

Abstract
Biodegradable–biocompatible polyurethanes were prepared with fixed hexamethylene diisocyanate and varying ratios of isomannide and poly(ϵ-caprolactone) diol using a simple one-step polymerization without a catalyst. The polyurethane structures were confirmed by 1H-nuclear magnetic resonance, Fourier transform infrared spectroscopy, and gel permeation chromatography. The glass transition temperatures were determined by thermal analysis to be between 25°C and 30°C. Degradation tests performed at 37°C in phosphate buffer produced mass losses of 5%–10% after 8 weeks. After 5 days of culture, using osteoblastic cells, the relative cell number on all the polyurethane films was only slightly lower than that of an optimized tissue culture plastic. These polymers offer significant promise with a simplistic synthesis and controlled degradation.
Keywords: Biodegradable, biobased monomer, polyurethane, biocompatible, isomannide
Introduction
Tissue engineering is the future medical treatment for reconstructing defective tissues over the last decade.1 Tissue regeneration is a relatively simple tissue engineering treatment compared with other treatments, such as in vitro tissue reconstruction; for example, in periodontitis, new tissue was reconstructed using a membrane to prevent other tissues from invading the affected areas.2 Recently, tissue regeneration, and the biomaterials used in tissue regeneration, has undergone a significant expansion both academically and commercially.3
The major materials used in tissue regeneration are biodegradable polymers. Biodegradable polymers, with good biocompatibility, have significant potential in various tissue engineering and drug delivery fields.4 Virtually, all tissue engineering devices are implanted in a mechanically active site in the body; thus, these implants are subjected to dynamic stress; therefore, it is very important that the polymers do not physically degrade, as their by-products can cause inflammatory effect to the surrounding tissues. Several families of biodegradable polymers have been developed, such as polyurethanes, poly(aliphatic esters), and poly(phosphate esters).5 These polymers have dominated the field as they are relatively cheap and they have Food and Drug Administration approval for clinical use.
Polyurethanes are a class of biodegradable polymers that have been applied as tissue engineering scaffolds as they show low in vitro and in vivo cytotoxicity.6 Polyurethanes created by reacting dialcohol with diisocyanate result in the highly hydrogen-bonded urethane linkages with good physical and mechanical properties and with the haemocompatibility and flexibility needed for use in biomedical applications.6 In fact, many non-biodegradable urethanes have been utilized in blood contact applications such as breast implants, aortic grafts, bone adhesives, dialysis membranes, and heart valves.6 Biodegradable polyurethanes can be obtained using biodegradable soft segments and isomannide hard segments. In the biodegradable soft segment, polyurethanes, such as those containing poly(ϵ-caprolactone) (PCL), have been obtained7 along with poly(ethylene oxide) (PEO) and poly(l-lactide) PLA.8 In the biodegradable hard segments, the diisocyanate and the chain extender can be designed from a variety of biologically relevant molecules.
Several possibilities were considered to produce biodegradable polyurethanes. Biodegradable hard segments have been obtained with aliphatic diisocyanate, methylene lysine diisocyanate, and hexamethylene diisocyanate (HDI). Good examples of such raw materials are those produced from glucose as the natural feedstock.9 Isomannide is a promising material for biobased monomers as it is nontoxic and has appropriate chirality and bicyclic ring structure, which imparts rigidity.10 Isomannide has been used in various polymer systems such as polyester, polyether, and polyurethane.
In this study a polyurethane series was made with isomannide, PCL diol, and HDI compounds to satisfy the demands of tissue regeneration technology. Aliphatic diisocyanates are usually chosen to synthesize biocompatible and biodegradable polyurethanes because their degradation product is nontoxic. Polyester diols, such as PCL diol, are also chosen as they hydrolytically degrade to caproic acid. Isomannide was chosen as the dialcohol monomer for its chiral, rigid, and nontoxic properties. The typically polyurethanes are made in a two-step polymerization.11 However, a bulk, one-pot synthetic method without catalyst, was employed in this study.
Experimental section
Materials and instruments
Isomannide (98%) and HDI (99%) were purchased from Sigma–Aldrich (St Louis, MO, USA). PCL diol 2000 (CAPA 2201A; Mw = 2000) was purchased from Kangnam Chemical Co., Ltd (Seoul, South Korea), and phosphate-buffered saline (approximately pH 7.3) was obtained from Oxoid Ltd (Hampshire, England). All chemicals were used without further purification.
