

Abstract
Naturally autoantibodies are part of the normal human immunoglobulin repertoire. These antibodies react to self antigens, are usually polyreactive with relatively low affinity and typically of the IgM isotype. Mouse natural IgMs that stimulated remyelination in CNS demyelinating disease all shared the characteristics of binding to the surface of live oligodendrocytes and myelinated tracts in living slices of CNS tissue. A screen for human IgMs with similar character resulted in two human natural antibodies that, when injected peripherally into animal models of demyelination induced remyelination. A recombinant human IgM was constructed (rHIgM22) that also promoted remyelination in vivo. Very small doses of this IgM are required for the promotion of remyelination—the EC50 is 460 ng per 20 gram mouse. It is clear that after peripheral delivery rHIgM22 enters the CNS and accumulates in CNS lesions. rHIgM22 was tracked in living mice using ferritin-labeled anti-human mu chain antibodies visualized by MRI and traditional immunocytochemistry. Although the exact antigen recognized by rHIgM22 is not clear, all mouse IgMs that promote remyelination bind to myelin membrane lipids, suggesting the antigen for rHIgM22 is similar. We propose that the IgMs bind to CNS cells and reorganize the membrane initiating a signal that results in oligodendrocyte proliferation and/or protection the end result being more myelin. Recombinant natural human antibodies are potentially important therapeutic molecules that may modulate a wide spectrum of human disease.
Introduction
Naturally occurring autoantibodies are part of our human immunoglobulin repertoire (Coutinho et al., 1995). Natural autoreactive (NA) monoclonal IgM antibodies can promote central nervous system (CNS) protection and repair. These repair-promoting IgMs have characteristics of classic NA antibodies. For example, they are generally of the IgM isotype; encoded by germline genes with few somatic mutations; and polyreactive with low affinity with a range of structurally unrelated, self and non-self antigens, specifically cytoskeleton, nuclear proteins and DNA (Avrameas et al., 2007). They proposed that the molecules play a natural physiological function either to stimulate cell processes or to remove cellular debris.
Naturally occurring autoantibodies react to self antigens, whereas conventional antibodies react to exogenous antigens, and compared to conventional antibodies, natural autoantibodies are of relatively low affinity. They are derived from our germline immunoglobulin genes but can also contain somatic mutations. They are frequently poly reactive. They are more frequently IgMs rather than IgGs and are usually physiologic unlike conventional antibodies, which are blocking or pathologic.
Role of Immunoglobulins in the Promotion of CNS Remyelination
Theiler’s murine encephalomyelitis virus (TMEV)-mediated disease results in spinal cord lesion pathology similar to that observed in human MS with progressive neurologic deficits (Rodriguez et al., 1987b). Spinal cord demyelination begins at 21 days post infection and plateaus at 90 days post infection. From this point on, the disease involves primarily neuronal injury and the progressive dropout of large caliber axons accounting for progressive clinical deficits (McGavern et al., 2000). This provides an ideal model in which to screen drugs designed to promote remyelination as well as protect axons. Our discovery of natural antibodies for CNS reparative was serendipity. To test the hypothesis of virus-induced autoimmunity and molecular mimicry mice with TMEV induced demyelination were also immunized with myelin. However, when mice were immunized with CNS antigens months after TMEV infection, rather than more extensive demyelination significant spinal cord remyelination was observed (Rodriguez et al., 1987a). Classical passive transfer studies were performed in which antisera or purified immunoglobulins from uninfected mice immunized with myelin antigens were transferred into mice with extensive TMEV mediated spinal cord demyelination (Rodriguez and Lennon, 1990). Mice that received hyper-immune sera or immunoglobulin directed against myelin demonstrated extensive remyelination after 5 weeks in contrast to mice that received control antisera.
