

Abstract
ApoAII, the second most abundant protein of the human plasma HDLs, was discovered nearly 50 years ago. Over the subsequent years, nearly 2000 studies – epidemiological, cell-based, biochemical, mouse and human – have attempted to unravel its role in human lipid metabolism. On the basis of these studies, apoAII has been described as an activator and inhibitor of various plasma activities, and as both pro- and anti-atherogenic. Here, we summarize the studies of apoAII, use the preponderance of evidence to propose that the apoAII compass can be reset towards an antiatherogenic course, and suggest ways to stay the course.
Keywords: atherosclerosis, cholesterol, epidemiology, lipid metabolism, lipoprotein therapeutics, reverse cholesterol transport
Human plasma HDLs are a heterogeneous particle population comprising cholesterol and its esters, triglycerides (TGs), phospholipids and specialized proteins, comprising mostly apoAI and apoAII, and smaller amounts of apoCs and apoE [1]. Human plasma HDL cholesterol (HDL-C) concentrations are negatively correlated with the occurrence of atherosclerosis [2,3]. Conventional thought has been that the cardioprotective effect of HDL is mediated by its role in reverse cholesterol transport (RCT), the transfer of macrophage cholesterol in the subendothelial space of the arterial wall to the liver for disposal. RCT comprises macrophage cholesterol efflux producing nascent HDL, cholesterol esterification by plasma LCAT and, finally, the selective hepatic disposal of HDL cholesteryl esters [4–6]. The major HDL protein, apoAI, is a key RCT player because it is an avid acceptor of macrophage cholesterol efflux [7], an activator of cholesterol esterification by LCAT [8], and the major protein of mature forms of HDL [1]. Given that atherosclerosis is a chronic inflammatory process, it is notable that both apoAI and HDL are anti-inflammatory [9]. Moreover, other studies have shown that apoAII suppresses concanavalin A-induced hepatitis and that apoAII administration suppressed hepatitis after its induction, suggesting a therapeutic role for apoAII in patients with viral hepatitis [10]. Since its identification as a component of HDL nearly 50 years ago, there have been numerous contradictory data and attendant opinions regarding its role, if any, in lipid metabolism and atherosclerosis. The contradictions have arisen, in part, from studies using different species and a variety of experimental models, which demonstrated many apoAII activities, but in summary, have failed to provide a clear and consistent role for apoAII in atherogenesis.
ApoAII structure & function
Human apoAII, the second most abundant protein of human plasma HDL [11], is a dimer of two identical polypeptide chains joined by a disulfide bond at residue 6 [12]. ApoAII belongs to a family of exchangeable proteins comprising multiple lipid-binding regions of amphipathic helices. Like the other members of the gene family, the gene structure for apoAII comprises four exons, two of which compose the coding region that contains a prosequence that is cleaved by the thiol protease, elastase [13,14]. The exchangeable apolipoproteins associate with phospholipids giving reconstituted HDL (rHDL), which have been characterized by numerous physicochemical methods that were the basis of a general model. While HDLs are spherical, the rHDLs are discoidal particles in which several, largely helical apolipoproteins surround a phospholipid bilayer [15]. The discoidal rHDL are models for early forms of HDL that are found intrahepatically and in response to the apolipoprotein-mediated efflux of cholesterol from macrophages. The number and arrangement of apolipoproteins, including apoAII, have been determined. These studies reveal apoAII in a belt-like structure around the edge of a phospholipid disc. According to the pattern of lysine cross-links found, Silva et al. concluded that in forming rHDL, four apoAII molecules occur in a double-hairpin conformation on the disc edge (Figure 1) [16].
Figure 1. Model of the double-hairpin structure of apoAII in reconstituted HDL discs.
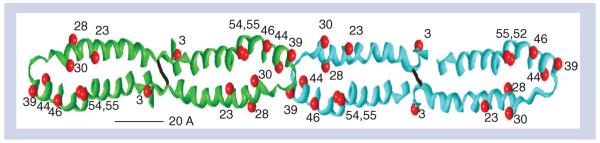
Based upon chemical cross-links identified by mass spectrometry, the C-terminal 38–40 residues of each strand double back onto the N-terminal half of the strand with an intervening turn sequence. Reproduced with permission from [16].
Human plasma levels of apoAII are approximately 40 mg/dl (~25 μM as the homodimer). Compared with apoAI, apoAII is the less studied apolipoprotein, approximately 10,000 versus approximately 2000 PubMed citations, respectively. The reasons for this are varied. ApoAI is more abundant and more easily isolated, so that studies were more easily supported. In addition, early studies unambiguously showed that apoAI had roles that went beyond simple lipid binding, the most important being the activation of plasma LCAT, an important intermediate step in RCT. Perhaps this and its greater availability produced a bandwagon effect that induced more academic and industrial investigators to study the broad science of apoAI rather than apoAII. As a consequence, there are numerous corroborated studies of the structure, properties, activities and physio logical role of apoAI. The same cannot be said of apoAII, for which there is, with a notable exception, little convincing epidemiological evidence identifying this apolipoprotein as atheroprotective or atherogenic. The atheroprotective versus atherogenic value of apoAII has been studied in the EPIC-Norfolk cohort, by determining whether plasma apoAII concentrations predicted the occurrence of future cardiovascular disease (CVD) events [17]. This study showed that plasma apoAII concentrations were lower in CVD subjects than in control subjects, and were inversely associated with CVD risk. Even after adjusting for confounders, including apoAI and HDL-C concentrations, apoAII and the risk of future CVD remained correlated. Although the difference between CVD and control plasma apoAII concentrations was only approximately 2%, the data supported an antiatherogenic role for apoAII. Thus, in this context, a recent report, discussed below, will likely increase interest in apoAII as an antiatherogenic therapeutic target.