Preparation of the polyurethanes
The monomers were mixed together and allowed to react.11 Synthesis was carried out in a 250 mL round-bottomed four-neck flask with reactant stoichiometry of 1:1 with diisocyanate:diol (PCL diol and isomannide) under a dry nitrogen atmosphere. A nitrogen flushed four-neck flask equipped with a mechanical stirrer, thermometer, and condenser was charged with 12.5 mmol of PCL diol (25 g) and 12.5 mmol of isomannide (41.83 g) (reactant stoichiometry and precursor weights are given as an example for polyurethane (PU) 1; Table 1). The flask was maintained at 80°C and stirred under a nitrogen atmosphere. After the solid contents were melted completely, 25 mmol of HDI (4.21 g) was added and stirred for 5 min. The sample was allowed to stand in a Teflon dish to react for 12 h at 150°C. The synthesized polyurethanes were dissolved in N,N-dimethylformamide (DMF). After the resulting solution was poured into a large amount of isopropyl alcohol, a polymer precipitate was formed and filtered. The product was dried at 40°C for 72 h under vacuum and stored in a desiccator. PU 2, PU 3, and PU 4 were prepared by similar procedures. Polymer yields of 87%, 78%, 71%, and 59% were obtained. The formulation used is described in Table 1.
Table 1.
Polyurethanes with different composition contents and their yields.
| Polyurethane | HDI | Isomannide | PCL diol (Mw = 2 kDa/mol) | Yield (%) |
|---|---|---|---|---|
|
| ||||
| Mole ratio | ||||
| PU 1 | 8 | 3 | 5 | 87 |
| PU 2 | 8 | 4 | 4 | 78 |
| PU 3 | 8 | 5 | 3 | 71 |
| PU 4 | 8 | 6 | 2 | 59 |
HDI: hexamethylene diisocyanate; PCL: poly(ϵ-caprolactone).
Polymer characterization
1H-nuclear magnetic resonance (NMR) spectrum was recorded for the synthesized polyurethanes with a Bruker Avance 600 spectrometer at 600 MHz (Bruker Co., Germany) and performed at ambient temperature with 5% (w/v) polymer solution in CDCl3. Fourier transform infrared (FT-IR) spectra were obtained at room temperature using a Bio-Rad Excaliber TS-3000MX (Bio-Rad, Tokyo, Japan) in the range of 800–4000 cm−1. A 2.5% solution of polymer in chloroform was placed directly onto a KBr pellet (Sigma–Aldrich), and the chloroform was evaporated at 50°C under vacuum for 2 h. The spectra were also checked for evidence of residual solvent.
Molecular weight
The weight average (Mw) and number average (Mn) molecular weights of the polyurethanes were measured by gel permeation chromatography (GPC) using a Futecs NP-4000 instrument (Futecs, Seoul, South Korea) equipped with a model P-4000 pump, a model AT-4000 column oven, a GPC KF-804 column, and a Shodex (Shodex, Tokyo, Japan) R1-101 refractive index detector. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL min−1, and a sample concentration of 2.5 mg mL−1 was used. Polystyrene (Mw = 2000, 7000, 12,000, 65,000, and 120,000) was used as the standard.
Thermal analysis
Differential scanning calorimetry (DSC) data were recorded with DSC (SEIKO Exstar 7000, Tokyo, Japan) instrument. Specimens (approximately 10 mg) were sealed in a DSC aluminum pan before being placed in the calorimeter. The samples were cooled to −70°C and then heated to 250°C at a rate of 10°C min−1 under a nitrogen atmosphere.
Mechanical properties
Following synthesis of the polymer, polymer films for mechanical testing and degradation testing were prepared by solvent casting in DMF at 10% polymer concentration into a polytetrafluoroethylene (PTFE) dish followed by air drying to produce films of 0.2 mm thickness. Tensile strength and elongation at break of the polyurethanes were measured on an Instron universal testing machine (Model 3344; Instron Engineering Corp., Canton, MA, USA) at a crosshead speed of 10 mm min−1 at room temperature. The specimens were prepared with a dog bone–shaped cutter. The thickness and width of the specimens were 0.2 and 5 mm, respectively. The length of the sample between the grips of the testing machine was 15 mm. Five specimen measurements were conducted for each polyurethane, and the results were averaged to obtain a mean and standard deviation.
Contact angle and surface energy
The wetting ability of the polymer surface was evaluated based on contact angle measurements using a Phoenix 300 contact angle equipment (SEO, Seoul, South Korea). Tests were carried out using the sessile drop mode with ultrapure water, which was used to represent polar characteristics. Approximately 5 µL droplets of the test liquid were placed on polymer specimens using a manual syringe. The drop profile was then recorded at an interval of 1 s for 1 min, and the measurements were carried out on triplicate samples. The contact angle values were evaluated using Phoenix 300 software. The polyurethane series was dried for 24 h prior to contact angle measurement. The calculation of the surface energy was conducted using the Girifalco–Good–Fowkes–Young method and Phoenix software.
