

Abstract
Candida albicans is a major fungal pathogen that causes serious systemic and mucosal infections in immunocompromised individuals. In yeast, histone H3 Lys56 acetylation (H3K56ac) is an abundant modification regulated by enzymes that have fungal-specific properties, making them appealing targets for antifungal therapy. Here we demonstrate that H3K56ac in C. albicans is regulated by the RTT109 and HST3 genes, which respectively encode the H3K56 acetyltransferase (Rtt109p) and deacetylase (Hst3p). We show that reduced levels of H3K56ac sensitize C. albicans to genotoxic and antifungal agents. Inhibition of Hst3p activity by conditional gene repression or nicotinamide treatment results in a loss of cell viability associated with abnormal filamentous growth, histone degradation and gross aberrations in DNA staining. We show that genetic or pharmacological alterations in H3K56ac levels reduce virulence in a mouse model of C. albicans infection. Our results demonstrate that modulation of H3K56ac is a unique strategy for treatment of C. albicans and, possibly, other fungal infections.
H3K56ac is an abundant post-translational modification found in newly synthesized H3 molecules deposited throughout the genome during DNA replication1,2. Originally discovered in yeast2–6, H3K56ac also occurs in human cells7–10. In Saccharomyces cerevisiae, H3K56ac peaks when new histones are synthesized during S phase2 and is mediated by the histone acetyltransferase Rtt109p11–13. H3K56ac is removed genome-wide by the histone deacetylases Hst3p and Hst4p during the G2 and M phases1,14. Tightly regulated H3K56ac plays a key part in the DNA damage response, as mutants that cannot acetylate or deacetylate Lys56 are extremely sensitive to genotoxic agents1,2,12,14,15.
C. albicans is a major human fungal pathogen in terms of both its clinical importance and its use as an experimental model for fungal pathogenesis16,17. It is one of the leading causes of infections affecting immunodeficient individuals, including HIV-infected individuals and patients undergoing cancer therapy. C. albicans is the fourth most common cause of nosocomial bloodstream infections in the US and is associated with high mortality rates18–20. Treatment of candidiasis involves the use of azole or echinocandin drugs that target ergosterol and cell wall biosynthesis, respectively, but their efficacy can be compromised by the emergence of drug-resistant strains21,22. As the fungal enzymes that regulate H3K56ac have diverged considerably from their human counterparts23,24, we sought to determine whether these enzymes could be potential targets for treatment of C. albicans infections.
RESULTS
H3K56ac modulates fungicidal agent sensitivity in C. albicans
We first verified the presence of H3K56ac in C. albicans. Immunoblots of total lysates from SC5314 and SN152 strains probed with an antibody against H3K56ac1,2 revealed a band of the expected molecular weight (~15 kDa) (Fig. 1a). We also isolated histones from C. albicans cells and analyzed them by mass spectrometry. The product ion spectrum of a doubly protonated precursor peptide (m/z 638.92+) shows a y-type fragment ion series that confirms acetylation of Lys56 on the basis of the the 42-Da mass shift between the y7 and y8 fragment ions (Fig. 1b). We also estimated the stoichiometry of H3K56ac by derivatization of nonmodified lysine ε-amino groups with deuterated acetic anhydride before trypsin digestion1,2. In asynchronously growing cultures, the Lys56-acetylated peptide was present in 20% to 27% of H3 molecules (Fig. 1c,d). Thus, H3K56ac is an abundant modification in C. albicans. This is in stark contrast to the low stoichiometry of H3K56ac (1%) in human cells7,9.
Figure 1.

H3K56 acetylation modulates genotoxic and fungicidal agent sensitivity in C. albicans. (a) Immunoblot of H3K56ac and total H3 in whole-cell lysates from exponentially growing populations of each strain. (b) Fragmentation spectrum of precursor ion m/z 638.92+ showing the presence of H3K56ac. (c) Extracted ion chromatograms for m/z 638.92+ (H3K56ac) and its deuterated analog m/z 640.42+ (H3K56-D3ac). (d) Stoichiometry of H3K56ac determined by mass spectrometry for histones purified from exponentially growing cells. ε-NH2 of Lys56 is modified with either an acetyl (ac) or a deuterated acetyl (pr) group. (e) Colony formation assays (tenfold serial dilutions of each strain). YPD, control; HU, hydroxyurea; MMS, methyl methane sulfonate; FLC, fluconazole; CSF, caspofungin; MCF, micafungin; NAC, N-acetyl-L-cysteine; VitC, vitamin C.
