2010 when a large-scale DNA methylation analysis of human leukemia found that the expression of mutated IDH, by increasing 2-HG levels, led to DNA hyper-methylation, a broad epigenetic change associated with poor hematopoietic differentiation. Of note, such a peculiar change in DNA methylation was dependent on the inhibition of TET2 caused by 2HG [34]. A similar epigenetic fingerprint has also been observed in a subset of breast tumors where 2HG was found to accumulate to millimolar levels. Interestingly, however, in these tumors, the accumulation of 2HG was not caused by overt IDH mutations but, rather, by a particular metabolic rewiring instigated by Myc overexpression [35]. These results suggest that 2HG has an important role in tumorigenesis and that it can accumulate in cancer cells not only upon IDH mutations but also as a consequence of metabolic derangements, including hypoxia [36]. More recent results revealed that, besides inhibiting DNA demethylases, 2HG accumulation also causes profound changes in histone methylation [37], indicating that this metabolite has multiple and well-defined epigenetic roles. The inhibitory effects of 2HG toward PHDs are instead more controversial and appear isomer-specific. In fact, while the S-enantiomer of 2HG (S-2HG) was shown to inhibit PHDs, R-2HG activates them, leading to accelerated degradation of HIFs [38]. Although initially unclear, the paradoxical activation of PHDs by R-2HG can be explained by its non-enzymatic oxidation to OG, the natural substrate of these enzymes [39]. Of note, these results imply that HIF is not required for R-2HG-induced tumorigenesis and, on the other hand, suggest that this transcription factor might act as a tumor suppressor in this specific context.
Succinate dehydrogenase
Succinate dehydrogenase (SDH) is an enzyme complex bound to the inner mitochondrial membrane that converts succinate into fumarate, in a reaction coupled to the reduction of flavin adenine dinucleotide (FAD) to FADH2. SDH represents a unique link between the TCA cycle and the mitochondrial respiratory chain, where it is also known as respiratory chain complex II (Figure 1A,B). SDH is the only known enzyme of the respiratory chain completely encoded by nDNA and is devoid of proton pumping activity. Inactivating mutations of SDH subunits and assembly factors have been linked to different types of hereditary and sporadic forms of cancer, including hereditary paraganglioma and pheochromocytoma (PGC/PCC) [40], renal carcinoma [41], gastrointestinal stromal tumor [42], and breast cancer [43]. SDH can behave as a classic tumor suppressor gene since the mutated allele is inherited in a heterozygous fashion, while the remaining wild type allele is lost in tumor samples. Similarly to mutant IDHs, most of the oncogenic activity of SDH mutations has been attributed to a metabolite, succinate, which accumulates in SDH-deficient cells. The oncogenic role of succinate was initially linked to the inhibition of PHDs and the subsequent stabilization of HIF [44]. More recently, succinate was found to be a prototypical ‘epigenetic hacker’ [45], capable of inhibiting both DNA [46,47] and histone demethylases [48], leading to epigenetic changes that overlap with those observed in mutant IDH cancers [49].
Fumarate hydratase
FH catalyzes the reversible conversion of fumarate to malate (Figure 1A). Germline mutations of FH were originally discovered in hereditary leiomyomatosis and renal cell cancer (HLRCC) [50]. More recently FH germline mutations were also found in a subset of PGC/PCC [49,51]. FH was also found to be downregulated in glioblastoma [52] and sporadic clear cell carcinoma [53] and deleted in non-Myc-amplified neuroblastoma [54]. Similarly to SDH, FH behaves as a classic tumor suppressor. Part of its tumorigenic activity has been attributed to the abnormal accumulation of fumarate, which peaks to high millimolar levels in FH-deficient cancer cells [55]. Fumarate shares some similarities with succinate and 2HG in that it can inhibit several OG-dependent enzymes, including PHDs [56], and histone and DNA demethylases [46]. Interestingly, however, fumarate possesses another unique property linked to its chemical structure. In fact, fumarate is a moderately reactive α,β-unsaturated electrophilic metabolite that, under physiological conditions, can covalently bind to cysteine residues of proteins in a process called succination[57,58]. Several proteins are succinated in FH-deficient cells, including aconitase [20], and Kelch-like ECH-associated protein 1 (Keap1) [57,58]. Of note, the succination of Keap1 abrogates its inhibitory activity toward the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcription factor, leading to the activation of several antioxidant genes thought to play key roles in supporting tumor formation [57,58]. Interestingly, also, the reactive thiol residue of GSH is subject to succination, and this phenomenon is linked to increased oxidative stress in FH-deficient cancer cells UOK262 [59].