In vitro degradation test
Polyurethane film (Φ = 12 mm) degradation was quantified by the changes in dry weight. The polyurethane samples were allowed to degrade for 2, 4, and 8 weeks. Dry films were weighed (w0) and immersed in a conical tube containing 10 mL phosphate-buffered solution (approximately pH = 7.3). The degradation was conducted at 37°C ± 1.5°C in a water bath (WB-6; DAIHAN Scientific Co., Ltd, Seoul, South Korea). Samples were taken at intervals, rinsed with water, dried in a vacuum oven for 2 days at 50°C, and weighed (wt), after which they were discarded. The weight remaining was calculated as
Cell culture
The polyurethane films were sterilized by soaking in 50%, 70%, and 100% ethanol for 30 min prior to use, and then they were dried for 2 h. MC3T3-E1 cells were maintained in standard T75 tissue culture flasks in normal growth medium composed of alpha-modified minimum essential medium (α-MEM) (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum (Gibco, Daejeon, South Korea) and 1% penicillin/streptomycin (Gibco). Prior to cell seeding, sections were cut from the synthesized polyurethane films, placed into 96-well plates, and seeded with 5 × 103 cells suspended in 1 mL normal growth medium and maintained at 37°C with 5% CO2 for a subsequent time course analysis of cell number.
Cell proliferation
Cells were cultured on the polyurethane films (n = 3) (the films were held down in the culture plates with PTFE inserts) in 96-well plates for 1, 3, and 7 days. Cell proliferation was determined at these time periods using the cell counting kit-8 (CCK-8) (2-(methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) assay, according to the manufacturer’s instructions (CCK-8; Dojindo Laboratories, Kumamoto, Japan). The cells were then incubated for 1, 3, and 7 days, and the cell numbers were measured using the CCK-8 reagent. The CCK-8 solution was freshly prepared in growth medium, and 200 µL of CCK-8 reagent was added to each well, followed by incubation in a humidified atmosphere with 5% CO2. Absorbance was measured at 450 nm using a spectrophotometer (Bio-Rad, Seoul, South Korea). A blank experiment to detect cell-free background absorbance was also performed in parallel. Results are expressed as relative CCK-8 activity compared to control conditions (cell-free background absorbance).
Results
A family of biocompatible and biodegradable polyurethanes was synthesized by a simple one-pot polymerization of PCL diol (Mw = 2000) and isomannide, as shown in Scheme 1. The reaction was carried out without a catalyst or solvent, which are usually required, in order to decrease potential toxicity.13 The chemical structures of the polyurethanes obtained were characterized by 1H-NMR and FT-IR spectroscopy.
Scheme 1.
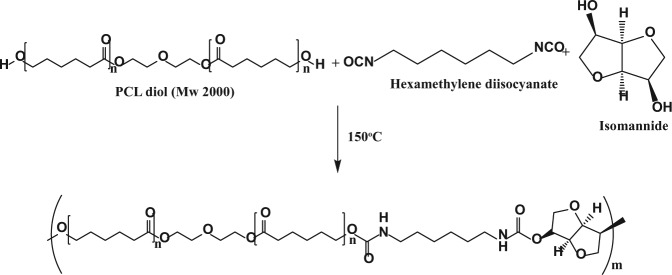
Synthetic of the route and structure of the polyurethane series.
PCL: poly(ϵ-caprolactone).
Figure 1 displays the 1H-NMR spectrum of polyurethane in CDCl3, in which all proton signals of isomannide, PCL diol 2000, and HDI segments were confirmed. The signals occurring from 3.5 to 5.2 were assigned to methylene protons of isomannide. In addition, the peak at 3.1 parts per million (ppm) was assigned to the amine proton of the urethane linkage.
Figure 1.

1H-nuclear magnetic resonance (NMR) spectra of polyurethane.