To determine whether monoclonal antibodies could promote remyelination we generated a panel of monoclonal clones against spinal cord antigens. Supernatants from groups of 10 spinal cord homogenate binding clones were tested at a time in TMEV infected mice for the ability to promote remyelination. The first remyelination-promoting mAb identified was spinal cord homogenate 94.03 (SCH 94.03) (Miller et al., 1994). This IgM, when injected peritoneally, induced almost complete remyelination in 30 percent of spinal cord lesions in TMEV infected mice. This is in contrast to control monoclonal clones where less than 5 percent of spinal cord lesions were remyelinated. In characterizing SCH94.03 we found it bound to the surface of living oligodendrocytes. Several well characterized mouse IgMs used to characterize the oligodendrocyte lineage and bound to glycolipids on to the surface of live oligodendrocytes, (A2B5, O1, O4, HNK-1) also promoted remyelination (Asakura et al., 1998). These IgMs had had relatively conserved germ line sequences. We hypothesized that IgM-mediated remyelination required recognition of oligodendrocyte plasma membrane lipids.
Identification of Human Antibodies that Promote Remyelination
Based on these observations, we asked whether natural antibodies were present in the human population that bound to oligodendrocytes and could promote remyelination. We employed a novel strategy to identify potential therapeutic human molecules by screening for auto reactive human mAbs from the sera of individual’s that carry the mAbs in high concentration (Warrington et al., 2000). Searching through the over 140,000 samples archived over 50 years in the Mayo Clinic serum bank we sought samples from patients with monoclonal gammopathies—diseases that result in an immortalized b cell clone and a high level of serum monoclonal protein. Specifically samples from patients with multiple myeloma, Waldenstrom’s syndrome and monoclonal gammopathy of unknown significance were identified. We screened for Ab binding to myelin in slices of live mouse central nervous system and to the surface of oligodendrocytes in culture without regard for antigen. Six of 52 serum-derived human IgMs (sHIgM) and zero of 50 serum-derived human IgGs (sHIgG) bound in these assays. The histories of the patients synthesizing positive mAbs were examined to ensure that there were no neurologic or antibody-associated pathologies. Positive mAbs were then tested for the ability to modulate in vivo demyelinating disease. Two human IgMs (sHIgM22 and sHIgM46) promoted significant remyelination in TMEV infected mice. We also tested IVIg and polyclonal human IgM for the ability to promote remyelination in the TMEV model—both were effective, but polyclonal IgM and the two human monoclonal IgMs were superior. IVIg may be effective in the TMEV model by modulating the immune response while the IgMs may act directly upon the cells of the nervous system (Warrington et al., 2000). The recognition of the surface of appropriate tissues or cells appears to be an important defining characteristic of therapeutic IgMs. The IgMs demonstrate specificity only when cells and tissue are maintained under live physiological conditions(Asakura et al., 1998; Miller et al., 1996; Miller and Rodriguez, 1995).
Using oligodendrocytes isolated from human tissue from patients undergoing temporal lobectomy for epilepsy, we were able to demonstrate that both human IgMs bound to the surface of human oligodendrocytes in vitro providing evidence that these IgMs may be effective in promoting remyelination human patients. This method of identifying mAbs with biologic efficacy is very different from the traditional methodology used by the pharmaceutical industry (Rodriguez et al., 2009). Each patient has carried high concentrations of monoclonal Ig in their blood for many years without neurologic disease. Each IgM isolated has, in a certain sense, already been tested for toxicology by the individual that carried the antibody.