Speciation of HDL
In human plasma, apoAII occurs largely as a homodimer on HDL, with smaller amounts existing as heterodimers with apoE and apoD [18,19]. Although apoAII is HDL-associated, not all HDL particles contain apoAII. Rather, approximately a third of all HDL contain apoAI but no apoAII, called lipoprotein (Lp)A-I, whereas the remainder, LpA-I/A-II, contain both apoAI and apoAII [20]. LpA-I comprises two particle sizes with Stokes diameters of 10.8 and 8.5 nm. LpA-I/A-II comprises particles with Stokes diameters of 9.6, 8.9 and 8.0 nm. ApoC, D and E, and LCAT are associated with both particle types. LpA-I/A-II have an A-I:A-II molar ratio of approximately 2:1. According to mass spectrophotometric studies of the cross-linked products of LpA-I, there are more apoAI molecules on the large than on the small LpA-I particles. By contrast, LpA-I/A-II particles contain only two proximal apoAI molecules per particle, while the number of apoAII molecules increases from one to two, and then to three, with increasing size. LpA-I comprises particles that contain one or two molecules of apoC-III, whereas LpA-I/A-II particles contain no more than one apoC-III each.
Monomeric versus dimeric apoAII
Most studies indicate only minor differences in the biophysics of monomeric and dimeric apoAII. The lipid–protein complexes formed by monomeric and dimeric apoAII are very similar [21]. According to in vitro studies, monomeric and dimeric apoAII displace equal amounts of the less lipophilic apoAI from HDL in the same dose-dependent way [22]. This would seem to suggest that monomeric and dimeric apoAII are similarly lipophilic, but this is not likely the case. A direct-binding assay of monomeric apoAII to lipid vesicles or HDL gives a free energy of association of approximately −7 kcal/mol, whereas the binding affinity of dimeric apoAII was too great to quantify (hypothetically 2 × −7 kcal/mol = approximately −14 kcal/mol) [23]. This is an important distinction that likely explains the lower levels of monomeric apoAII in mice versus the dimeric species in man [1,24]. The lower lipophilicity and size of monomeric versus dimeric apoAII makes it more likely to be renally cleared.
ApoAII dimerization, production & secretion
In humans and mice, apoAII is synthesized mainly by the liver [25]. Human apoAII gene transcription is controlled by several regulatory elements within and outside the promoter. The first intron reduces the transcription driven by the apoAII promoter to 15–18% of its original value [26]. The apoAII promoter is also trans-activated by RXR ligands, which include fatty acids and fibrates in vivo via PPAR-γ activation [27,28], suggesting that the RXR–PPAR heterodimer positively regulates transcription. According to these and other data, plasma apoAII levels are altered by gene transcription changes in response to intra- and extra-cellular stimuli and, unlike apoAI, plasma apoAII levels are determined by synthesis, not catabolism [29]. Pharmacologically, apoAII synthesis in mice and humans is controlled differently. Fibrates increase human apoAII synthesis via RXR–PPAR, whereas in mice, a decrease is observed [28].
ApoAI and apoAII follow distinct itineraries for synthesis, association with phospholipids and secretion (Figure 2). According to pulse-chase studies, only approximately 20% of newly synthesized apoAI is lipidated in the endoplasmic reticulum and Golgi compartment prior to secretion [30–32]. Early apoAI lipidation in the endoplasmic reticulum is ABCA1-independent, whereas lipidation in the Golgi and at the plasma membrane is ABCA1-dependent [32]. By contrast, apoAII is completely lipidated and dimeric within 2 h of synthesis [31]. The intrahepatic concentration of apoAII is not known, but is likely lower than the plasma concentration of approximately 10−5 M, and at this concentration, the rate of dimerization is expected to be slow; as expected, the observed rate of dimerization of isolated apoAII has a half-time on the order of weeks [33]. Gillard et al. resolved the discrepancy between the slow in vitro and rapid intracellular rate by showing that apoAII lipidation and dimerization are linked [31,33]. Mechanistically, phospholipids catalyze dimerization by lipidation, which concentrates two or more apoAII molecules on a lipid surface, thereby greatly increasing the local concentration of apoAII over that in the aqueous solutuion. This, in turn, produces a commensurate increase in the rate of dimerization of nearly four orders of magnitude [33]. Although apoAI and apoAII follow different secretory mechanisms, in plasma they occur on a common particle, LpA-I/A-II. Both biochemical and cell studies implicate LCAT in the formation of LpA-I/A-II. Biochemical studies showed that formation of spherical rHDL containing both apoAI and apoAII is mediated by LCAT. This finding is consistent with studies with hepatocytes, which concurrently secrete LCAT and two kinds of nascent HDL particles: those containing apoAI and dimeric apoAII, respectively. ApoE, another HDL apolipoprotein, is hepatically secreted on VLDL particles and subsequently transfers to HDL [31].
Figure 2. Hepatic HDL formation and secretion.

Apolipoprotein synthesis and its initial lipidation occur in the ER. In the Golgi, apoAII particles are further lipidated and approach the size of HDL, while apoAI particles are smaller, with more than half of the intracellular apoAI remaining lipid-free. After secretion, apoAI acquires lipid via ABCA1, and nascent apoAI HDL matures to spherical LpA-I by action of LCAT. ApoAII, which dimerizes after lipidation, is secreted as a lipidated dimer on particles without apoAI. Shortly after secretion, LCAT promotes fusion of these particles with spherical apoAI–HDL. Monomeric apoE in the Golgi associates with VLDL, which are remodeled after secretion (double arrow) to LDL-sized particles and some apoE transfers to HDL, where it dimerizes with apoAII. Apolipoproteins are colored as: gray (apoAI), green (apoAII) and orange (apoE) rods to show helix structure; cysteine groups are red spheres.
ER: Endoplasmic reticulum; Lp: Lipoprotein.
Reproduced with permission from [31].
