

Abstract
Purpose
The cilium in photoreceptors appears ultrastructurally very similar to the nasal ciliated epithelium. The purpose of this study was to evaluate the nasal ciliary beat frequency and beat pattern in patients with retinitis pigmentosa (RP) and Usher syndrome type II and compare it with that of healthy control subjects.
Methods
A prospective, comparative control study. Fresh samples of nasal mucosa were obtained from 13 patients with typical forms of RP, and from 4 patients with Usher syndrome type II. The nasal ciliary beat frequency (CBF) and beat pattern were determined using high-resolution digital high-speed video imaging (DHSV). The control group included 32 fresh nasal mucosa samples from 32 healthy volunteers without any other confounding diseases.
Results
The nasal CBF was lower in patients with Usher syndrome than in control subjects (Mann-Whitney U test, P = 0.01). The nasal CBF was 9.28 ± 0.4 (mean ± SD) Hz in patients with Usher syndrome and 10.82 ± 1.39 Hz in patients of the control group. No significant difference was observed in the nasal CBF between the RP (10.59 ± 1.54 Hz) and control group (Mann-Whitney U test, P = 0.64). Normal ciliary beat pattern was observed in all the patients and healthy volunteers.
Conclusions
The nasal CBF is diminished in patients with Usher syndrome type II, whereas it remains normal in simplex RP patients. These results add evidence to the fact that Usher syndrome could be a primary ciliary disorder.
The ciliary beat frequency of the nasal ciliated epithelium of patients with retinitis pigmentosa (RP) and Usher syndrome type II was compared with that of healthy control subjects. The nasal ciliary beat frequency is reduced in patients with Usher syndrome type II, whereas it remains normal in simplex RP.
Introduction
Retinitis pigmentosa (RP) is the most common cause of retinal degeneration, with a prevalence of approximately 1 in 4000.1 Visual impairment in RP is primarily a result of loss of photoreceptors, which leads to subsequent damage of the retinal pigment epithelium (RPE) and other layers of the retina.2 The group of diseases included under the term RP is genetically heterogeneous, but phenotypically similar, with more than 50 different genes having been identified (http:// www.sph.uth.tmc.edu/Retnet/). RP can be transmitted as an autosomal dominant, autosomal recessive, or X-linked trait. Most commonly, RP occurs as isolated cases (simplex) in which no other family member is affected.3,4 Usher syndrome is a form of autosomal recessive sensorineural hearing impairment associated with RP, and is considered the most common cause of combined blindness and deafness.5 It is classified into three main categories according to the age of onset and the degree of hearing loss and vestibular involvement. Type I is characterized by early childhood onset of RP associated with profound congenital deafness. Vestibular function is also absent, causing delayed motor milestones and clumsiness. Type II is characterized by later onset of RP, often in the late second decade of life, with moderate to severe congenital deafness but normal vestibular function. In Type III, the onset of vision loss is variable, whereas often both vision and hearing start normal or near normal and progressively deteriorate over decades.6
The term “retinal ciliopathies” started to be used with the observation that patients with X-linked RP and Usher syndrome showed that motile cilia were morphologically abnormal (nasal ciliated epithelium and sperm).7–11 Photoreceptors, inner ear receptors (choclear and vestibular), sperm flagellum, and nasal ciliated epithelium share a characteristic organelle: the cilium or flagellum. The cilia are widely distributed in the human body and they recover diverse functions, but they share a similar ultrastructure and molecular composition.
The connecting cilium of photoreceptors is the stalk that separates the outer segment from the inner segment. This connecting cilium is essential for the function of the photoreceptors, as the cilium possesses a system of intraflagellar transport, responsible for moving cargo toward the axonemal cilia tip and away from it. A defective connecting cilium could impede transport of outer segment proteins from their site of synthesis in the inner segment, leading to outer segment degeneration, as occurs in RP.11–15 There are numerous products of genes implicated in retinal degenerations that have been found to be specifically localized in the retinal cilia and basal bodies, highlighting the importance of this organelle in retinal disease. The cilium in photoreceptors appears ultrastructurally very similar to the nasal ciliated epithelium and the flagellum of spermatozoa. This similarity led to the hypothesis that an abnormality in ciliary function may be linked to the sperm and nasal cilia abnormalities, as well as to the retinal degeneration.