Malic enzyme
Malic enzyme (ME) catalyzes the oxidative decarboxylation of malate into pyruvate and CO2 (Figure 1A). In mammalian cells, two NADP+-dependent MEs, the cytosolic ME1 and the mitochondrial ME3, and the mitochondrial NAD+-dependent ME2 have been described. The first link between mitochondrial MEs and cancer traces back to the 1970s, when Lehninger's laboratory observed that mitochondria isolated from leukemia-derived ascites cancer cells carried unexpectedly high rates of conversion of malate into pyruvate [60]. Ten years later, the same lab suggested that malate metabolism is compartmentalized: malate generated from glutamine oxidation in the mitochondria proceeds through the TCA cycle, whereas cytosolic malate is converted into pyruvate by the mitochondrial ME2. The authors also observed that extra-mitochondrial malate, after conversion into pyruvate and then citrate, could fuel fatty acids and cholesterol biosynthesis, supporting tumor growth [61]. More recent evidence underscored the role of this enzyme in leukemia cells, where the silencing of ME2 led to diminished proliferation and increased apoptosis [62]. Interestingly, the expression of ME1 and ME2 has been found to be regulated by p53 and to tightly control NADPH homeostasis, corroborating the connection between these enzymes and oncogenic metabolic rewiring [63].
Mitochondrial DNA mutations and cancer
Mitochondria contain a circular chromosome of 16,596 base pairs, coding for 37 genes translated into 13 subunits of the respiratory chain and ATPase complexes, 22 tRNAs and 12S and 16S ribosomal RNAs. Mammalian cells contain thousands of copies of mitochondrial DNA (mtDNA) [64]. In contrast to nDNA, mtDNA mutations coexist with normal mtDNA in a heterogeneous mixture known as heteroplasmy. Importantly, by varying the level of heteroplasmy, a single mtDNA mutation might result in a wide range of bioenergetics defects, from mild mitochondrial dysfunction to a severe bioenergetic impairment and cell death [65]. Somatic mtDNA mutations have been found in a wide array of human cancers including tumors of colon, breast, lung, prostate, liver, pancreas, kidney, thyroid and brain as well as in gastric carcinoma and ovarian cancer [66] and are usually associated with bioenergetics defects. Nevertheless, a complete loss of mtDNA seems detrimental for cancer cells. For instance, experiments with mtDNA-deficient cells (ρ0 cells) have clearly shown that cancer cells need functional mitochondria for their survival and proliferation [67,68]. A thorough description of mtDNA mutations in cancer has been given in other excellent reviews (see for instance [66] and [9]). In our review, we will summarize the most recent findings and propose a unifying theory of the role of mtDNA mutations in cancer.
Complex I
Among mtDNA mutations associated with cancer initiation and progression, those affecting complex I (CI) of the respiratory chain are the most common. CI, also known as NADH:ubiquinone oxidoreductase, catalyzes the transfer of two electrons from NADH to ubiquinone via flavin mononucleotides, producing NAD+ and four protons, which are pumped in the intermembrane space (Figure 1B) [11]. CI is the first site of the electron transport chain and active site of reactive oxygen species (ROS) production. Therefore, mutations in CI can significantly alter cell bioenergetics and redox homeostasis [69]. Mutations in mitochondrial genes encoding for CI have been linked to the development of colon, thyroid, pancreas, breast, bladder, and prostate cancer as well as of head and neck tumors and medulloblastoma (reviewed in [66]). Furthermore, mtDNA mutations that affect CI have been linked to increased ROS-dependent metastatic potential in Lewis lung carcinoma and breast cancer cells [70,71]. The contribution of CI mutations to cancer largely depends on the corresponding bioenergetics dysfunction that they cause. In fact, cancer cells affected by severe CI deficiency exhibited decreased tumorigenic potential both in vitro and in vivo, if compared to cells with a mild CI dysfunction [72] and CI activity is required for the induction of aerobic glycolysis in osteosarcoma cells [73]. In line with these finding, a recent study showed that intact CI activity is essential for cancer cell survival at low glucose levels, a condition commonly found in tumor microenvironment [74].