Development of a Recombinant Expression Vector for Human IgMs
To definitively prove that the IgM and no other serum factors promoted remyelination we developed a recombinant version of sHIgM22. We developed an expression vector incorporating a methotrexate amplification system capable of receiving any human heavy chain sequence and light chain sequence to generate large quantities of pure, recombinant mAb. rHIgM22 promoted myelin repair in the Theiler’s virus infection-induced model of MS equal to or better than the serum-derived form. rHIgM22 binds to the surface of live oligodendrocytes and myelin and promotes CNS remyelination in virus (Mitsunaga et al., 2002) and toxin-induced models of MS (Bieber et al., 2002). Spinal cord remyelination is induced after a single dose low dose (25 μg/ml) of rHIgM22 (Warrington et al., 2007). It is remarkable that one peripheral intraperitoneal injection of a short lived molecule (15 hr half life in the mouse circulation) promotes maximal tissue repair within 5 weeks in a model of MS that normally presents with little spontaneous repair. The properties of rHIgM22 are similar to that of a targetable growth factor
To determine whether non-invasive imaging could be used to measure IgM mediated repair of the CNS we measured the total volume of spinal lesions by MRI before treatment with rHIgM22 and 5 weeks after treatment. The entire mouse spinal cord was scanned by MRI, lesions were represented by voxels, and the total resulting pixels were calculated in three dimensions in both T1 and T2-weighted MRI scans (Pirko et al., 2004). Comparing initial spinal cord lesion load with that five weeks later, we found that mice receiving control saline showed slightly increasing lesions volumes. In contrast, mice treated with rHIgM22 showed a remarkable decrease in lesion volume load in the spinal cord. Again, these results were obtained after a single dose of the IgM, and nearly every mouse responded with decreased lesion size. We correlated spinal cord areas of reduction of MRI signal with histology in 1 micron thick araldite-embedded sections of the spinal cord and discovered there was a precise correlation between areas of reduction of MRI signal and areas of remyelination.
We tested whether rHIgM22 promoted spinal cord remyelination by altering an aspect of the immune response. We measured no change in immune function in TMEV infected mice or in mice with EAE induced by adoptive transfer of myelin basic protein specific T cells (Ciric et al., 2004). Another possibility was that rHIgM22 reduced the level of virus in TMEV infected mice, allowing normal repair to proceed. Immunosuppressive treatment of TMEV infected mice leads to remyelination, but also an increase in virus replication in the CNS (Rodriguez and Lindsley, 1992). We found that a 500 ug dose of rHIgM22 given to chronically TMEV infected mice did not reduce CNS virus load as measured northern blot or immunocytochemistry (Ciric et al., 2004). Therefore rHIgM22 promotes remyelination without affecting the virus replication.
The development of rHIgM22 is at the point of GMP commercial production, purification and animal toxicology. Gram quantities of GMP-grade rHIgM22 have been purified for formal toxicology and a potential Phase I clinical trial for safety in 2010. The studies in this disease model suggest that very little rHIgM22 will be required to modulate human disease. A mouse weighing 20 grams responding to a 500-ng dose corresponds to a 2 mg dose in humans weighing 50 or 70 kg. Over the last 5 years we have developed the infrastructure at Mayo Clinic and commercial vendors necessary to rapidly translate other human antibodies from the point of discovery, through cloning, manufacturing, toxicology to clinical testing.
IgMs Can Enter the CNS After Peripheral Administration
It has been generally accepted that IgMs with a molecular weight of close to 1 million are too large to cross the blood brain barrier (BBB) from the circulation to enter the CNS (Kozlowski et al., 1992). However, there is accumulating evidence some IgMs can cross the BBB. A study by our laboratory (Hunter et al., 1997) examined the tissue localization of a mouse monoclonal IgM shown to promote remyelination, SCH94.03. 35S-methionine labeled SCH94.03 was administered intraperitoneally to TMEV-infected and uninfected SJL mice. In infected mice radiolabel accumulated in the brain, spinal cord, heart, lung and muscle and plateaued by 24 hr. In uninfected mice, radiolabel accumulated at similar levels, but required 7 days to plateau. It was calculated that 0.4% of the administered 35S counts accumulated in the brain and spinal cord of both uninfected and TMEV-infected mice. We performed a MRI based study using a 7-Tesla magnet to localize rHIgM22 in Theiler’s virus infected mice (Pirko et al., 2004). rHIgM22 entry into the CNS and accumulation in lesions was demonstrated prominently by MRI. In mice that were not infected with virus or did not have demyelination, rHIgM22 did not accumulate in the central nervous system (Pirko et al., 2004). A control human IgM that does not bind to myelin or oligodendrocytes did not accumulate in CNS lesions.