The purpose of our study was to evaluate the ciliary beat frequency (CBF) and beat pattern using high-resolution digital high-speed video imaging (DHSV) of the nasal ciliated epithelium in patients with RP and Usher syndrome type II and compare it with that of healthy control subjects to elucidate if there is also a functional disorder in the nasal ciliated epithelium, apart from the already described ultrastructural abnormalities.
Methods
This prospective, comparative control study investigates the nasal CBF and beat pattern in patients with RP and Usher syndrome type II. Nasal ciliated epithelium samples from healthy control subjects without any other ocular, ear, nose, throat, or systemic diseases were collected as controls. The study protocol complied with the Declaration of Helsinki and was reviewed and approved by the Ethics Committee of a tertiary referral hospital. Informed consent was obtained from all subjects.
Fresh samples of nasal mucosa were obtained from 13 patients with typical forms of RP, and from 4 patients with Usher syndrome type II with no other confounding ocular or systemic disease. Patients were at least 18 years of age and displayed the typical forms of RP in the eye, which are characterized by an elevated final dark-adaptation threshold, retinal arteriolar narrowing, and a reduced and delayed electroretinogram. Patients with syndromic forms of RP, such as Usher syndrome type II, also had moderate to severe congenital deafness but normal vestibular function. The ages of patients are shown in Table 1. All patients underwent a full ophthalmic examination that included best-corrected visual acuity, automated visual field, and optical coherence tomography. Nasal fossae were examined by endoscopy and be observed as normal in all subjects
Table 1.
Detailed Data of Patients
|
Age (y) |
Sex |
Nasal Ciliary Beat Frequency (Hz) |
|
| RP patients | |||
| 1 | 38 | F | 11 |
| 2 | 35 | M | 8.5 |
| 3 | 44 | F | 11 |
| 4 | 22 | F | 9 |
| 5 | 51 | F | 11 |
| 6 | 61 | F | 12.5 |
| 7 | 55 | F | 14 |
| 8 | 36 | M | 9 |
| 9 | 62 | M | 10 |
| 10 | 62 | M | 11.5 |
| 11 | 56 | M | 9.1 |
| 12 | 61 | M | 10.6 |
| 13 | 47 | M | 10 |
| Usher patients | |||
| 1 | 44 | M | 9.9 |
| 2 | 33 | M | 9.25 |
| 3 | 38 | F | 9 |
| 4 | 56 | M | 9 |
| Control patients | |||
| 1 | 20 | F | 11.58 |
| 2 | 26 | M | 10 |
| 3 | 61 | M | 10 |
| 4 | 33 | F | 12 |
| 5 | 42 | F | 11.35 |
| 6 | 21 | M | 10.2 |
| 7 | 20 | F | 13 |
| 8 | 53 | F | 8.5 |
| 9 | 54 | M | 9 |
| 10 | 24 | F | 10.6 |
| 11 | 32 | M | 12.3 |
| 12 | 42 | F | 11 |
| 13 | 59 | F | 12 |
| 14 | 22 | M | 12.5 |
| 15 | 39 | M | 10.8 |
| 16 | 27 | F | 9.1 |
| 17 | 39 | F | 14 |
| 18 | 23 | F | 10.7 |
| 19 | 44 | M | 9.5 |
| 20 | 45 | F | 9.5 |
| 21 | 46 | F | 11.8 |
| 22 | 29 | M | 11 |
| 23 | 28 | M | 9.5 |
| 24 | 38 | F | 9 |
| 25 | 25 | M | 10.8 |
| 26 | 52 | F | 14 |
| 27 | 38 | F | 9.75 |
| 28 | 27 | F | 9.75 |
| 29 | 20 | M | 11.75 |
| 30 | 60 | M | 10.5 |
| 31 | 22 | F | 10.5 |
| 32 | 22 | F | 10.5 |
RP, retinitis pigmentosa; M, male; F, female.