In the FT-IR spectra, the complete disappearance of the isocyanate peaks in the polyurethane samples occurred at 2250 cm−1 (Figure 2),14 indicating that no unreacted isocyanate groups were present in the polyurethane samples. After polymerization, the –OH peaks in the area around 3446–3558 and 3427 cm−1 of PCL diol and isomannide, respectively, disappeared. A new peak at 1529 cm−1 appeared, which was caused by the amide (N–H) bending vibration of the polyurethanes. The FT-IR spectrum confirmed that the polyurethane structures had a pronounced free amide group (–NH–) peak at 3322 cm−1, and hydrogen-bonded NH stretching bands were seen at 1529 cm−1,15 and a carbonyl group (C=O) stretching transmittance at 1721 cm−1. Methylene group polymer chains were represented by peaks between 2938 and 2860 cm−1 in the polyurethanes and PCL diol. C-H bending and C-O stretching vibration bands were seen in the area from 1500 to 600 cm−1.
Figure 2.
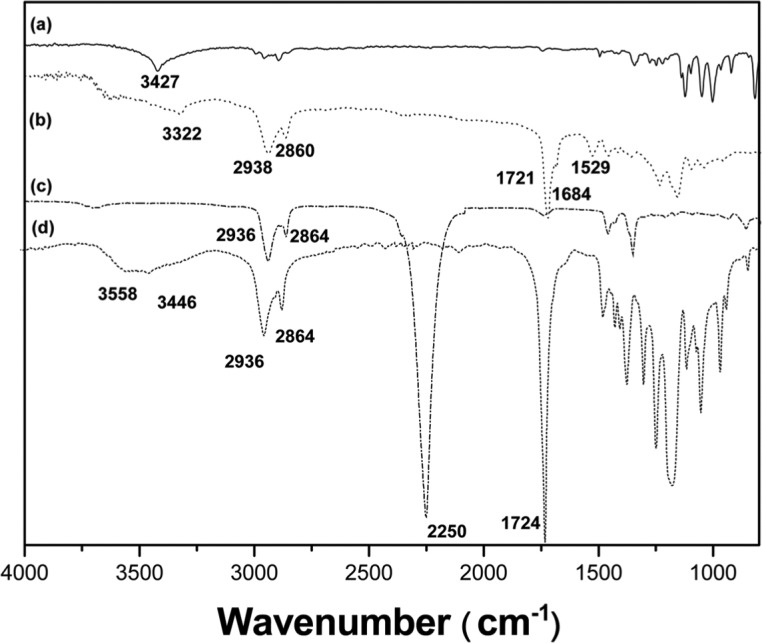
Fourier transform infrared (FT-IR) spectra of (a) isomannide, (b) polyurethane, (c) HDI, and (d) PCL diol 2000.
HDI: hexamethylene diisocyanate; PCL: poly(ϵ-caprolactone).
Molecular weights and thermal properties
The thermal properties and molecular weights of the polyurethanes are listed in Table 2. The weight average molecular weights (Mw) ranged from 16,892 to 145,070. Polyurethanes with the higher levels of isomannide yielded lower Mw compared to polyurethanes with low levels of isomannide may be due to the presence of a secondary hydroxyl group, which is lower in reactivity compared to the primary hydroxyl group12 of the PCL diol. It is important to understand the thermal behavior of polymers developed for biomedical applications, as it determines the physical properties of the materials and the processability. The DSC data of the synthesized polyurethane series are listed in Table 2. Only one Tg was detected for all samples. The values of Tg of PU 1, PU 2, PU 3, and PU 4 were 32°C, 30.9°C, 26.4°C, and 25.3°C, respectively. A melting peak was not observed for any of the polymers examined.
Table 2.
Molecular weights, thermal properties, and surface energy of the synthesized polyurethanes.
| Mw a(kDa)/mol | Mn b(kDa)/mol | PDIc | Tg (°C)d | Water (°)e | Surface energy (mN m−1) | |
|---|---|---|---|---|---|---|
| PU 1 | 145.0 | 101.8 | 1.4257 | 32.00 ± 0.32 | 61.34 ± 1.48 | 49.19 ± 1.50 |
| PU 2 | 68.2 | 44.6 | 1.5284 | 30.90 ± 0.21 | 60.67 ± 0.84 | 49.71 ± 1.13 |
| PU 3 | 30.8 | 20.2 | 1.5242 | 26.43 ± 0.45 | 60.44 ± 0.63 | 50.10 ± 0.64 |
| PU 4 | 16.9 | 12.6 | 1.2960 | 25.30 ± 0.22 | 58.00 ± 0.52 | 52.59 ± 0.53 |
PDI: polydispersity index.
The weight average molecular weights.
The number average molecular weights.
PDI = Mw/Mn.
Glass transition temperature.
Water contact angle.