Other independent investigators have also demonstrated that certain human IgMs can cross the blood-brain barrier (Banks et al., 2007). The ability of two human anti-beta amyloid (Aβ) IgMs to cross the BBB in normal and SAMP8 mice (a model of Alzheimer’s disease AD) were compared. 125I labeled human IgMs were injected intravenously. One anti-Aβ IgM (L11.3) accumulated in the brain of normal and diseased mice two hours later. The L11.3 anti-Aβ IgM reversed cognitive impairment in the AD mice whether the IgM was delivered by intracerebral injection or intravenously. It is well established that anti-Aβ Abs delivered to the brain of mice that over express Aβ can reverse histologic and cognitive impairment and underlie the Aβ immunization strategies in human trials.
These data support the concept that some IgMs can cross the blood-brain-barrier with low efficiency enter CNS and accumulate within injured regions of the CNS. It should be stressed that very little drug is required in the CNS to produce a therapeutic effect. Only 0.02% of a peripheral dose of morphine enters the brain, yet this is sufficient for analgesia. For most CNS therapeutics on the market less than 0.2% of a peripheral dose enters the CNS.
Mechanism of Action: Clustering of Specific Membrane Domains
We propose the mechanism of action of the IgMs is through lipid microdomain signaling. The large pentameric structure of the IgM can easily cross-link antigens on the cell surface bringing molecules that normally do not interact to come together into signaling complex. In fact, the pentameric structure of rHIgM22 is necessary for in vivo remyelination. Even though remyelination promoting IgMs bind to the surface of oligodendrocytes, not all human IgMs that bind to oligodendrocytes promoted remyelination. Therefore, IgM-mediated repair requires binding to specific antigens on the oligodendrocyte membrane. All of the IgMs that promote remyelination induce a transient calcium influx in glial cells (Paz Soldan et al., 2003). Transient elevation of Ca++ in cells is associated with cell survival, whereas persistent elevation of Ca++ with cell death. Disruption of the lipid microdomain by the detergent B-methyl cyclodextrin prevents Ca++ signaling (Howe et al., 2004).
The exact antigen bound by rHIgM22 on myelin is not required to move forward to FDA approval, if reasonable data exists supporting an in vivo mechanism of action. We have recently discovered that rHIgM22 does not bind to live CNS tissue slices from glycolipid knock out mice. Myelinated tracts in mice lacking the enzymes cerebroside sulfotransferase (CST) or cerebroside galactosyltransferase (CGT) and therefore lacking sulfatide or galactosylcerebroside (GalC) are not recognized by rHIgM22. However, rHIgM22 does not bind to other sulfatide-expressing tissues such peripheral nervous system myelin and Schwann cells. These data support the hypothesis that rHIgM22 binding depends upon sulfated antigens present exclusively in the CNS
There are currently no effective treatments to prevent or reverse deficits in MS. Available drugs target immune aspects of the disease and have no effect on the chronic progressive form of the disease. rHIgM22 will be the first therapeutic designed to enhance repair in an MS lesion by targeting cells of the brain and spinal cord. If successful this will increase interest in natural antibodies as safe low dose therapeutic reagents.
Fig. 1. Theiler’s murine encephalomyelitis virus induced disease in the SJL mouse: a model of human multiple sclerosis.

The TMEV model offers a platform in which to develop strategies to promote remyelination and prevent axon loss. TMEV-mediated disease in susceptible strains of mice results in spinal cord lesion pathology very similar to that observed in human MS. The disease is characterized by an acute encephalitis phase that resolves. The virus takes up long term residence in glial cells of the spinal cord where chronic inflammation leads to demyelination and axon loss with progressive neurologic deficits. Spinal cord demyelination begins at 21 days post infection and plateaus at 90 days post infection. From this point on, the disease involves primarily progressive neuronal injury where the dropout of large caliber axons accounts for increasing clinical deficits. The more widely used model of human MS, experimental autoimmune encephalomyelitis (EAE), is primarily autoimmune mediated and therefore a large number of immune modulating reagents improve clinical disease. In the TMEV model immunosuppression increases virus load as immune surveillance of the CNS is reduced. TMEV mediated disease would likely improve with effective anti-viral therapy. Such options are limited in humans. Remyelination promoting IgMs are the only identified reagents that promote CNS repair without modulating the immune system or altering viral load in this model.