Association of apoAII with lipids
Many lipid–apolipoprotein association studies have been performed with dimyristoylphosphatidylcholine (DMPC), a synthetic phospholipid that readily associates with many of the exchangeable apolipoproteins, giving a model rHDL having a discoidal structure similar to those of nascent HDL produced by hepatocytes. Given that hepatocytes assemble apoAII in nascent HDL particles that lack apoAI and apoE, studies of the mechanisms of apoAII–DMPC association and the structures of their products are relevant to metabolism in vivo. Moreover, the rHDL formed from a single lipid and a single protein species are useful for the identification structure–function relationships between specific proteins within HDL, without the confounding effects of other proteins. Both apoAI and apoAII spontaneously associate with DMPC. The addition of up to approximately 12.5 mol% cholesterol to DMPC increases the rate of formation of rHDL containing apoAI. However, as the cholesterol content approaches 20 mol%, the rate decreases to nil, an effect that may be due to kinetic (rate) or thermodynamic (affinity) factors [34]. Similar studies were conducted with apoAII, which has a greater affinity for lipids than apoAI. Again, the rate of rHDL association decreased to nil at 20 mol% cholesterol [35]. In the mechanistic model based on these data (Figure 3), apolipoproteins bind to DMPC via surface defects that disappear at 20 mol% cholesterol. These data suggest apoAII-containing HDL formed intrahepatically are likely cholesterol-rich, compared with the smaller intracellular lipid-poor apoAI HDL.
Figure 3. Lipid packing of free cholesterol in dimyristoylphosphatidylcholine and reconstituted HDL formation.
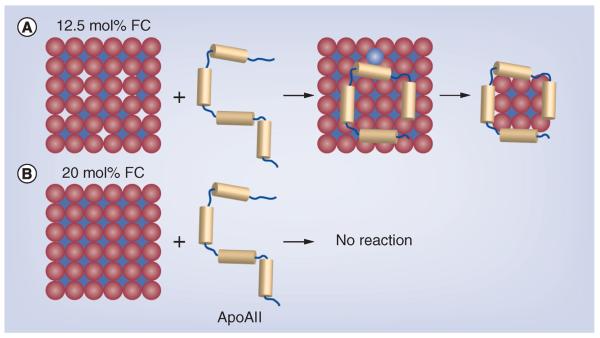
(A) Packing at approximately 15 mol% cholesterol showing the defects (gaps), which permit insertion of apoAII leading to reconstituted HDL formation. (B) Close packing at 20 mol% cholesterol, showing the absence of major defects and no reaction with apoAII.
FC: Free cholesterol.
Reproduced with permission from [35].
ApoAII lipophilicity & metabolic fate
ApoAII and apoAI have distinct metabolic fates that are a function of the much higher lipophilicity of the former. Although turnover studies indicate similar plasma lifetimes for both apolipoproteins [36], the metabolic pathways are different. Physicochemical studies of HDL using thermal and chemical perturbation with the denaturant, guanidine hydrochloride, led to a new and biologically relevant model of HDL stability [37]. These studies showed that heating HDL or incubating HDL with guanidine hydrochloride was associated with the release of lipid-free apoAI, but not apoAII, followed by fusion of the apoAI-poor HDL into a larger apoAII-rich particle. Thus, contrary to previously held assumptions, the underlying cause of HDL particle stability is kinetic rather than thermodynamic in origin; HDL resides in a kinetic trap from which it escapes given the appropriate chemical or biological perturbation. In vitro studies have shown that this model is highly relevant to the effects of several HDL-modifying activities. These include CETP [38], phospholipid transfer protein [39], LCAT [40], hepatic lipase [41] and streptococcal serum opacity factor [42]. The activities of these proteins introduce two common secondary effects: the release of lipid-free apoAI, but not apoAII, from the particle surface, and subsequent fusion of the remaining apoAI-depleted HDL that is obligatorily apoAII-enriched. Lipid-free apoAI is the putative ligand for the ABCA1 transporter, which mediates macrophage cholesterol efflux. Although apoAII has been shown to support macrophage cholesterol efflux [43], this is not likely one of its physiological roles because there are no known plasma activities that catalyze production of lipid-free apoAII. However, the apoAII-rich HDL that is likely formed by these plasma activities are candidates for cholesterol efflux via ABCG1. Thus, despite their similar plasma lifetimes, apoAI and apoAII appear to have distinct metabolic itineraries that are dictated by the activities of several HDL-modifying activities in plasma.
Atherogenesis in mouse models of apoAII metabolism
An early epidemiological study showed that like apoAI, plasma apoAII concentrations are negatively associated with CVD [44]. Moreover, human apoAII deficiency is not associated with any profound lipoprotein abnormalities [45]. However, conclusions about apoAII-associated CVD risk based on these studies were limited by the analytical technology then available, which could not measure many of the possible confounders or cofactors in the former case, and in the latter case, by the small number of patients with this phenotype. Thus, later studies turned to cell- and mouse-based models in which variables are more easily controlled and the statistical power of large numbers is much greater.
Genetically altered mice in which specific proteins are either overexpressed (transgenic models) or ablated (knockout [KO] models) frequently uncover the mechanistic roles of proteins in health and disease. The susceptibility of apoAI KO mice to atherosclerosis confirmed previous notions about this apolipoprotein in atheroprotection. Similarly, the susceptibility of LDL-receptor KO and apoE KO mice to atherosclerosis substantiated previous hypotheses about the atheroprotective qualities of these proteins. The case has not been so clear for genetically altered mice in which apoAII has been overexpressed.
The earliest studies of apoAII overexpression on a background of human apoAI indicated that apoAII ablated the atheroprotective effects of apoAI [46]. On the other hand, other studies suggested that expression of human apoAII in mice is atheroprotective [47]. Subsequent studies used overexpression of human apoAII to identify its effects on atherogenesis. Transgenic overexpression of human (dimeric) apoAII in mice resulted in exclusive hepatic mRNA expression and the appearance of the protein in plasma [24]. The plasma cholesterol concentration, mostly as HDL-C, was lower in transgenic versus control mice. Plasma apoAI and mouse apoAII concentrations were inversely correlated with the levels of human plasma apoAII. The highest expressers of apoAII were mildly hypertriglyceridemic. Compared with control mice, those expressing the highest plasma apoAII levels (74 mg/dl) exhibited an additional small HDL species.