Complex III
Complex III, also known as coenzyme Q:cytochrome c oxidoreductase, or cytochrome bc1, catalyzes the electron transfer from reduced ubiquinone or coenzyme Q 10 to cytochrome c followed by the pumping of four protons into the intermembrane space (Figure 1B). mtDNA mutations that affect CIII have been found in various cancers, including colorectal [75], ovarian [76], thyroid [77], breast [78], and bladder [79] cancers. In support to an oncogenic function of CIII dysfunctions, it was demonstrated that the expression of a truncated subunit of CIII in MB49 bladder cancer cells increases cell growth and invasion both in vitro and in vivo[80]. Interestingly, this oncogenic phenotype was accompanied by lactate secretion, increased ROS production, and resistance to apoptosis via activation of NF-κB2 pathway [80]. In line with these findings, the expression of a mutated form of CYTB in SV40-immortalized human uroepithelial cells induced an antiapoptotic signaling cascade that sustained cancer cell growth [81]. Together, these results suggest that mtDNA mutations that affect CIII activity are sufficient to drive tumorigenesis via a mechanism that involves ROS production and the inhibition of apoptosis.
Complex IV
Cytochrome c oxidase, also known as complex IV (CIV) is the terminal complex of the respiratory chain. CIV is composed of 12 subunits, of which 3 (I, II, and III) are encoded by mtDNA and 9 (IV–XIII) by nDNA. CIV receives four electrons from cytochrome c and reduces molecular oxygen into water and four protons, which are pumped in the intermembrane space (Figure 1B). CIV is the rate-limiting step of respiratory chain and a well-characterized site of ROS production [82]. The link between CIV activity and cancer is controversial. Mutations of the mtDNA-encoded CIV subunit 1 (COX1) have been associated with ovarian cancer [83] and prostate cancer [84]. On the other hand, nDNA-encoded subunits of CIV are generally upregulated in cancer. For instance, the overexpression of the antiapoptotic protein Bcl-2 in leukemia cells increased the mitochondrial localization of the subunit Va of CIV (cytochrome oxidase (COX) Va) and COX Vb, leading to increased respiration and high intracellular ROS [85]. In line with these findings, the expression of oncogenic Ras in immortalized human bronchial epithelial cells increases CIV activity and the inhibition of Ras in A549 lung adenocarcinoma cells reduces COX Vb expression [86]. Finally, hypoxia, an environmental cue experienced by cancer cells, can also increase CIV efficiency by regulating the ratio between two CIV subunits (COX4-1 and COX4-2) in HIF1-dependent fashion [87]. These results seem to suggest that mtDNA-encoded subunits are generally tumor-suppressing, whereas nDNA encoded-subunits are tumor-promoting.
Complex V
Adenosine triphosphate (ATP) synthase, also known as complex V (CV), is the final enzyme of oxidative phosphorylation. CV exploits the electrochemical potential gradient across the inner mitochondrial membrane to generate ATP from ADP and inorganic phosphate (Figure 1B). Of note, the ATP synthase has recently been found to be part of the permeability transition pore (PTP) [88], a membrane-embedded mitochondrial complex involved in several mitochondria-dependent processes, including calcium buffering and apoptosis [89]. Mutations in CV subunits encoded by mtDNA have been found in thyroid [77], pancreatic [90], and prostate [84] cancer. To investigate the oncogenic activity of CV mutations, Shidara and colleagues introduced two different point mutations in the mtDNA gene encoding for the CV subunit 6 (MTATP6) [91]. Interestingly, mutant ATP6 increased cell proliferation in 2D cultures and led to higher oncogenic potential in xenografts. Importantly, the reintroduction of a nuclear-encoded wild-type ATP6 suppressed tumor formation in these cells. Several factors could explain the link between CV mutations and tumorigenesis. For instance, mutant cells displayed reduced apoptosis, suggesting that the oncogenic function of mutant ATP6 could involve inhibition of programmed cell death, which is consistent with the role of CV in the regulation of the PTP [88]. Also, ATP6 mutations were associated with increased ROS production, suggesting that, even if the ATP synthase is not directly involved in the transport of electrons, its inhibition could cause electron leak from the respiratory chain, inducing ROS generation. In contrast with the link between low CV and cancer, a recent work showed that a functional ATP synthase is instead required for cell survival in the presence of overt dysfunction of oxidative phosphorylation. Indeed, it was recently found that the loss of the ATPase inhibitory factor ATPIF1 protected from antimycin-induced cell death, in a human haploid cells. Interestingly, it was demonstrated that the ablation of ATPIF1 is required to allow the reversal of ATP synthase, a process whereby ATP synthase hydrolyses ATP to maintain a mitochondrial membrane potential [92]. These observations underscore the plasticity of CV, which can shape its activity to maintain mitochondrial potential and, eventually, to support survival.