Samples of ciliary airway epithelial cells were obtained in both groups from the middle nasal concha using curettage16 without local anesthesia, in a period during which acute infection was absent. The tissue sample was immersed in 1 mL Dulbecco's modified Eagle's medium (DMEM, Cambrex Bio Science, Verviers, Belgium) supplemented with 10% fetal calf serum, 2 mM glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL), and turned over until partial dissolution occurred. Then, 150 mL of each nasal epithelium biopsy were plated on tissue culture plates (12-well Corning Costar 3513, Corning, NY), which had been coated with 0.5% gelatin to promote adherence of the ciliated cells. The CBF of healthy individuals, and RP and Usher patients was measured at room temperature (23–27°C) within 30 minutes after biopsy. Ciliary beat pattern was also studied in the same conditions.
To corroborate the results, each nasal biopsy was plated on tissue culture plates coated with human collagen (type IV, Vitrogen-100; Cohesion Technologies, Palo Alto, CA) and incubated in DMEM at 37°C in a humidified atmosphere of 5% CO2 in air for 24 hours, and the CBF and pattern was measured again.
Samples were imaged with a DHSV imaging technique using a Nikon Eclipse TS100 microscope (Nikon, Tokyo, Japan) with a ×40 Nikon phase-contrast objective, providing a final optical gain of ×400. The method used for CBF measurement was based on previously published methods.17 A normal ciliary beat pattern was characterized by a planar motion with a forward power stroke and a backward recovery stroke in a coordinated mode.
Demographic characteristics of the patients were summarized with descriptive statistics by SPSS for Windows (SPSS Inc., Chicago, IL). The Mann-Whitney U test for independent samples was used to compare the nasal CBF of the groups. P less than 0.05 was considered significant.
Results
A total of 49 fresh samples of nasal mucosa were collected from 13 RP patients, 4 Usher syndrome type II patients, and 32 control patients. All participants were Caucasian. Table 1 shows detailed information for every patient.
The nasal CBF was 10.59 ± 1.54 (mean ± SD) Hz in RP patients, 9.28 ± 0.4 Hz in Usher syndrome type II patients, and 10.82 ± 1.39 Hz in the control group (Table 2). The nasal CBF did not differ significantly between the RP group and the Usher group (Mann-Whitney U test, P = 0.11); the difference was also not significant between the RP group and the control group (Mann-Whitney U test, P = 0.64). The nasal CBF was significantly different between the Usher group and the control group (Mann-Whitney U test, P = 0.01), with those of the Usher patients proving to be significantly lower than those of controls (Fig. 1). Normal ciliary beat pattern was observed both in patients and in healthy volunteers.
Table 2.
Patient Baseline Characteristics and Nasal Ciliary Beat Frequency
|
Disease Group |
Age (y), Mean ± SD (range) |
Sex (M:F) |
Nasal Ciliary Beat Frequency (Hz), Mean ± SD |
| RP (n = 13) | 48.4 ± 12.7 (22–62) | (7:6) | 10.59 ± 1.54 |
| Usher (n = 4) | 42.75 ± 9.9 (33–56) | (3:1) | 9.28 ± 0.4 |
| Control (n = 32) | 10.82 ± 1.39 |
RP, retinitis pigmentosa; M, male; F, female.
Figure 1.