Notably, endogenous plasma LCAT activity was profoundly reduced in the apoAII-overexpressing mice, whereas activity against exogenous substrate was not and, remarkably, replacing the chow diet with a high-fat diet normalized plasma HDL concentrations. These two observations provoke a question about the atherogenicity of high-level apoAII expression. Would the inhibition of a key RCT step lead to more atherosclerosis or would the HDL-C-increasing effects of the high-fat diet be atheroprotective? This was tested by comparing the apoAII-overexpressing mice with control mice following an atherogenic diet. These tests showed that the transgenic mice presented with greater atherosclerotic lesion areas, which were up to sevenfold more extensive than those of nontransgenic control mice. Moreover, the severity of atherosclerosis increased with increasing apoAII expression, thereby supporting the hypothesis that apoAII is an atherogenic lipoprotein. Paradoxically, the lesion areas of transgenic and control mice fed a regular chow diet were similar. The essence of this finding was later corroborated by others [48].
Effects of apoAII on VLDL metabolism
CETP activity sometimes makes mice a poor model for studying lipoprotein metabolism, including that of apoAII. Interestingly, mice expressing human apoAII and CETP have an unexpected phenotype. On a chow diet, the addition of the apoAII transgene to a human CETP background produced significant changes in plasma concentrations of HDL-C (−60%), HDL–TG (+130%), TG (+~500%), free fatty acids (+14%) and endogenous LCAT activity (−80%). On a high-fat atherogenic diet, addition of the apoAII transgene to a human CETP background produced the following changes in plasma concentrations: total cholesterol (+100%), HDL-C (−50%), HDL–TG (+1000%), TG (+~10,000%), free fatty acids (+100%) and endogenous LCAT activity (−55%). On both diets, the fraction of cholesterol occurring in the unesterified form was increased by the addition of the apoAII transgene. Importantly, expression of the apoAII transgene in CETP transgenic mice delayed TG clearance (−30%). Addition of the apoAII transgene to the CETP transgene produced a twofold increase in atherosclerotic lesion areas in male mice. A similar comparison of female mice showed no significant differences. The observed phenotype likely stems from the elevation in plasma free fatty acids, which are extracted by the liver for VLDL–TG production. However, the increase in plasma free fatty acids is small (+28 mg/dl; +100%) compared with the increase in plasma TG (+500 mg/dl; +10,000%). This difference is readily understood considering that many pools of fatty acid, which have a plasma life time in the order of minutes, pass through the liver during the plasma life time of VLDL–TG (approximately 1 h). In the presence of hypertriglyceridemia, the HDL are made TG-rich and cholesterolpoor by CETP activity. In the context of this model, one open question is, `What is the underlying cause of the elevation in plasma free fatty acids in the double KO mice?' Although the authors cited several possible mechanisms as the underlying cause of the phenotype, the most attractive possibility best supported by data is that apoAII increases adipose tissue lipolysis by direct and/or indirect mechanisms.
ApoAII atheroprotects in rabbits
Economics and mature genetic technologies have made studies in mice the first option in determining the roles of various proteins in many diseases, including atherosclerosis. However, mice have several well-known model deficiencies. First, mice, like most other rodents, have high endogenous HDL concentrations, an obvious athero protective confounder. Second, mice express apoAI and apoAII; the plasma concentrations of both are reduced by apoAII overexpression; moreover, mouse apoAII is monomeric. Lastly, mice do not express CETP, so that any athero protective mechanism in which the transfer of cholesteryl esters from HDL to the apoB-containing lipoproteins is important would be missed. An alternative model is the apoAII overexpression in rabbits [49]. Rabbit plasma contains CETP but not apoAII, so overexpression of apoAII in rabbits is likely a more valid test of apoAII-induced effects on atherogenesis. The phenotype for rabbits overexpressing apoAII has been reported [50]. The plasma apoAII levels are similar to those of humans (30 mg/dl); plasma concentrations of TG, total cholesterol and phospholipids were increased, while those for HDL-C were similar to those of nontransgenic littermates. In addition, the apoAII-overexpressing rabbits had mild dyslipidemia with elevated VLDL, and intermediate-density lipoprotein concentrations and post heparin lipoprotein lipase activity was profoundly decreased compared with nontransgenic rabbits. Collectively, this lipoprotein profile emulates that for human combined hyperlipidemia, which is associated with atherosclerosis. A similar phenotype was observed for apoE KO mice overexpressing apoAII [51], perhaps via inhibition of lipoprotein lipase activity. ApoAII was found in all HDL fractions of the apoAII-overexpressing rabbits, and was most enriched in smaller HDL particles and with a lower apoAI content. Interestingly, unlike in normal rabbits, the plasma HDL-C concentration was not reduced when the apoAII-overexpressing rabbits were placed on a high-fat diet. Under these conditions, much of the HDL had pre-β mobility.
The relative susceptibility of human apoAII transgenic versus control rabbits to CVD was tested by feeding each an atherogenic diet containing approximately 0.3% cholesterol and 3% soybean oil for 16 weeks [52]. By nearly every measure of atherosclerosis – aortic and coronary lesion area, and lesion macrophage and smooth muscle cell counts – the apoAII-overexpressing rabbits had less atherosclerotic disease. Interestingly, apoAII immunoreactive proteins were readily detected in the lesions, although the relative amounts were not quantified. According to the well-accepted experimental criteria, the functionality of HDL from apoAII-transgenic rabbits was only modestly better than that of nontransgenic sibling controls. When normalized to HDL protein concentration, cholesterol efflux from macrophages to isolated HDL2 and HDL3 was slightly better for transgenic rabbits versus controls. The higher content of HDL–phospholipids (the essential cholesterol-binding component of all lipoproteins) and the higher plasma levels of pre-β-HDL, which are superior acceptors of cellular cholesterol efflux, could account for the improved efflux. HDL from transgenic rabbits was also somewhat better than HDL from control rabbits in suppressing cytokine mRNA levels in lipopolysaccharide-induced macrophages. Moreover, plasma CRP levels and neutrophil and monocyte counts in the blood of transgenic rabbits on the high-fat diet were much lower than those of control rabbits, suggesting that apoAII reduces inflammatory status. ApoAII overexpression also reduced the oxidizability of β-VLDL, an effect that may be mediated by the association of some apoAII with apoB lipoproteins.