Conclusions
In this review, we have explored the link between defects in mitochondrial metabolism, caused by mtDNA or nDNA mutations, and tumorigenesis. We have also discussed the hypothesis that mitochondrial dysfunction not only perturbs cellular bioenergetics, supporting the metabolic transformation of cancer cell, but that it also triggers tumor-promoting (epi)genetic changes mediated by the small molecule metabolites that they release. Given the importance of mitochondria in tumorigenesis, it is not surprising that canonical oncogenes and tumor suppressors exert their functions by regulating mitochondrial function [7]. For instance, Trap1 [93] and the endocytic adaptor protein β-arrestin [94] were shown to alter SDH expression and activate a succinate-dependent pseudoxypoxic response in support of their tumorigenic program. Hence, deregulation of mitochondrial function plays a key role not only in tumor initiation but also during tumor progression, where secondary mitochondrial dysfunction would enable cancer cells to adapt to a constantly evolving tumor microenvironment. In this scenario, however, mtDNA mutations, by virtue of their tunable bioenergetic outcome, would represent a more efficient way to adapt to novel metabolic niches than nDNA mutations. We propose that nDNA and mtDNA mutations are co-selected to finely shape the metabolic efficiency of cancer cell during tumor evolution: mtDNA mutations would enable fast and reversible explorations of different metabolic niches, whereas nDNA mutations would permanently fix an advantageous metabolic configuration and pass this information to the daughter cells (Figure 2). Considering the long-standing evolutionary cooperation between mitochondria and the host cells, it is not surprising that their two genomes are hard-wired for cell survival and proliferation.
Figure 2.
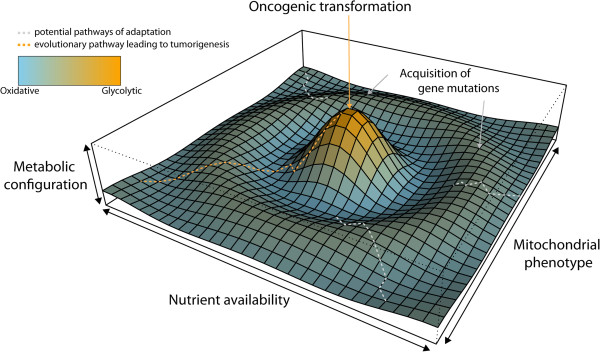
The evolving metabolic landscape of a cell. Schematic representation of the evolutionary process of a cancer cell driven by metabolic cues. The high bioenergetic flexibility of mitochondria allows cells to adapt to ever-changing environments, acquiring different metabolic configurations within the metabolic landscape. This metabolic flexibility is achieved by mutations of mtDNA and further shaped by the degree of heteroplasmy of the mutations itself. According to pre-existing metabolic adaptations (mitochondrial phenotypes) and to nutrient availability, there might be a selective pressure on the acquisition of genetic mutations that can sustain a certain metabolic configuration (gray dashed lines). The nDNA mutation is then passed to the progeny. The fixation of a specific metabolic configuration (e.g. aerobic glycolysis) could then lead to tumorigenic transformation (orange dashed lines) by yet unidentified mechanisms. This scenario could be used to trace the metabolic evolution of cancer based on an evolving metabolic landscape.