Fig. 2. Alternative approach to the identification and production of therapeutic human mAbs.

There is a critical need for toxicity testing early in drug development. Safety issues are the most common cause of drug candidate attrition from the pipeline and the marketplace. This alternative approach to identify antibody based drugs starts with a screen of the hundreds of thousands of samples collected over 45 years in the Mayo Clinic sera bank for patients with a monoclonal spike in the sera. These are typically patients with lymphoproliferative disorders where a B cell expansion results in high levels of monoclonal protein in their circulation. The strength of this approach to human mAb identification is that candidate antibodies are, in a sense, tested for toxicity by the patient who carries it. Samples from patients with antibody based disease are excluded. However, mabs with characteristics of interest are identified without regard to antigen or mechanism of action. Antigen discovery or a reasonable understanding of the mechanism of action is necessary to translate reagent such a mAb to a marketable drug.
Fig. 3. rHIgM22, a recombinant human IgM, promotes remyelination of spinal cord demyelination.
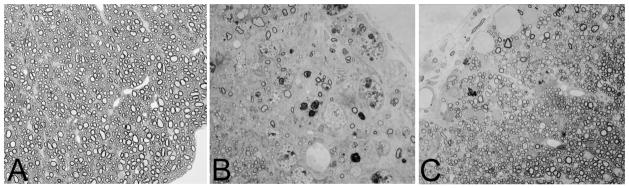
Light photomicrographs of normal spinal cord white matter showing densely packed myelinated axons (A). A typical demyelinated lesion with minimal remyelination as observed in the spinal cord of a susceptible mouse infected with Theiler’s murine encephalomyelitis virus for 5 months. This mouse was treated five weeks earlier with a control human IgM that does not bind to the surface of oligodendrocytes. The area contains only a few remyelinated axons. Infiltrating macrophages in the demyelinating lesion are characterized by the presence of darkly stained debris-laden vesicles (B). In contrast, much remyelination is observed in areas of chronic spinal cord demyelination of mice treated with rHIgM22, a recombinant human IgM that binds to myelin and the surface of oligodendrocytes (C). An area of nearly complete remyelination is present in the ventral lateral spinal cord after a single 50 μg dose intraperitoneally of rHIgM22 five weeks earlier. Remyelination is characterized by densely packed thin myelin sheaths in relation to axon diameter. Panels A, B and C are the same magnification.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 NS 24180, R01 NS 32129, P01 NS 38468, R01 CA104996, R01 CA096859), the National Multiple Sclerosis Society (RG 317 2-B-8, CA 1011 A8-3), the Multiple Sclerosis Society of Canada, the Applebaum Foundation, the Hilton Foundation, the Peterson Foundation and Acorda Therapeutics, Inc. (Hawthorne, NY). Patents for IgM mediated repair of the central nervous system are issued and are owned by Mayo Clinic. Therefore, the authors have a potential financial conflict of interest.