Mechanical properties
The ultimate tensile strength (UTS) and Young’s modulus for the polyurethanes are shown in Figure 3. The higher molecular weight polyurethane was obtained with PU 1, and this produced a mean stiffness (called Young’s modulus) of 27.19 ± 0.5 MPa, which was similar to that of ethylene–methyl acrylate. The UTS was 20.4 ± 1.09 MPa. Lower molecular weight polyurethane was obtained with PU 4, and this produced a mean stiffness of 50.19 ± 5.7 MPa and a UTS of 6.12 ± 1.00 MPa.
Figure 3.

(a) Ultimate tensile strength (UTS), (b) Young’s modulus, (c) strain at breaking (%), and (d) example of tensile stress–strain curves for the polyurethane series.
In vitro degradation test
The degradation characteristics of the polyurethane series were studied in vitro using a phosphate buffer solution at 37°C. Degradation profiles of the polyurethanes are presented in Figure 4 with the percentage weight loss of the polymers with time. It is evident that the polyurethane series was undergoing slow degradation. The changes in the polyurethane film surface after degradation (Figure 5) were determined by scanning electron microscope (SEM) (model JSM-6510; JEOL, Tokyo, Japan) photographs. All surfaces appear relatively smooth initially with a few defects. However, all of the polyurethanes had ~7% weight lost over 2 weeks. A continual slow weight loss of ~12% was observed for all of polyurethanes between weeks 2 and 8.
Figure 4.
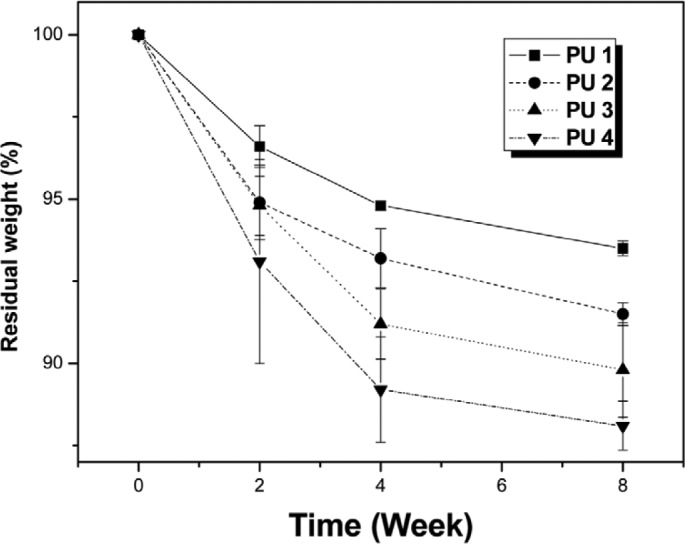
Residual weight (%) of the polyurethane films after degradation in phosphate buffer solution: (a) PU 1, (b) PU 2, (c) PU 3, and PU 4.
Figure 5.

(a and b) SEM of PU 1 prior to degradation and at 2 months, (c and d) SEM of PU 2 prior to degradation and at 2 months, (e and f) SEM of PU 3 prior to degradation and at 2 months, and (g and f) SEM of PU 4 prior to degradation and at 2 months.
SEM: scanning electron microscope.
CCK-8 assay
The results of CCK-8 absorbance values for MC3T3-E1 cells (CCK-8) adhesion to and proliferation on the polyurethane films compared to that of PCL are shown in Figure 6. The CCK-8 assay was implemented to measure mesenchymal stem cell (MSC) growth, and as it is specific to viable cells, it reflects not only the ability of the polyurethane film to promote cell adhesion and proliferation, but also its cytotoxicity. In comparison with PCL (Mw = 60,000, Sigma–Aldrich), relative cell numbers on the polymer films between days 1 and 7 of culturing on polyurethane films proliferated at least as well as those on PCL. Lower cell viability was observed from 1 to 7 days on the polyurethane surfaces in comparison with that of the control.
Figure 6.

CCK-8 assay of rat MC3T3-E1 cultured on polyurethane wells showed more metabolically active cells on polyurethanes during the first 7 days.
ANOVA: analysis of variance; CCK: cell counting kit; PCL: poly(ϵ-caprolactone).
Data are mean ± standard deviation. Significantly different from the control (*p < 0.05, **p < 0.01, ANOVA, n = 3). Normalized values are shown.