The RTT109 gene encodes an acetyltransferase responsible for H3K56ac11–13; its ortholog in C. albicans is orf19.7491 (RTT109). Deletion of both alleles of RTT109 in strain SN152 (Supplementary Fig. 1) led to nearly complete disappearance of H3K56ac on the basis of immunoblots and mass spectrometry (Fig. 1a,d and Supplementary Table 1). Deacetylation of H3K56 requires the sirtuins Hst3p and Hst4p in S. cerevisiae. Deletion of both genes results in DNA damage sensitivity and thermosensitivity, but the double mutant remains viable1,14,15. We only found one gene in C. albicans (orf19.1934; HST3) encoding a potential ortholog of S. cerevisiae HST3 and HST4. We generated a heterozygous deletion mutant for HST3 in SN152, but were unable to obtain an hst3Δ/hst3Δ (hst3Δ/Δ hereafter) homozygous mutant, suggesting that the C. albicans HST3 gene is essential. Although immunoblots of whole-cell extracts from hst3Δ/HST3 (HST3+/Δ hereafter) cells failed to detect an increase in H3K56ac (Fig. 1a), the stoichiometry of H3K56ac monitored by quantitative mass spectrometry was twofold higher than in wild-type cells (Fig. 1d and Supplementary Table 1).
We studied the survival of C. albicans rtt109 and hst3 mutants treated with genotoxic and antifungal agents. Deletion of both alleles of RTT109 conferred sensitivity to hydroxyurea and methyl methane sulfonate (Fig. 1e). HST3+/Δ heterozygous mutants were also sensitive to these agents (Fig. 1e). We also tested the susceptibility of these mutants to two families of antifungal agents: azoles (fluconazole) and echinocandins (caspofungin, micafungin). We found that rtt109Δ/ rtt109Δ (rtt109Δ/Δ hereafter) and HST3+/Δ cells were no more susceptible to fluconazole than wild-type cells, but the rtt109Δ/Δ strain was very sensitive to both echinocandins (Fig. 1e). Because echinocandins induce oxidative stress25, we tested whether the echinocandin sensitivity of rtt109Δ/Δ mutants could be suppressed by antioxidants. The antioxidants N-acetyl-L-cysteine and vitamin C restored viability of rtt109Δ/Δ mutant cells exposed to micafungin (Fig. 1e). These results show that genetic perturbation of H3K56ac sensitizes C. albicans to genotoxic and antifungal drugs.
H3K56 deacetylation by Hst3p is essential in C. albicans
Because we were unable to obtain a homozygous hst3Δ/Δ mutant, we generated strains in the CaSS1 background26, in which we deleted one copy of the HST3 gene and placed the other copy under the control of a promoter that can be repressed with doxycycline (hst3Δ/pTET-HST3, Supplementary Methods and Supplementary Fig. 2c–e). Addition of doxycycline to hst3Δ/pTET-HST3 cells resulted in a strong decrease in HST3 mRNA levels and a concomitant increase in H3K56ac (Fig. 2a,b). To confirm this result, we isolated histones either before or after doxycycline addition and determined the stoichiometry of modification by mass spectrometry. We found that the fraction of H3 molecules that were Lys56-acetylated increased from 48% to 87% when we treated the hst3Δ/pTET-HST3 strain with doxycycline for 7.5 h (Fig. 2c and Supplementary Table 1). The degree of acetylation of other lysine residues in the N-terminal tail of H3 did not increase upon HST3 repression with doxycycline (Supplementary Table 1). To obtain equal amounts of histone H3 for immunoblotting, we needed to load increasing amounts of total protein as a function of time after doxycycline addition (Fig. 2b). Immunoblots of whole-cell lysates from equal numbers of cells confirmed that the amounts of both H3 and H4 declined over time after doxycycline addition to hst3Δ/pTET-HST3 cells (Fig. 2b). This time-dependent loss of H3 and H4 suggests that HST3 repression leads to pronounced changes in chromatin structure.
Figure 2.