Abbreviations
2HG: 2-hydroxyglutarate; AcCoA: acetyl coenzyme A; Aco: aconitase; ADP: adenosine diphosphate; ATP: adenosine triphosphate; ATPIF: ATPase inhibitory factor; CI–V: respiratory chain complex I–V; CS: citrate synthase; COX: cytochrome oxidase; CYT: cytochrome; EMT: epithelial to mesenchymal transition; FAD: flavin adenine dinucleotide; FH: fumarate hydratase; GSH: reduced glutathione; HIF: hypoxia-inducible factor; HLRCC: hereditary leiomyomatosis and renal cell cancer; IDH: isocitrate dehydrogenase; Keap1: Kelch-like ECH-associated protein 1; ME: malic enzyme; mtDNA: mitochondrial DNA; NADH: nicotinamide adenine dinucleotide; NADPH: nicotinamide adenine dinucleotide phosphate; nDNA: nuclear DNA; Nrf2: nuclear factor (erythroid-derived 2)-like 2; OG: 2-oxoglutarate; PGC/PCC: hereditary paraganglioma and pheochromocytoma; PHD: prolyl hydroxylases; PTP: permeability transition pore; ROS: reactive oxygen species; SDH: succinate dehydrogenase; TCA: tricarboxylic acid; TET: ten-eleven translocation.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EG wrote the manuscript and prepared the figures. CF supervised the design of the review and wrote the manuscript. Both authors read and approved the final manuscript.
Authors’ information
EG is a PhD student of the University of Cambridge in the laboratory of CF. CF is a group leader at the MRC Cancer Unit.
Contributor Information
Edoardo Gaude, Email: eg418@MRC-CU.cam.ac.uk.
Christian Frezza, Email: cf366@MRC-CU.cam.ac.uk.
Acknowledgements
EG and CF would like to thank the members of the Frezza lab for discussing some of the key concepts proposed in the review and Dr. Mike Murphy for carefully reading the manuscript.
References
- Martin W, Muller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392:37–41. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- Margulis L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp Soc Exp Biol. 1975. pp. 21–38. [PubMed]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 2004;5:123–135. doi: 10.1038/nrg1271. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Aravind L. Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ. 2002;9:394–404. doi: 10.1038/sj.cdd.4400991. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Frezza C. The role of mitochondria in the oncogenic signal transduction. Int J Biochem Cell Biol. 2014;48:11–17. doi: 10.1016/j.biocel.2013.12.013. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–1834. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Salway J. Metabolism at a Glance. Oxford, U.K: Wiley-Blackwell; 2004. [Google Scholar]
- Schlichtholz B, Turyn J, Goyke E, Biernacki M, Jaskiewicz K, Sledzinski Z, Swierczynski J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas. 2005;30:99–104. doi: 10.1097/01.mpa.0000153326.69816.7d. [DOI] [PubMed] [Google Scholar]
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, Yeh CW, Chen YC, Liao CC, Chang WT. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. 2012;2:785. doi: 10.1038/srep00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Kloog Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014;5:e1065. doi: 10.1038/cddis.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh JP. Sodium, potassium, calcium, magnesium, zinc, citrate and chloride content of human prostatic and seminal fluid. J Reprod Fertil. 1985;75:35–41. doi: 10.1530/jrf.0.0750035. [DOI] [PubMed] [Google Scholar]
- Singh KK, Desouki MM, Franklin RB, Costello LC. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol Cancer. 2006;5:14. doi: 10.1186/1476-4598-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui KH, Chung LC, Wang SW, Feng TH, Chang PL, Juang HH. Hypoxia upregulates the gene expression of mitochondrial aconitase in prostate carcinoma cells. J Mol Endocrinol. 2013;51:131–141. doi: 10.1530/JME-13-0090. [DOI] [PubMed] [Google Scholar]
- Kurhanewicz J, Vigneron DB, Nelson SJ, Hricak H, MacDonald JM, Konety B, Narayan P. Citrate as an in vivo marker to discriminate prostate cancer from benign prostatic hyperplasia and normal prostate peripheral zone: detection via localized proton spectroscopy. Urology. 1995;45:459–466. doi: 10.1016/S0090-4295(99)80016-8. [DOI] [PubMed] [Google Scholar]