The reduction in atherosclerosis by apoAII overexpression is profound. Gross lesion areas were reduced in both the thoracic and abdominal aorta, with the total reduction being more than 50%. Stenosis of the left and right coronary arteries was also impressively reduced, as were the macrophage- and smooth muscle cell-positive areas. The preponderance of data suggest that apoAII has potential therapeutic value at the level of the arterial wall. Given the profound reduction in atherosclerosis and the small improvement in HDL functionality, one might validly hypothesize that the effects of apoAII on lesion reduction do not involve the direct interaction of HDL or any other lipoprotein with the arterial wall. Conversely, the appearance of apoAII in the lesions suggests that some other mechanism involving the direct interaction of apoAII with the proatherogenic components of the subendothelial space may be important. This, of course, is readily testable by biochemical and immunological means.
ApoAII as a therapeutic target
In 1994, Hedrick and Lusis reviewed the apoAII literature in a paper titled, `Apolipoprotein A-II: a protein in search of a function' [53]. In 2001, Blanco-Vaca et al. also reviewed the apoAII literature, this time with a paper titled, `Role of apoAII in lipid metabolism and atherosclerosis: advances in the study of an enigmatic protein' [54]. Although some progress has been made in the intervening years, our perspective on the role of apoAII in lipid metabolism and especially athero genesis, has been changed little, and remains confused by conflicting data from various laboratories, models and mice. As a consequence, there is little momentum in any direction, whether dyslipidemic versus normolipidemic, atheroprotective versus atherogenic or mouse versus man. Nevertheless, the atheroprotective effects of apoAII merit testing. As with studies of apoAI and apoAIMilano [55–57], one could reasonably begin with tests in which isolated apoAII or apoAII in rHDL is infused into a mouse model of atherosclerosis. Alternatively, one might consider infusion of an apoAII fragment. The essential and distinctive physiological properties of the exchangeable apolipoproteins are coded by exon 4. This exon in the genes for apoAI, apoC-II and apoE, codes respectively for the protein regions that activate LCAT, stimulate lipoprotein lipase activity and target apoE-containing lipoproteins to multiple hepatic receptors. Exon 4 of apoAII codes for amino acid residues 40–77 [58], which corresponds to a 38-residue peptide that could be readily produced by solid-phase peptide synthesis. Previous studies have shown that this region of apoAII, but not residues 1–26, associate with phospholipids [59,60]. Moreover, this region of the protein has both observed and predicted a-helical content and hydrophobic moment as do other phospholipid-binding regions of plasma apolipoproteins [13,59–61]. On the other hand, even though plasma apoAII concentrations predict CVD events, there is no assurance that increasing apoAII plasma concentration will be therapeutic. ApoAII, like HDL-C, may be a biomarker, a weak one at that, for a parallel or underlying mechanism that is atheroprotective. Like HDL-C, the level of apoAII may not be as important as how it got that way; mechanism is important and the mechanism(s) that determine apoAII plasma concentration may be the compass to setting the course to develop the therapeutic potential of this apolipoprotein.
Conclusion
Unlike apoAII from most other species, human plasma apoAII is a dimer that is more lipophilic than apoAI and apoAII monomers. ApoAII follows a biosynthetic pathway of lipidation and dimerization that is distinct from those of the other major HDL apolipoproteins, apoAI, apoE and apoCs. Despite some conflicting data using a variety of approaches – cell based, epidemiological, biochemical and mouse models – the preponderance of evidence supports the hypothesis that dimeric apoAII is antiatherogenic. Given the dearth of successful HDL therapies and the need to develop such therapies to complement statin therapy in some individuals, or in place of statin therapy in statin-sensitive and -resistant patients, it is worth testing apoAII and its mimetics as lipid-free species or in rHDL in nonmouse models. As the second most abundant protein of human HDL, apoAII should not be ignored as a therapeutic target for atherosclerosis and the appropriate tests should be completed.
Future perspective
With few other options, apoAII will be subjected to additional tests that determine how its bio synthesis can be controlled. This would begin with cell-based studies in hepatic cell lines and, more importantly, in human hepatocytes. While the effects on plasma lipid and lipoprotein analytes were modest, given the profound antiatherogenic effects of apoAII overexpression on atherosclerosis in a nonmouse model, the rabbit, it is worth testing whether apoAII has direct effects on the biochemistry of cells comprising the arterial wall (i.e., endothelial cells, smooth muscle cells and monocyte-derived macrophages). Moreover, the profound effect of apoAII on plasma TG levels begs the question of apoAII effects on adipocyte lipolysis. Initial tests would compare apoAII and its rHDL, and if warranted, subsequent studies would determine whether the antiatherogenic effects are confined to a specific region of the protein. This would be followed by infusion of apoAII or its peptides as isolated protein or as rHDL into rabbits to test whether the findings with apoAII transgenic rabbits are duplicated. Lastly, if the cell and animal-based studies, including studies of safety and efficacy in nonhuman primates, support an antiatherogenic role for apoAII, human studies of atheroregression with apoAII and rHDL (apoAII) could be justified. Initiation of these studies would require a better expression system for apoAII that is appropriate to the larger amounts that would eventually be needed.
Executive summary.
ApoAII structure
-
■
Unlike apoAII from most other species, human apoAII, the second most abundant HDL protein, occurs in plasma largely as a homodimer of 17.4 kDa.
ApoAII properties
-
■
The lipid-associating region of apoAII resides in the region of the protein that is coded by exon 4; this region comprises residues 40–77.
-
■
ApoAII is the most lipophilic of the exchangeable apolipoproteins and, unlike apoAI, is not converted to its lipid-free form by plasma lipid transfer proteins and enzyme activities associated with HDL.
ApoAII biosynthesis
-
■
ApoAII plasma levels are determined by synthesis, in response to PPAR/RXR elements.
-
■
ApoAII dimerization is catalyzed by phospholipids, and all apoAII are dimeric and lipidated intrahepatically.