Cell attachment
Attachment of cells to a material is one of the prerequisites for evaluating biocompatibility for possible utilization in biomedical applications. The morphology of MC3T3-E1 cells after they were cultured on the polyurethane polymer films for 1–7 days was examined by SEM. The morphology of MC3T3-E1 cells attached to the wells were exhibited in Figure 7. The polyurethane films provided a suitable environment for cell attachment and spreading. The SEM images of the MC3T3-E1 cell morphology after being cultured on polyurethane film during 7 days PU 1, PU 2, PU 3, and PU 4 are shown in Figure 7(a) to (d), respectively. The cell culture results indicate that these polyurethanes are biocompatible and nontoxic.
Figure 7.

SEM images illustrating morphology of MC3T3-E1 cells after being cultured on polyurethane film during 7 days (a) PU 1, (b) PU 2, (c) PU 3, and (d) PU 4.
SEM: scanning electron microscope.
Discussion
A great deal of research over the last decade has been conducted to develop biodegradable polyurethanes for medical applications.1,2 It would be advantageous for some applications if implants were produced from biodegradable and/or bioresorbable materials. Continuous degradation of the implant material may then be accompanied by the formation of new organs and/or tissues.16 Biodegradable polyurethane elastomers are promising materials for such implants.17
The goal of this study was to evaluate the cytocompatibility of experimental polyurethanes with different isomannide-to-PCL diol content ratios in MC3T3-E1 cells. Our one-pot polymerization process is very simple and reduces the risk of toxicity from a surfactant or catalyst. In the molecular weight study, we should consider the reactivity of isomannide. Isomannide may have lower reactivity. The effect of the lower reactivity secondary hydroxyl of isomannide appeared to be quite evident with PU 1, which had the lowest isomannide content, and showed the highest Mw. This was also supported by the polymer yield data. The variations in the Tg of the soft segment as a function of composition have been suggested as an indicator of the degree of microphase separation in thermoplastic polyurethane elastomers.18 In general, the Tg value of soft segments decreases with increasing soft-segment molecular weight and decreasing diisocyanate content, although the changes are quite small. The Tg value also increased with increasing molecular weight.19 Factors with an effect on Tg in polyurethanes based on aliphatic diisocyanate include crystallization of the soft-segment and hard-segment components, the steric hindrance of the hard-segment unit during hydrogen bonding, and the inherent solubility of the hard and soft components.20 If the Tg value is above body temperature, material will be in a rigid state.21 On the other hand, if the Tg of the polymer is below body temperature, the material would be in a rubbery state. For the polyurethanes in this study, the Tg decreased with decreasing molecular weight and Tg increased with decreasing contents of isomannide as expected. All four polyurethanes had a Tg below body temperature; thus, these polyurethanes could exhibit elastomeric properties at body temperature. Elastomeric properties at body temperature are an important quality for polymers in soft tissue engineering.21 The polyurethane films were flexible with tensile strengths from 6.12 ± 1.0 to 20.40 ± 1.09 MPa, stiffness from 27.2 ± 0.51 to 50.19 ± 5.7 MPa, and breaking strains from 208% to 1239%. The UTS of the films was comparable to that of the aorta (50%–100%),21 whereas the breaking strains were generally greater than that of the aorta. The mechanical properties of the film depended on the molecular weight, hard segments, and soft segments. As the molecular weight of a polymer increases, tensile strength increased. In addition, there was a relationship between hard segment and stiffness.22 The polyurethane stiffness increased with increasing isomannide (hard segment) levels. The main degradable polymers used in biomedicine are PCL, polyglycolic acid (PGA), and poly(l-lactic acid) (PLLA) and their derivatives. These polymers are all very stiff and are not suitable for utilization for tissue engineering of soft tissues, even in a fibrous or woven state. The development of softer, more flexible, and degradable polymers, as shown in this study, opens up a range of opportunities for soft tissue augmentation and regeneration, and the polymer family synthesized in this study offers such properties.
Much attention has been paid to the surface properties and structure of polymers, because the functional groups on the surface play an important role in the interactions of the materials with cells.23 Contact angles have been used to estimate surface properties. The measured contact angles are listed in Table 2, and all of the polyurethane water contact angles were <61°. It is generally agreed that hydrophilic surfaces have a contact angle with water in the range of 1°–30°, whereas hydrophobic surfaces have contact angles of >90°. Therefore, our polyurethanes had values somewhere between these ranges. As the isomannide content, which is a hydrophilic unit monomer, increased, the water contact angle of the polyurethanes decreased, indicating that the hydrophilicity of the polyurethanes can be controlled, although the changes were relatively small.