HST3 controls H3K56 deacetylation and is required for cell viability in C. albicans. (a) Repression of HST3 mRNA by doxycycline treatment, as measured by quantitative RT-PCR. Results are the mean of three experiments ± s.e.m. (b) Immunoblots of lysates from hst3Δ/ pTET-HST3 cells treated with doxycycline (+Doxy, 40 μg ml−1) or left untreated (−Doxy). Equal amounts of histone H3 (top) or of total proteins (bottom) were loaded for each time point. (c) Stoichiometry of H3K56ac determined by mass spectrometry of histones purified from hst3Δ/pTET-HST3 cells grown in the absence or presence of doxycycline (duplicates). (d) Growth of hst3Δ/pTET-HST3 cells on plates containing doxycycline (50 μg ml−1) or control plates. CASS1, wild-type strain. (e) Growth of hst3Δ/ pTET-HST3 cells transiently exposed to doxycycline (50 μg ml−1) for 24 h and plated on YPD lacking doxycycline. (f) Lethality of hst3Δ/pTET-HST3 cells caused by doxycycline treatment (20 μg ml−1 for 24 h), as assayed by fluorescence microsopy after propidium iodide staining. Control cells were either heat-killed or triggered to undergo filamention (spider medium). DIC, differential interference contrast. PI, propidium iodide. (g) Susceptibility to genotoxic agents of WT (SN152), single (rtt109Δ/Δ, hst3+/Δ) and double (rtt109Δ/Δ hst3+/Δ, rtt109Δ/Δ hst3Δ/Δ) mutants (tenfold serial dilutions), as determined by colony formation assays.
Repression of HST3 expression with doxycycline severely impaired the growth of hst3Δ/pTET-HST3 cells (Fig. 2d). To determine whether repression of HST3 was fungistatic or fungicidal, we performed colony formation assays after transient doxycycline exposure. We grew tenfold serial dilutions of hst3Δ/pTET-HST3 and control cells in liquid cultures in the presence or absence of doxycycline for 24 h and then transferred the cells to solid medium lacking doxycycline. We found that the number of colony-forming units in hst3Δ/pTET-HST3 cultures was much lower after a 24 h exposure to doxycycline (Fig. 2e) compared to control cells, suggesting that transient HST3 repression led to cell death. To confirm this, we used propidium iodide staining of nucleic acids as a measure of cell death; live cells are not permeable to propidium ions27. Almost all hst3Δ/pTET-HST3 cells killed with heat (65 °C for 30 min) or treated for 24 h with doxycycline stained with propidium iodide (Fig. 2f). We also observed that HST3 repression with doxycycline triggered anomalous filamentous growth (Fig. 2f). It was possible that filamentous growth in itself could increase propidium iodide staining, irrespective of cell viability. To rule out this possibility, we grew hst3Δ/pTET-HST3 cells in the absence of doxycycline but under conditions that induce a switch to filamentous growth. We found that the resulting filaments were negative for propidium iodide staining (Fig. 2f). These results demonstrate that, unlike in S. cerevisiae and Schizosaccharomyces pombe, repression of HST3 in C. albicans leads to a loss of cell viability.
We hypothesized that the high levels of H3K56ac induced by repression of HST3 were causing cell death. This model predicts that deletion of the RTT109 acetyltransferase gene should rescue the lethality of the hst3Δ/Δ mutant. Consistent with this, we were able to delete both alleles of the HST3 gene in the rtt109Δ/Δ genetic background (Fig. 2g and Supplementary Fig. 2f–h). The rtt109Δ/Δ hst3+/Δ and rtt109Δ/Δ hst3Δ/Δ mutants were not more susceptible to genotoxic agents than the rtt109Δ/Δ strain, although they grew more slowly than either rtt109Δ/Δ or hst3+/Δ mutants (Fig. 2g and Supplementary Fig. 2h). These results demonstrate that the lethality of hst3Δ/Δ mutants depends upon the RTT109 gene. Therefore, we conclude that H3 Lys56 hyperacetylation is toxic to C. albicans cells and that H3K56ac is a major physiological substrate of C. albicans Hst3p.