- Ternette N, Yang M, Laroyia M, Kitagawa M, O'Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler BM, Lappin T, Frizzell N, Soga T, Adam J, Pollard PJ. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013;3:689–700. doi: 10.1016/j.celrep.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Mai C, Wei YL, Zhao JJ, Hu YM, Zeng ZL, Yang J, Lu WH, Xu RH, Huang P. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med Oncol. 2013;30:552. doi: 10.1007/s12032-013-0552-5. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- Liu X, Kato Y, Kaneko MK, Sugawara M, Ogasawara S, Tsujimoto Y, Naganuma Y, Yamakawa M, Tsuchiya T, Takagi M. Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med. 2013;2:803–814. doi: 10.1002/cam4.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borger DR, Goyal L, Yau T, Poon RT, Ancukiewicz M, Deshpande V, Christiani DC, Liebman HM, Yang H, Kim H, Yen K, Faris JE, Iafrate AJ, Kwak EL, Clark JW, Allen JN, Blaszkowsky LS, Murphy JE, Saha SK, Hong TS, Wo JY, Ferrone CR, Tanabe KK, Bardeesy N, Straley KS, Agresta S, Schenkein DP, Ellisen LW, Ryan DP, Zhu AX. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res. 2014;20:1884–1890. doi: 10.1158/1078-0432.CCR-13-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TY, Kurley SJ, Putluri V, Sonavane R, Edelman DC, Wulff J, Starks AM, Yang Y, Kittles RA, Yfantis HG, Lee DH, Ioffe OB, Schiff R, Stephens RM, Meltzer PS, Veenstra TD, Westbrook TF, Sreekumar A, Ambs S. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O'Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG Jr. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarhonskaya H, Rydzik AM, Leung IK, Loik ND, Chan MC, Kawamura A, McCullagh JS, Claridge TD, Flashman E, Schofield CJ. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5:3423. doi: 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, Pediatric NIH, Wild-Type GIST C, O'Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim DH, Jung WH, Koo JS. Succinate dehydrogenase expression in breast cancer. Springerplus. 2013;2:299. doi: 10.1186/2193-1801-2-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Yang M, Pollard PJ. Succinate: a new epigenetic hacker. Cancer Cell. 2013;23:709–711. doi: 10.1016/j.ccr.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI Jr, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O'Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera AM, Bayley JP, Devilee P, McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer. 2009;8:89. doi: 10.1186/1476-4598-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reyniès A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomäki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA. Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Currás-Freixes M, Franco-Vidal V, Guillaud-Bataille M, Simian C, Morin A, Letón R, Gómez-Graña A, Pollard PJ, Rustin P, Robledo M, Favier J, Gimenez-Roqueplo AP. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. doi: 10.1093/hmg/ddt639. [DOI] [PubMed] [Google Scholar]
- Khalil AA. Biomarker discovery: a proteomic approach for brain cancer profiling. Cancer Sci. 2007;98:201–213. doi: 10.1111/j.1349-7006.2007.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarshan S, Shanmugasundaram K, Naylor SL, Lin S, Livi CB, O'Neill CF, Parekh DJ, Yeh IT, Sun LZ, Block K. Reduced expression of fumarate hydratase in clear cell renal cancer mediates HIF-2alpha accumulation and promotes migration and invasion. PLoS One. 2011;6:e21037. doi: 10.1371/journal.pone.0021037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieuw A, Kumps C, Schramm A, Pattyn F, Menten B, Antonacci F, Sudmant P, Schulte JH, Van Roy N, Vergult S, Buckley PG, De Paepe A, Noguera R, Versteeg R, Stallings R, Eggert A, Vandesompele J, De Preter K, Speleman F. Identification of a novel recurrent 1q42.2-1qter deletion in high risk MYCN single copy 11q deleted neuroblastomas. Int J Cancer. 2012;130:2599–2606. doi: 10.1002/ijc.26317. [DOI] [PubMed] [Google Scholar]
- Zheng L, Mackenzie ED, Karim SA, Hedley A, Blyth K, Kalna G, Watson DG, Szlosarek P, Frezza C, Gottlieb E. Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 2013;1:12. doi: 10.1186/2049-3002-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinié V, Richard S, Tan PH, Teh BT, Furge KA. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Adam J, Hatipoglu E, O'Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford RG, Lehninger AL. Active oxidative decarboxylation of malate by mitochondria isolated from L-1210 ascites tumor cells. Biochem Biophys Res Commun. 1973;51:480–486. doi: 10.1016/0006-291x(73)91282-5. [DOI] [PubMed] [Google Scholar]
- Moreadith RW, Lehninger AL. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P) + −dependent malic enzyme. J Biol Chem. 1984;259:6215–6221. [PubMed] [Google Scholar]