The rate of degradation of polyurethanes in aqueous media by hydrolysis is controlled mainly by the presence of urethane and ester bonds and varies significantly with the polymer chemical structure.24 The acidic carboxyl group accelerates the hydrolysis of the polyester segments.18 The biodegradability of polyurethanes also depends on the molecular weight, degree of crystallinity, chemical structure, as well as susceptibility to microbial attack.26 The number of carbons in the dialcohol seems to play an important role during hydrolytic degradation. The isomannide gives higher hydrophilicity to the polyurethane segment than does PCL diol, making hydrolytic attack easier. The rate of hydrophilicity and hard-segment contents had a more dominant effect on biodegradation rate rather than crystallinity for the polyurethanes presented here.25
Many studies have ascertained biocompatibility using the CCK-8 or 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay.18 When cell proliferation on a polymer is maintained for 1–2 weeks,26 then the polymer can be considered cytocompatible and biocompatible. MC3T3-E1 cells were used as they are highly sensitive to culture conditions. The cell number was assessed via the CCK-8 assay due to its reliability and sensitivity.27 The cells showed lower numbers from 1 to 7 days compared with the control, when seeded on polyurethane surfaces (Figure 7); however, the cells began to proliferate and increase in number. In comparison with PCL, relative cell number on polyurethane films in 7 days of culture was approximately the same as PCL. Although the cells showed a lower rate of proliferation than that of the control, the polyurethane films were considered to be cytocompatible. Furthermore, the synthesized polyurethanes had good mechanical strength, thermal stability, cytocompatibility, and biodegradability.
Conclusion
Polyurethane biobased isomannide polymers were synthesized from HDI via a simple one-pot polymerization. The important advantage was not to require a catalyst, solvent, or surfactant during the reaction, which offered clinical advantages related to toxicity. Based on the in vitro mouse osteoblast cultures, the cells were suppressed slightly compared to the control but they grew well on the polyurethane films for 5 days. The CCK-8 cytotoxicity assay indicated that the polyurethanes had good biocompatibility. Taking into account the biocompatibility, degradability, and mechanical properties, these biobased polyurethanes have potential for soft tissue reconstruction, such as bladder, tracheal, and cardiovascular applications.
Footnotes
Declaration of conflicting interests: The authors declare that there is no conflict of interest.
Funding: This study was supported by Priority Research Centers Program (no. 2009-0093829) and the World Class University (WCU) Program through the National Research Foundation of Korea (no. R31-10069) and funded by the Ministry of Education, Science, and Technology.
References
- 1. Kikuchi M, Koyama Y, Yamada T, et al. Development of guided bone regeneration membrane composed of β-tricalcium phosphate and poly (l-lactide-co-glycolide-co-ϵ-caprolactone) composites. Biomaterials 2004; 25: 5979–5986 [DOI] [PubMed] [Google Scholar]
- 2. Chen FM, Zhang J, Zhang M, et al. A review on endogenous regenerative technology in periodontal regenerative medicine. Biomaterials 2010; 31: 7892–7927 [DOI] [PubMed] [Google Scholar]
- 3. Rezwan K, Chen QZ, Blaker JJ, et al. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006; 27: 3413–3431 [DOI] [PubMed] [Google Scholar]
- 4. Tomas E, Lubomir K, Vladimir S, et al. High-molecular-weight polymers containing biodegradable disulfide bonds: synthesis and in vitro verification of intracellular degradation. J Bioact Compat Polym 2010; 25: 5–26 [Google Scholar]
- 5. Kawase T, Tanaka T, Nishimoto T, et al. An osteogenic grafting complex combining human periosteal sheets with a porous poly(l-lactic acid) membrane scaffold: biocompatibility, biodegradability, and cell fate in vivo . J Bioact Compat Polym 2012; 27: 107–121 [Google Scholar]
- 6. Marcos-Fernández A, Abraham GA, Valentín JL, et al. Synthesis and characterization of biodegradable non-toxic poly(ester-urethane-urea)s based on poly(ϵ-caprolactone) and amino acid derivatives. Polymer 2006; 47: 785–798 [Google Scholar]