-
■
Following hepatic secretion, LCAT catalyzes the fusion of nascent apoAII-only HDL with apoAI–HDL.
ApoAII & atheroprotection
-
■
Early studies of apoAII overexpression in mice gave rise to an LCAT-deficient state.
-
■
According to various models and laboratory methodology, apoAII has been shown to be both pro- and anti-atherogenic.
-
■
Recent studies in rabbits demonstrated that apoAII overexpression is antiatherogenic, without producing profound changes in plasma lipid and lipoprotein concentrations usually associated with such an outcome.
-
■
Studies of the antiatherogenic effects of apoAII in cells and animal models (including nonhuman primates) would have to be completed and show that apoAII has an antiatherogenic effect before studies in humans could be proposed.
Acknowledgments
This work was supported by grants-in-aid from the NIH (HL-30914 and HL-56865 to HJ Pownall) and by the Bass Foundation.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Havel RJ, Goldstein JL, Brown MS. Lipoproteins and lipid transport. In: Bondy PK, Rosenberg LE, editors. The Metabolic Control of Disease. Saunders Publishing, PA, USA; 1980. pp. 398–494. [Google Scholar]
- 2.Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34(4):679–697. doi: 10.1161/01.cir.34.4.679. [DOI] [PubMed] [Google Scholar]
- 3.Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988;8(6):737–741. doi: 10.1161/01.atv.8.6.737. [DOI] [PubMed] [Google Scholar]
- 4.Glomset JA, Parker F, Tjaden M, Williams RH. The esterification in vitro of free cholesterol in human and rat plasma. Biochim. Biophys. Acta. 1962;58:398–406. doi: 10.1016/0006-3002(62)90050-1. [DOI] [PubMed] [Google Scholar]
- 5.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 1968;9(2):155–167. [PubMed] [Google Scholar]
- 6.Rosenson RS, Brewer HB, Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125(15):1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendez AJ, Anantharamaiah GM, Segrest JP, Oram JF. Synthetic amphipathic helical peptides that mimic apolipoprotein A-I in clearing cellular cholesterol. J. Clin. Invest. 1994;94(4):1698–1705. doi: 10.1172/JCI117515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fielding CJ, Shore VG, Fielding PE. A protein cofactor of lecithin:cholesterol acyltransferase. Biochem. Biophys. Res. Commun. 1972;46(4):1493–1498. doi: 10.1016/0006-291x(72)90776-0. [DOI] [PubMed] [Google Scholar]
- 9.Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Anti-inflammatory properties of HDL. Circ. Res. 2004;95(8):764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 10.Yamashita J, Iwamura C, Sasaki T, et al. Apolipoprotein A-II suppressed concanavalin A-induced hepatitis via the inhibition of CD4 T cell function. J. Immunol. 2011;186(6):3410–3420. doi: 10.4049/jimmunol.1002924. [DOI] [PubMed] [Google Scholar]
- 11.Shore B, Shore V. Heterogeneity in protein subunits of human serum high-density lipoproteins. Biochemistry. 1968;7(8):2773–2777. doi: 10.1021/bi00848a011. [DOI] [PubMed] [Google Scholar]
- 12.Lux SE, John KM, Brewer HB., Jr Isolation and characterization of apoLp-Gln-II (apoA-II), a plasma high density apolipoprotein containing two identical polypeptide chains. J. Biol. Chem. 1972;247(23):7510–7518. [PubMed] [Google Scholar]
- 13.Li WH, Tanimura M, Luo CC, Datta S, Chan L. The apolipoprotein multigene family: biosynthesis, structure, structure–function relationships, and evolution. J. Lipid Res. 1988;29(3):245–271. [PubMed] [Google Scholar]
- 14.Byrne RE, Polacek D, Gordon JI, Scanu AM. The enzyme that cleaves apolipoprotein A-II upon in vitro incubation of human plasma high-density lipoprotein-3 with blood polymorphonuclear cells is an elastase. J. Biol. Chem. 1984;259(23):14537–14543. [PubMed] [Google Scholar]
- 15.Davidson WS, Silva RA. Apolipoprotein structural organization in high density lipoproteins: belts, bundles, hinges and hairpins. Curr. Opin. Lipidol. 2005;16(3):295–300. doi: 10.1097/01.mol.0000169349.38321.ad. [DOI] [PubMed] [Google Scholar]
- 16.Silva RA, Schneeweis LA, Krishnan SC, Zhang X, Axelsen PH, Davidson WS. The structure of apolipoprotein A-II in discoidal high density lipoproteins. J. Biol. Chem. 2007;282(13):9713–9721. doi: 10.1074/jbc.M610380200. [DOI] [PubMed] [Google Scholar]
- 17.Birjmohun RS, Dallinga-Thie GM, Kuivenhoven JA, et al. Apolipoprotein A-II is inversely associated with risk of future coronary artery disease. Circulation. 2007;116(18):2029–2035. doi: 10.1161/CIRCULATIONAHA.107.704031. [DOI] [PubMed] [Google Scholar]
- 18.Blanco-Vaca F, Via DP, Yang CY, Massey JB, Pownall HJ. Characterization of disulfide-linked heterodimers containing apolipoprotein D in human plasma lipoproteins. J. Lipid Res. 1992;33(12):1785–1796. [PubMed] [Google Scholar]
- 19.Weisgraber KH, Shinto LH. Identification of the disulfide-linked homodimer of apolipoprotein E3 in plasma. Impact on receptor binding activity. J. Biol. Chem. 1991;266(18):12029–12034. [PubMed] [Google Scholar]
- 20.Cheung MC, Albers JJ. Characterization of lipoprotein particles isolated by immunoaffinity chromatography. Particles containing A-I and A-II and particles containing A-I but no A-II. J. Biol. Chem. 1984;259(19):12201–12209. [PubMed] [Google Scholar]