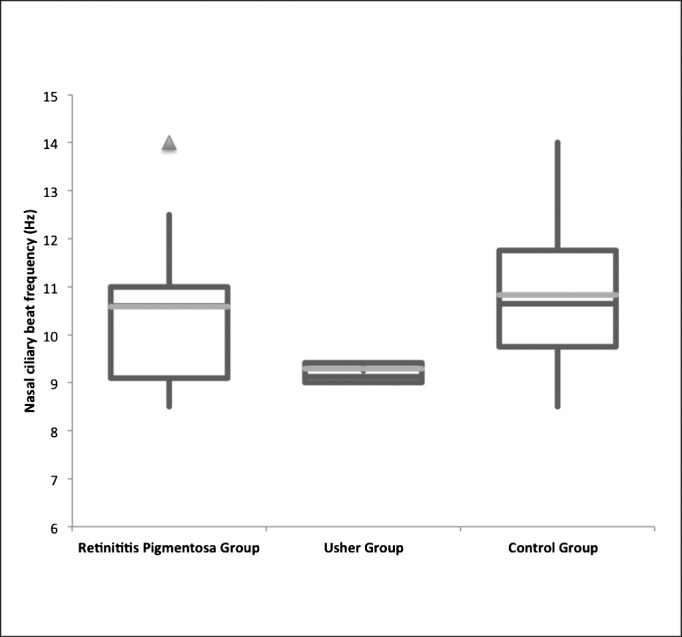
Nasal CBF (Hz) in 13 retinitis pigmentosa (RP) patients, 4 Usher syndrome type II patients, and 32 control patients. The nasal CBF was significantly different between the Usher patients and the control patients (Mann-Whitney U test, P = 0.01), with those of Usher patients being significantly lower than those of controls.
Discussion
In this study, we demonstrated that the nasal ciliated epithelium of Usher syndrome type II patients has a lower CBF than that of healthy control subjects. At the same time, RP patients with a simplex heredity pattern failed to demonstrate significant differences in the nasal CBF when compared with control subjects.
The DHSV method used for CBF measurement has high sensitivity for the detection of abnormalities in the CBF and pattern, with high specificity for differentiating between several ciliary beat abnormalities. This is a rapid technique that can reliably guide the diagnosis of ciliary dyskinesias.17,18
Defects in cilia cause a broad spectrum of human diseases known collectively as ciliopathies. Although all ciliopathies arise from defective cilia, the range of symptoms can vary significantly and only a small subset of possible ciliary disease symptoms may be present in any given syndrome. There is evidence that immotile nasal cilia can be associated with Usher syndrome type I,19 and that ultrastructural defects of both motile and photoreceptors connecting cilia, are present in patients with Usher syndrome.2,10 In the past 2 decades, five genes have been identified as responsible for Usher syndrome type I, three genes for Usher syndrome type II, and one for Usher syndrome type III. The proteins encoded by the Usher genes have very different functions and represent different classes of proteins; however, there is growing evidence that suggests that the unifying feature of these proteins is that they form an integrated protein network known as the “Usher interactome” that is present at the connecting cilium in the photoreceptors and in the hair cells of the organ of Corti, including the proteins encoded by the three genes associated with Usher type II (USH2A, GPR98, and WHRN). The USH2A and the GPR98 genes are thought to play a role in the maintenance of periciliary structure of the photoreceptors, and account for virtually all the Usher type II patients. The protein encoded by WHRN, whirlin, is a scaffolding protein of the connecting cilium and inner ear that has been only reported to be causative in four USH2 families worldwide.20–22 Such defected genes could also be responsible for the lower beat frequency of the cilia in the nasal ciliated epithelium; however, in the case of the four patients in this series suffering from Usher syndrome type II, ciliary activity, although slightly diminished, is sufficient for the mucociliary system to operate normally so they do not suffer from chronic respiratory infections. In the respiratory cilia this represents molecular alterations with no clinical consequences.