Under conditions that normally maintain the yeast form of C. albicans, HST3 repression with doxycyline triggered a transition to filamentous growth (Fig. 3a). True hyphae typically lack constrictions at sites of septation and have narrow (~2-μm) parallel cell walls along their entire length28. In contrast, pseudohyphae have visible constrictions at sites of septation28. To determine whether HST3 repression produced true hyphae, we stained hst3Δ/pTET-HST3 cells grown in doxycycline-containing medium with DAPI and calcofluor white to visualize DNA, cell walls and septa. Cells initially appeared to divide normally, as we observed many budded cells after 3 h of doxycycline treatment (Fig. 3b). After 6 h, we observed pseudohyphae-like cells with septal junctions (Fig. 3b). Notably, at later time points (9 and 24 h), most cells appeared as abnormal ‘V-shaped’ filaments (Fig. 3b). In contrast to HST3 repression, hst3Δ/pTET-HST3 cells in spider medium lacking doxycycline (conditions that preserve HST3 expression) produced hyphae with parallel-sided, narrow germ tubes and no clear septal junctions (Fig. 3a). These features are clearly different from the morphological changes observed after repression of HST3 (Fig. 3b). After 9 h and 24 h of doxycycline treatment, hst3Δ/pTET-HST3 cells showed diffuse or absent DAPI staining (Fig. 3b), suggesting that HST3 repression resulted in DNA fragmentation and degradation. This is consistent with the presence of high levels of spontaneous DNA damage previously reported in S. cerevisiae hst3Δ hst4Δ mutants1. These results indicate that inappropriately high levels of H3K56ac lead to DNA fragmentation, histone H3 and H4 degradation and an anomalous morphological transition that culminate in cell death. Remarkably, all of these events occurred when the stoichiometry of H3K56ac increased from about 48% to 87% after HST3 gene repression (Fig. 2c and Supplementary Table 1). Some of these pronounced phenotypes were not reported in S. cerevisiae and S. pombe cells that lack H3K56 deacetylases1,14,15,29,30. In addition, we observed that a fraction of rtt109Δ/Δ cells (about 10%) spontaneously formed pseudohyphae with abnormal nuclear DNA staining (Supplementary Fig. 1g), indicating that tightly regulated levels of H3K56ac are crucial for proper chromatin structure and morphogenesis in C. albicans.
Figure 3.
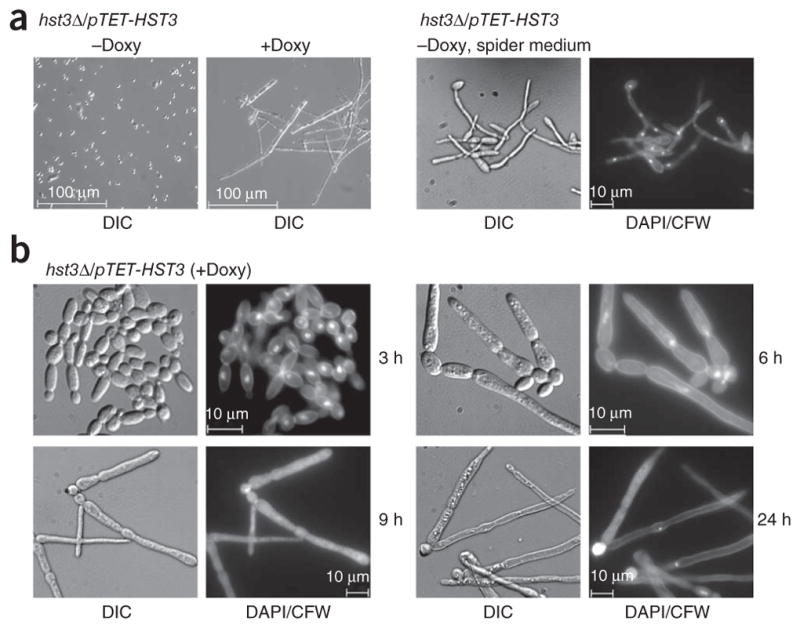
HST3 repression triggers abnormal changes in cell morphology and DNA staining. (a) Left, differential interference contrast (DIC) images of hst3Δ/pTET-HST3 cells grown in YPD medium at 30 °C in the absence or presence of doxycycline (20 μg ml−1 for 24 h). Right, cells grown in spider medium at 37 °C (hyphae-inducing conditions), stained with DAPI and calcofluor white (CFW) to mark DNA and cell walls, respectively, and visualized by epifluorescence microscopy. (b) DIC and epifluorescence images showing the morphology, DNA, septa and cell walls of hst3Δ/pTET-HST3 cells monitored at various times after doxycycline addition.
Hst3p inhibition with nicotinamide is cytotoxic to C. albicans
Hst3p is a member of a family of NAD+-dependent histone deacetylases known as sirtuins (Supplementary Fig. 3). These enzymes are inhibited by nicotinamide, a product of the NAD+-dependent deacetylation reaction. We reasoned that nicotinamide treatment of C. albicans cells should phenocopy HST3 repression. Addition of nicotinamide to wild-type cells caused an increase in H3K56ac (Fig. 4a) and led to robust growth inhibition (Fig. 4b,c). Furthermore, growth inhibition was not observed with nicotinic acid, which does not inhibit Hst3p1, and was RTT109 dependent (Fig. 4b,c). This suggests that nicotinamide exerts its effect through inhibition of H3K56 deacetylation. Finally, incubation of wild-type cells for 24 h with nicotinamide triggered the formation of V-shaped filaments with abnormal DNA staining (Fig. 4d), similar to those observed in doxycycline-treated hst3Δ/pTET-HST3 cells (Fig. 3b).