- 21.Lund-Katz S, Murley YM, Yon E, Gillotte KL, Davidson WS. Comparison of the structural and functional effects of monomeric and dimeric human apolipoprotein A-II in high density lipoprotein particles. Lipids. 1996;31(11):1107–1113. doi: 10.1007/BF02524284. [DOI] [PubMed] [Google Scholar]
- 22.Edelstein C, Halari M, Scanu AM. On the mechanism of the displacement of apolipoprotein A-I by apolipoprotein A-II from the high density lipoprotein surface. Effect of concentration and molecular forms of apolipoprotein A-II. J. Biol. Chem. 1982;257(12):7189–7195. [PubMed] [Google Scholar]
- 23.Pownall HJ, Hickson D, Gotto AM., Jr Thermodynamics of lipid-protein association. The free energy of association of lecithin with reduced and carboxymethylated apolipoprotein A-II from human plasma high density lipoprotein. J. Biol. Chem. 1981;256(19):9849–9854. [PubMed] [Google Scholar]
- 24.Marzal-Casacuberta A, Blanco-Vaca F, Ishida BY, et al. Functional lecithin:cholesterol acyltransferase deficiency and high density lipoprotein deficiency in transgenic mice overexpressing human apolipoprotein A-II. J. Biol. Chem. 1996;271(12):6720–6728. doi: 10.1074/jbc.271.12.6720. [DOI] [PubMed] [Google Scholar]
- 25.Hussain MM, Zannis VI. Intracellular modification of human apolipoprotein AII (apoAII) and sites of apoAII mRNA synthesis: comparison of apoAII with apoCII and apoCIII isoproteins. Biochemistry. 1990;29(1):209–217. doi: 10.1021/bi00453a029. [DOI] [PubMed] [Google Scholar]
- 26.Bossu JP, Chartier FL, Vu-Dac N, Fruchart JC, Laine B. Transcription of the human apolipoprotein A-II is down-regulated by the first intron of its gene. Biochem. Biophys. Res. Commun. 1994;202(2):822–829. doi: 10.1006/bbrc.1994.2004. [DOI] [PubMed] [Google Scholar]
- 27.Vu-Dac N, Schoonjans K, Kosykh V, et al. Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J. Clin. Invest. 1995;96(2):741–750. doi: 10.1172/JCI118118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vu-Dac N, Schoonjans K, Kosykh V, et al. Retinoids increase human apolipoprotein A-11 expression through activation of the retinoid X receptor but not the retinoic acid receptor. Mol. Cell Biol. 1996;16(7):3350–3360. doi: 10.1128/mcb.16.7.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikewaki K, Zech LA, Kindt M, Brewer HB, Jr, Rader DJ. Apolipoprotein A-II production rate is a major factor regulating the distribution of apolipoprotein A-I among HDL subclasses LpA-I and LpA-I:A-II in normolipidemic humans. Arterioscler. Thromb. Vasc. Biol. 1995;15(3):306–312. doi: 10.1161/01.atv.15.3.306. [DOI] [PubMed] [Google Scholar]
- 30.Chisholm JW, Burleson ER, Shelness GS, Parks JS. ApoA-I secretion from HepG2 cells: evidence for the secretion of both lipid-poor apoA-I and intracellularly assembled nascent HDL. J. Lipid Res. 2002;43(1):36–44. [PubMed] [Google Scholar]
- 31.Gillard BK, Lin HY, Massey JB, Pownall HJ. Apolipoproteins A-I, A-II and E are independently distributed among intracellular and newly secreted HDL of human hepatoma cells. Biochim. Biophys. Acta. 2009;1791(12):1125–1132. doi: 10.1016/j.bbalip.2009.07.004. ■ Demonstrates that apoAII occurs on HDL particles that have no apoAI, and that hepatically derived factors catalyze fusion of apoAII-containing particles with those containing apoAI.
- 32.Maric J, Kiss RS, Franklin V, Marcel YL. Intracellular lipidation of newly synthesized apolipoprotein A-I in primary murine hepatocytes. J. Biol. Chem. 2005;280(48):39942–39949. doi: 10.1074/jbc.M507733200. [DOI] [PubMed] [Google Scholar]
- 33.Gillard BK, Chen YS, Gaubatz JW, Massey JB, Pownall HJ. Plasma factors required for human apolipoprotein A-II dimerization. Biochemistry. 2005;44(2):471–479. doi: 10.1021/bi048591j. [DOI] [PubMed] [Google Scholar]
- 34.Massey JB, Pownall HJ. Cholesterol is a determinant of the structures of discoidal high density lipoproteins formed by the solubilization of phospholipid membranes by apolipoprotein A-I. Biochim. Biophys. Acta. 2008;1781(5):245–253. doi: 10.1016/j.bbalip.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bassett GR, Gillard BK, Pownall HJ. Cholesterol determines and limits rHDL formation from human plasma apolipoprotein A-II and phospholipid membranes. Biochemistry. 2012;51(43):8627–8635. doi: 10.1021/bi3011994. ■ Demonstrates that the free cholesterol content of membranes determines the size of reconstituted HDL particles formed by microsolubilization; increased free cholesterol in membranes leads to an increase in the size of reconstituted HDL.
- 36.Shepherd J, Packard CJ, Gotto AM, Jr, Taunton OD. A comparison of two methods to investigate the metabolism of human apolipoproteins A-I and and A-II. J. Lipid Res. 1978;19(5):656–661. [PubMed] [Google Scholar]
- 37.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J. Mol. Biol. 2003;328(1):183–192. doi: 10.1016/s0022-2836(03)00155-4. ■■ Reveals, by a variety of physicochemical methods, that plasma HDL particles reside in a high-energy kinetic trap from which they escape by chemical perturbations, which emulate the effects of plasma enzymes and transfer proteins.