References
- Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMkappa antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18:7700–8. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrameas S, Ternynck T, Tsonis IA, Lymberi P. Naturally occurring B-cell autoreactivity: a critical overview. J Autoimmun. 2007;29:213–8. doi: 10.1016/j.jaut.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M. Anti-amyloid beta protein antibody passage across the blood-brain barrier in the SAMP8 mouse model of Alzheimer’s disease: an age-related selective uptake with reversal of learning impairment. Exp Neurol. 2007;206:248–56. doi: 10.1016/j.expneurol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieber AJ, Warrington A, Asakura K, Ciric B, Kaveri SV, Pease LR, et al. Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia. 2002;37:241–9. doi: 10.1002/glia.10033. [DOI] [PubMed] [Google Scholar]
- Ciric B, Van Keulen V, Paz Soldan M, Rodriguez M, Pease LR. Antibody-mediated remyelination operates through mechanism independent of immunomodulation. J Neuroimmunol. 2004;146:153–61. doi: 10.1016/j.jneuroim.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–8. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- Howe CL, Bieber AJ, Warrington AE, Pease LR, Rodriguez M. Antiapoptotic signaling by a remyelination-promoting human antimyelin antibody. Neurobiol Dis. 2004;15:120–31. doi: 10.1016/j.nbd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Hunter SF, Miller DJ, Rodriguez M. Monoclonal remyelination-promoting natural autoantibody SCH 94. 03: pharmacokinetics and in vivo targets within demyelinated spinal cord in a mouse model of multiple sclerosis. J Neurol Sci. 1997;150:103–13. doi: 10.1016/s0022-510x(97)00080-4. [DOI] [PubMed] [Google Scholar]
- Kozlowski GP, Sterzl I, Nilaver G. Localization patterns for immunoglobulins and albumins in the brain suggest diverse mechanisms for their transport across the blood-brain barrier (BBB) Prog Brain Res. 1992;91:149–54. doi: 10.1016/s0079-6123(08)62329-8. [DOI] [PubMed] [Google Scholar]
- McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123(Pt 3):519–31. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DJ, Njenga MK, Parisi JE, Rodriguez M. Multi-organ reactivity of a monoclonal natural autoantibody that promotes remyelination in a mouse model of multiple sclerosis. J Histochem Cytochem. 1996;44:1005–11. doi: 10.1177/44.9.8773566. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Sanborn KS, Katzmann JA, Rodriguez M. Monoclonal autoantibodies promote central nervous system repair in an animal model of multiple sclerosis. J Neurosci. 1994;14:6230–8. doi: 10.1523/JNEUROSCI.14-10-06230.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunaga Y, Ciric B, Van Keulen V, Warrington AE, Paz Soldan M, Bieber AJ, et al. Direct evidence that a human antibody derived from patient serum can promote myelin repair in a mouse model of chronic-progressive demyelinating disease. Faseb J. 2002;16:1325–7. doi: 10.1096/fj.01-0994fje. [DOI] [PubMed] [Google Scholar]
- Paz Soldan MM, Warrington AE, Bieber AJ, Ciric B, Van Keulen V, Pease LR, et al. Remyelination-promoting antibodies activate distinct Ca2+ influx pathways in astrocytes and oligodendrocytes: relationship to the mechanism of myelin repair. Mol Cell Neurosci. 2003;22:14–24. doi: 10.1016/s1044-7431(02)00018-0. [DOI] [PubMed] [Google Scholar]
- Pirko I, Ciric B, Gamez J, Bieber AJ, Warrington AE, Johnson AJ, et al. A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. Faseb J. 2004;18:1577–9. doi: 10.1096/fj.04-2026fje. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Lennon VA. Immunoglobulins promote remyelination in the central nervous system. Ann Neurol. 1990;27:12–7. doi: 10.1002/ana.410270104. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Lennon VA, Benveniste EN, Merrill JE. Remyelination by oligodendrocytes stimulated by antiserum to spinal cord. J Neuropathol Exp Neurol. 1987a;46:84–95. doi: 10.1097/00005072-198701000-00008. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Lindsley MD. Immunosuppression promotes CNS remyelination in chronic virus-induced demyelinating disease. Neurology. 1992;42:348–57. doi: 10.1212/wnl.42.2.348. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Oleszak E, Leibowitz J. Theiler’s murine encephalomyelitis: a model of demyelination and persistence of virus. Crit Rev Immunol. 1987b;7:325–65. [PubMed] [Google Scholar]
- Rodriguez M, Warrington AE, Pease LR. Invited Article: Human natural autoantibodies in the treatment of neurologic disease. Neurology. 2009;72:1269–76. doi: 10.1212/01.wnl.0000345662.05861.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U S A. 2000;97:6820–5. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington AE, Bieber AJ, Ciric B, Pease LR, Van Keulen V, Rodriguez M. A recombinant human IgM promotes myelin repair after a single, very low dose. J Neurosci Res. 2007;85:967–76. doi: 10.1002/jnr.21217. [DOI] [PubMed] [Google Scholar]