On the other hand, more than 50 genes are responsible for RP. The RP genes encode proteins with a wide range of functions, including phototransduction, the visual cycle pathway, phagocytosis of outer segments, retinal development, mRNA splicing and others including photoreceptor ciliary structure. Among these 50 genes, only RP1, responsible for autosomal dominant RP, RPGRIP1 and LCA5, responsible for Leber congenital amaurosis, and RP2 and RPGR, responsible for X-linked RP (XLRP) are known to be expressed in the photoreceptor-connecting cilium.23,24 The sperm flagellum is a cilium that acts as spermatozoa propulsor. Defects in sperm motility and structure (microtubule abnormalities) have been observed in patients with X-linked RP and Usher syndrome.11 Defects in nasal cilia structure have been documented by some investigators,8 although others have failed to demonstrate such changes.9 We failed to show differences in the nasal CBF between simplex RP patients and healthy control subjects. This observation correlates with previous studies in which no abnormality was observed in nasal cilia motility of patients with simplex RP.11,25 Other studies have demonstrated abnormalities in the motile cilia of X-linked RP,7,8,11 giving sense to the idea that cilia are complex structures that perform multiple functions simultaneously, and when one function is altered, others are left intact. All this can be explained by the fact that ciliary axoneme is composed of more than 200 proteins, and defects may involve distinct axonemal proteins located on different chromosomes, giving rise to different clinical manifestations.
The main limitation of our study is the low number of patients in the Usher group, but we consider it is demonstrative of at least a tendency. We can hypothesize that the ciliary abnormalities are more extensive in Usher syndrome than in the simple RP, which could explain the involvement of ciliary sensory receptors in the inner ear in Usher syndrome but not in the simple RP. Interestingly, a mutation in the USH2A gene, c.2276G > T (p.C579F) in the homozygous state was found to be associated with isolated RP.26 Later it was demonstrated that p.C759F in combination with other USH2A mutations cause Usher syndrome type II, indicating that the USH2A gene is a link between RP and Usher syndrome.27,28
In conclusion, the nasal CBF is diminishing in patients with Usher syndrome type II, whereas it remains normal in simplex RP patients. These results add evidence to the fact that Usher syndrome could be a primary ciliary disorder. With the method used in this study, we have been able to develop a simple assay for ciliary function of human nasal samples, to better understand how different mutations affect the cilia of the nasal ciliated epithelium in several ciliopathies.
Footnotes
Supported by FEDER (European Funds for Regional Development), CIBER CB06/06/0027 from the Carlos III Health Institute of the Ministry of Health, and research grants from the Regional Government of Valencia, Spain. This work has not received financial support from drug companies.
Disclosure: M. Armengot, None; D. Salom, None; M. Diaz-Llopis, None; J.M. Millan, None; J. Milara, None; M. Mata, None; J. Cortijo, None
References
- 1. Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28(3):113–125. [DOI] [PubMed] [Google Scholar]
- 2. Barrong SD, Chaitin MH, Fliesler SJ, Possin DE, Jacobson SG, Milam AH. Ultrastructure of connecting cilia in different forms of retinitis pigmentosa. Arch Ophthalmol. 1992;110(5):706–710. [DOI] [PubMed] [Google Scholar]
- 3. Pagon RA. Retinitis pigmentosa. Surv Ophthalmol. 1988;33(3):137–177. [DOI] [PubMed] [Google Scholar]
- 4. Boughman JA, Fishman GA. A genetic analysis of retinitis pigmentosa. Br J Ophthalmol. 1983;67(7):449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vernon M. Usher's syndrome—deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. J Chronic Dis. 1969;22(3):133–151. [DOI] [PubMed] [Google Scholar]