- 38.Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J. Biol. Chem. 1997;272(7):3953–3960. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- 39.Lusa S, Jauhiainen M, Metso J, Somerharju P, Ehnholm C. The mechanism of human plasma phospholipid transfer protein-induced enlargement of high-density lipoprotein particles: evidence for particle fusion. Biochem. J. 1996;313(Pt 1):275–282. doi: 10.1042/bj3130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang HQ, Rye KA, Barter PJ. Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J. Lipid Res. 1996;37(9):1962–1970. [PubMed] [Google Scholar]
- 41.Clay MA, Newnham HH, Barter PJ. Hepatic lipase promotes a loss of apolipoprotein A-I from triglyceride-enriched human high density lipoproteins during incubation in vitro. Arterioscler. Thromb. 1991;11(2):415–422. doi: 10.1161/01.atv.11.2.415. [DOI] [PubMed] [Google Scholar]
- 42.Gillard BK, Courtney HS, Massey JB, Pownall HJ. Serum opacity factor unmasks human plasma high-density lipoprotein instability via selective delipidation and apolipoprotein A-I desorption. Biochemistry. 2007;46(45):12968–12978. doi: 10.1021/bi701525w. ■■ Demonstrates that a bacterial protein, serum opacity factor, catalyzes the escape of HDL from a kinetic trap, thereby forming a large cholesteryl ester-rich particle, a small cholesterol-deficient HDL and lipid-free apoAI.
- 43.Remaley AT, Stonik JA, Demosky SJ, et al. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 2001;280(3):818–823. doi: 10.1006/bbrc.2000.4219. [DOI] [PubMed] [Google Scholar]
- 44.Miller NE. Associations of high-density lipoprotein subclasses and apolipoproteins with ischemic heart disease and coronary atherosclerosis. Am. Heart J. 1987;113(2 Pt 2):589–597. doi: 10.1016/0002-8703(87)90638-7. [DOI] [PubMed] [Google Scholar]
- 45.Deeb SS, Takata K, Peng RL, Kajiyama G, Albers JJ. A splice-junction mutation responsible for familial apolipoprotein A-II deficiency. Am. J. Hum. Genet. 1990;46(4):822–827. [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz JR, Verstuyft JG, Gong EL, Nichols AV, Rubin EM. Protein composition determines the anti-atherogenic properties of HDL in transgenic mice. Nature. 1993;365(6448):762–764. doi: 10.1038/365762a0. [DOI] [PubMed] [Google Scholar]
- 47.Schultz JR, Rubin EM. The properties of HDL in genetically engineered mice. Curr. Opin. Lipidol. 1994;5(2):126–137. doi: 10.1097/00041433-199404000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Tailleux A, Bouly M, Luc G, et al. Decreased susceptibility to diet-induced atherosclerosis in human apolipoprotein A-II transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2000;20(11):2453–2458. doi: 10.1161/01.atv.20.11.2453. [DOI] [PubMed] [Google Scholar]
- 49.Remaley AT. Apolipoprotein A-II: still second fiddle in high-density lipoprotein metabolism? Arterioscler. Thromb. Vasc. Biol. 2013;33(2):166–167. doi: 10.1161/ATVBAHA.112.300921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koike T, Kitajima S, Yu Y, et al. Expression of human apoAII in transgenic rabbits leads to dyslipidemia: a new model for combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2009;29(12):2047–2053. doi: 10.1161/ATVBAHA.109.190264. [DOI] [PubMed] [Google Scholar]
- 51.Escola-Gil JC, Julve J, Marzal-Casacuberta A, Ordonez-Llanos J, Gonzalez-Sastre F, Blanco-Vaca F. Expression of human apolipoprotein A-II in apolipoprotein E-deficient mice induces features of familial combined hyperlipidemia. J. Lipid Res. 2000;41(8):1328–1338. [PubMed] [Google Scholar]
- 52.Wang Y, Niimi M, Nishijima K, et al. Human apolipoprotein A-II protects against diet-induced atherosclerosis in transgenic rabbits. Arterioscler. Thromb. Vasc. Biol. 2013;33(2):224–231. doi: 10.1161/ATVBAHA.112.300445. ■■ According to this paper, transgenic expression of apoAII in rabbits, which do not naturally express apoAII, induces profound reduction in almost every measure of atherosclerosis.
- 53.Hedrick CC, Lusis AJ. Apolipoprotein A-II: a protein in search of a function. Can. J. Cardiol. 1994;10(4):453–459. [PubMed] [Google Scholar]
- 54.Blanco-Vaca F, Escola-Gil JC, Martin-Campos JM, Julve J. Role of apoA-II in lipid metabolism and atherosclerosis: advances in the study of an enigmatic protein. J. Lipid Res. 2001;42(11):1727–1739. [PubMed] [Google Scholar]
- 55.Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant apoA-IMilano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290(17):2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 56.Nicholls SJ, Tuzcu EM, Sipahi I, et al. Relationship between atheroma regression and change in lumen size after infusion of apolipoprotein A-IMilano. J. Am. Coll. Cardiol. 2006;47(5):992–997. doi: 10.1016/j.jacc.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 57.Tardif JC, Gregoire J, L'Allier PL, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297(15):1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 58.Lackner KJ, Law SW, Brewer HB., Jr The human apolipoprotein A-II gene: complete nucleic acid sequence and genomic organization. Nucleic Acids Res. 1985;13(12):4597–4608. doi: 10.1093/nar/13.12.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson RL, Gotto AM, Jr, Lux SE, John KM, Fleischer S. Human plasma high density lipoprotein. Interaction of the cyanogen bromide fragments from apolipoprotein glutamine II (A-II) with phosphatidylcholine. J. Biol. Chem. 1973;248(24):8449–8456. [PubMed] [Google Scholar]
- 60.Mao SJ, Sparrow JT, Gilliam EB, Gotto AM, Jr, Jackson RL. Mechanism of lipid-protein interaction in the plasma lipoproteins: lipid-binding properties of synthetic fragments of apolipoprotein A-II. Biochemistry. 1977;16(19):4150–4156. doi: 10.1021/bi00638a003. [DOI] [PubMed] [Google Scholar]
- 61.Pownall HJ, Gotto AM, Jr, Knapp RD, Massey JB. The helical hydrophobic moment avoids prolines in phospholipid-binding proteins. Biochem. Biophys. Res. Commun. 1986;139(1):202–208. doi: 10.1016/s0006-291x(86)80099-7. [DOI] [PubMed] [Google Scholar]