- Ren JG, Seth P, Everett P, Clish CB, Sukhatme VP. Induction of erythroid differentiation in human erythroleukemia cells by depletion of malic enzyme 2. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013;493:689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- Cavalli LR, Varella-Garcia M, Liang BC. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997;8:1189–1198. [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36:976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. Suppression of mitochondrial complex I influences cell metastatic properties. PLoS One. 2013;8:e61677. doi: 10.1371/journal.pone.0061677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iommarini L, Kurelac I, Capristo M, Calvaruso MA, Giorgio V, Bergamini C, Ghelli A, Nanni P, De Giovanni C, Carelli V, Fato R, Lollini PL, Rugolo M, Gasparre G, Porcelli AM. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet. 2014;23:1453–1466. doi: 10.1093/hmg/ddt533. [DOI] [PubMed] [Google Scholar]
- Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, Marzano F, Manzari C, Betts CM, Carelli V, Ceccarelli C, Attimonelli M, Romeo G, Fato R, Rugolo M, Tullo A, Gasparre G, Porcelli AM. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013;1:11. doi: 10.1186/2049-3002-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- Liu VW, Shi HH, Cheung AN, Chiu PM, Leung TW, Nagley P, Wong LC, Ngan HY. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61:5998–6001. [PubMed] [Google Scholar]
- Maximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simoes M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. Am J Pathol. 2002;160:1857–1865. doi: 10.1016/S0002-9440(10)61132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens KM, Kulawiec M, Desouki MM, Vanniarajan A, Singh KK. Impaired OXPHOS complex III in breast cancer. PLoS One. 2011;6:e23846. doi: 10.1371/journal.pone.0023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008;68:700–706. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Forced cytochrome B gene mutation expression induces mitochondrial proliferation and prevents apoptosis in human uroepithelial SV-HUC-1 cells. Int J Cancer. 2009;125:2829–2835. doi: 10.1002/ijc.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadenbach B, Ramzan R, Vogt S. High efficiency versus maximal performance–the cause of oxidative stress in eukaryotes: a hypothesis. Mitochondrion. 2013;13:1–6. doi: 10.1016/j.mito.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Permuth-Wey J, Chen YA, Tsai YY, Chen Z, Qu X, Lancaster JM, Stockwell H, Dagne G, Iversen E, Risch H, Barnholtz-Sloan J, Cunningham JM, Vierkant RA, Fridley BL, Sutphen R, McLaughlin J, Narod SA, Goode EL, Schildkraut JM, Fenstermacher D, Phelan CM, Sellers TA. Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol Biomarkers Prev. 2011;20:1131–1145. doi: 10.1158/1055-9965.EPI-10-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZX, Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010;17:408–420. doi: 10.1038/cdd.2009.132. [DOI] [PubMed] [Google Scholar]
- Telang S, Nelson KK, Siow DL, Yalcin A, Thornburg JM, Imbert-Fernandez Y, Klarer AC, Farghaly H, Clem BF, Eaton JW, Chesney J. Cytochrome c oxidase is activated by the oncoprotein Ras and is required for A549 lung adenocarcinoma growth. Mol Cancer. 2012;11:60. doi: 10.1186/1476-4598-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH, Kern SE. Detection of mitochondrial DNA mutations in pancreatic cancer offers a "mass"-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001;61:1299–1304. [PubMed] [Google Scholar]
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- Chen WW, Birsoy K, Mihaylova MM, Snitkin H, Stasinski I, Yucel B, Bayraktar EC, Carette JE, Clish CB, Brummelkamp TR, Sabatini DD, Sabatini DM. Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep. 2014;7:27–34. doi: 10.1016/j.celrep.2014.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, Calabrese F, Laudiero G, Esposito F, Landriscina M, Defilippi P, Bernardi P, Rasola A. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013;17:988–999. doi: 10.1016/j.cmet.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchini V, Madhu B, Russell R, Pértega-Gomes N, Warren A, Gaude E, Borlido J, Stark R, Ireland-Zecchini H, Rao R, Scott H, Boren J, Massie C, Asim M, Brindle K, Griffiths J, Frezza C, Neal DE, Mills IG. Nuclear ARRB1 induces pseudohypoxia and cellular metabolism reprogramming in prostate cancer. EMBO J. 2014;33(12):1365–1382. doi: 10.15252/embj.201386874. [DOI] [PMC free article] [PubMed] [Google Scholar]