- 6. Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006; 15 Spec No 2:R262–R270. [DOI] [PubMed] [Google Scholar]
- 7. Arden GB, Fox B. Increased incidence of abnormal nasal cilia in patients with retinitis pigmentosa. Nature. 1979;279(5713):534–536. [DOI] [PubMed] [Google Scholar]
- 8. Fox B, Bull TB, Arden GB. Variations in the ultrastructure of human nasal cilia including abnormalities found in retinitis pigmentosa. J Clin Pathol. 1980;33(4):327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Finkelstein D, Reissig M, Kashima H, Massof R, Hillis A, Proctor D. Nasal cilia in retinitis pigmentosa. Birth Defects Orig Artic Ser. 1982;18(6):197–206. [PubMed] [Google Scholar]
- 10. Hunter DG, Fishman GA, Mehta RS, Kretzer FL. Abnormal sperm and photoreceptor axonemes in Usher's syndrome. Arch Ophthalmol. 1986;104(3):385–389. [DOI] [PubMed] [Google Scholar]
- 11. Hunter DG, Fishman GA, Kretzer FL. Abnormal axonemes in X-linked retinitis pigmentosa. Arch Ophthalmol. 1988;106(3):362–368. [DOI] [PubMed] [Google Scholar]
- 12. Rayborn ME, Moorhead LC, Hollyfield JG. A dominantly inherited chorioretinal degeneration resembling sectoral retinitis pigmentosa. Ophthalmology. 1982;89(12):1441–1454. [DOI] [PubMed] [Google Scholar]
- 13. Szamier RB, Berson EL. Retinal ultrastructure in advanced retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1977;16(10):947–962. [PubMed] [Google Scholar]
- 14. Szamier RB, Berson EL, Klein R, Meyers S. Sex-linked retinitis pigmentosa: ultrastructure of photoreceptors and pigment epithelium. Invest Ophthalmol Vis Sci. 1979;18(2):145–160. [PubMed] [Google Scholar]
- 15. Milam AH, Jacobson SG. Photoreceptor rosettes with blue cone opsin immunoreactivity in retinitis pigmentosa. Ophthalmology. 1990;97(12):1620–1631. [DOI] [PubMed] [Google Scholar]
- 16. Caruso G, Gelardi M, Passali GC, de Santi MM. Nasal scraping in diagnosing ciliary dyskinesia. Am J Rhinol. 2007;21(6):702–705. [DOI] [PubMed] [Google Scholar]
- 17. Armengot M, Milara J, Mata M, Carda C, Cortijo J. Cilia motility and structure in primary and secondary ciliary dyskinesia. Am J Rhinol Allergy. 2010;24(3):175–180. [DOI] [PubMed] [Google Scholar]
- 18. Chilvers MA, Rutman A, O'Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol. 2003;112(3):518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonneau D, Raymond F, Kremer C, Klossek JM, Kaplan J, Patte F. Usher syndrome type I associated with bronchiectasis and immotile nasal cilia in two brothers. J Med Genet. 1993;30(3):253–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ebermann I, Scholl HP. Charbel Issa P, et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121:203–211. [DOI] [PubMed] [Google Scholar]
- 21. Aller E, Jaijo T, van Wijk E, et al. Sequence variants of the DFNB31 gene among Usher syndrome patients of diverse origin. Mol Vis. 2010;16:495–500. [PMC free article] [PubMed] [Google Scholar]
- 22. Besnard T, Vaché C, Baux D, et al. Non-USH2A mutations in USH2 patients. Hum Mutat. 2012;33(3):504–510. [DOI] [PubMed] [Google Scholar]
- 23. Ayuso C y Millán JM. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2010;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Giménez-Pardo A, Ayuso C. An update on the genetics of Usher syndrome. J Ophthalmol. 2011;2011:417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roth Y, Kronenberg J. Nasal ciliary motility in retinitis pigmentosa. Auris Nasus Larynx. 1992;19(4):275–277. [DOI] [PubMed] [Google Scholar]
- 26. Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernal S, Medà C, Solans T, et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype–phenotype correlation. Clin Genet. 2005;68:204–214. [DOI] [PubMed] [Google Scholar]
- 28. Aller E, Jaijo T, Beneyto M. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]