- 7. Heijkants RGJC, van Calck RV, van Tienen TG, et al. Uncatalyzed synthesis, thermal and mechanical properties of polyurethanes based on poly(ϵ-caprolactone) and 1,4-butane diisocyanate with uniform hard segment. Biomaterials 2005; 26: 4219–4228 [DOI] [PubMed] [Google Scholar]
- 8. Izhar U, Schwalb H, Borman JB, et al. Novel synthetic selectively degradable vascular prostheses: a preliminary implantation study. J Surg Res 2001; 95: 152–160 [DOI] [PubMed] [Google Scholar]
- 9. Habeych DI, Benjamin Juhl P, Pleiss J, et al. Biocatalytic synthesis of polyesters from sugar-based building blocks using immobilized Candida antarctica lipase B. J Mol Catal B: Enzym 2011; 71: 1–9 [Google Scholar]
- 10. Hassan N, Asghar K. Synthesis of new prodrugs based on β-CD as the natural compounds containing β-lactam antibiotics. J Bioact Compat Polym 2007; 22: 77–88 [Google Scholar]
- 11. Gorna K, Gogolewski S. Biodegradable porous polyurethane scaffolds for tissue repair and regeneration. J Biomed Mater Res A 2006; 79: 128–138 [DOI] [PubMed] [Google Scholar]
- 12. Lisa T, Tim GM, Raju A, et al. Thermoplastic biodegradable polyurethanes: the effect of chain extender structure on properties and in-vitro degradation. Biomaterials 2007; 28: 5407–5417 [DOI] [PubMed] [Google Scholar]
- 13. Rivera-Armenta JL, Heinze Th, Mendoza-Martínez AM. New polyurethane foams modified with cellulose derivatives. Eur Polym J 2004; 40: 2803–2812 [Google Scholar]
- 14. Sarkar D, Yang JC, Gupta AS, et al. Synthesis and characterization of l-tyrosine based polyurethanes for biomaterial applications. J Biomed Mater Res A 2009; 90: 263–271 [DOI] [PubMed] [Google Scholar]
- 15. Luo N, Wang DN, Ying SK. Hydrogen-bonding properties of segmented polyether poly(urethane urea) copolymer. Macromolecules 1997; 30: 4405–4409 [Google Scholar]
- 16. Gogolewski S, Walpoth B, Rheiner P. Polyurethane microporous membranes as pericardial substitutes. Colloid Polym Sci 1987; 265: 971–977 [Google Scholar]
- 17. Dånmark S, Finne-Wistrand A, Wendel M, et al. Osteogenic differentiation by rat bone marrow stromal cells on customized biodegradable polymer scaffolds. J Bioact Compat Polym 2010; 25: 207–223 [Google Scholar]
- 18. Lelah MD, Cooper SL. Polyurethanes in medicine. Boca Raton, FL: CRC Press, 1986 [Google Scholar]
- 19. Katarzyna G, Sylwester G. Biodegradable polyurethanes for implants. II. In vitro degradation and calcification of materials from poly(ϵ-caprolactone)-poly(ethylene oxide) diols and various chain extenders. J Biomed Mater Res 2002; 60: 592–606 [DOI] [PubMed] [Google Scholar]
- 20. Zhang C, Zhang N, Wen X. Synthesis and characterization of biocompatible, degradable, light-curable, polyurethane-based elastic hydrogels. J Biomed Mater Res A 2007; 82: 637–650 [DOI] [PubMed] [Google Scholar]
- 21. Holzapfel GA. Biomechanics of soft tissue. Biomech preprint series, paper no. 7, November 2000. Graz: Institute of Structural Analysis-Computational Biomechanics, Graz University of Technology, Austria [Google Scholar]
- 22. Sharma B, Ubaghs L, Keul H, et al. Synthesis and characterization of alternating poly(amide urethane)s from epsilon-caprolactone, diamines and diphenyl carbonate. Polymer 2005; 46: 1775–1783 [Google Scholar]
- 23. Geoghegan M, Krausch G. Wetting at polymer surfaces and interfaces. Prog Polym Sci 2003; 28: 261–302 [Google Scholar]
- 24. Takahara A, Coury AJ, Hergenrother RW, et al. Effect of soft segment chemistry on the biostability of segmented polyurethanes. I. In vitro oxidation. J Biomed Mater Res 1991; 25: 341–356 [DOI] [PubMed] [Google Scholar]
- 25. Gualandi C, Soccio M, Govoni M, et al. Poly(butylene/diethylene glycol succinate) multiblock copolyester as a candidate biomaterial for soft tissue engineering: solid-state properties, degradability, and biocompatibility. J Bioact Compat Polym 2012; 27: 244–264 [Google Scholar]
- 26. Gupta D, Venugopal J, Prabhakaran MP, et al. Aligned and random nanofibrous substrate for the in vitro culture of Schwann cells for neural tissue engineering. Acta Biomater 2009; 5: 2560–2569 [DOI] [PubMed] [Google Scholar]
- 27. Koh T, Shinji N, Ryuichiro K, et al. Antitumor activity and macrophage nitric oxide producing action of medicinal herb, Crassocephalum crepidioides . BMC Complement Altern Med 2012; 78: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]