Figure 4.

HST3 repression and nicotinamide are cytotoxic to C. albicans strains that express RTT109. (a) Immunoblots of H3K56ac in wild-type (SN152) and rtt109Δ/Δ cells treated with 100 mM nicotinamide (+NAM). (b) Colony formation assays performed in the absence (YPD) or presence of either nicotinic acid (NA) or nicotinamide (NAM). (c) The growth of each strain (monitored by OD620 in a microtiter plate assay) at a given NAM concentration, as plotted as a percentage of its growth in the absence of NAM. (d) The effect of NAM on wild-type SN152 cells (50 mM NAM at 30 °C for 24 h), as monitored by DIC and fluorescence microscopy to detect either DNA (DAPI) or cell walls and septa (CFW).
These results prompted us to determine whether other clinically relevant isolates of C. albicans and fungal species are susceptible to growth inhibition by nicotinamide. Several azole- and echinocandin-resistant clinical isolates of C. albicans were as sensitive to nicotin-amide as a wild-type strain (Figs. 4c and 5a and Supplementary Fig. 4). We also found that other pathogenic Candida species were susceptible to nicotinamide. We observed robust growth inhibition of C. krusei at 25 mM nicotinamide, whereas growth of C. tropicalis, C. glabrata and C. parapsilosis was inhibited at higher nicotinamide concentrations (Fig. 5b). Nicotinamide also strongly inhibited the growth of Aspergillus fumigatus and Aspergillus nidulans, which are pathogenic to humans and plants, respectively (Fig. 5c). These results demonstrate that nicotinamide has broad antifungal properties.
Figure 5.

Nicotinamide inhibits growth of several clinically important pathogenic fungi. (a) Growth of azole-susceptible (AS) and azole-resistant (AR) strains (top) as well as echinocandin-susceptible (ES) and echinocandin-resistant (ER) strains (bottom), as tested in colony formation assays. Plates contained fluconazole (FLC; μg ml−1), micafungin (MCF; μg ml−1) or nicotinamide (NAM; mM). Ctl, control plates without NAM or antifugal agent. (b) Growth of two independent strains per Candida species on YPD medium containing NAM (mM). (c) Growth of two independent A. fumigatus or A. nidulans strains on complete medium containing NAM (mM).
Modulation of H3K56ac reduces C. albicans virulence in mice
We investigated the effect of H3K56ac modulation on C. albicans virulence in A/J mice, which are very sensitive to C. albicans infection31,32. This phenotype results, at least in part, from a deficiency in the C5 component of the complement system33. A/J mice accumulate high fungal loads within 24 h, providing a convenient model to rapidly and accurately measure fungal proliferation in vivo32. We first investigated the effect of HST3 repression on C. albicans virulence. A/J mice were injected with wild-type (SC5314 or KTY3) or hst3Δ/pTET-HST3 cells, and tetracycline was included in the drinking water of the mice to repress HST3. The kidney fungal loads of mice injected with hst3Δ/pTET-HST3 cells were markedly lower than those of mice injected with wild-type strains (Fig. 6a). In addition, clinical symptoms typical of a C. albicans infection were not visible in mice injected with hst3Δ/pTET-HST3 cells (data not shown). We next determined whether nicotinamide treatment would mimic the effect of HST3 repression in vivo. We gave mice nicotinamide 30 min before and 8 h after infection with C. albicans. Nicotinamide greatly lowered kidney fungal loads and clearly alleviated clinical signs of disease progression in mice injected with wild-type cells (Fig. 6b). In contrast, nicotinamide failed to protect mice injected with rtt109Δ/Δ cells, both in terms of fungal load and in terms of symptoms of candidiasis (Fig. 6b). As expected, introduction of a wild-type RTT109 gene into rtt109Δ/Δ cells reestablished sensitivity to nicotinamide (Fig. 6b). Nicotinamide has been reported to have complex immunomodulatory effects in mammals34. However, under our experimental conditions, nicotin-amide had no obvious side effects, as judged by the absence of various inflammation markers (Supplementary Table 4) and by careful monitoring of the mice. Furthermore, the fact that the anti-fungal effect of nicotinamide requires RTT109-dependent H3K56ac firmly establishes that nicotinamide exerts its therapeutic effect through inhibition of Hst3p-mediated H3K56 deacetylation, rather than by enhancing the host immune response. These results indicate that genetic or pharmacological inhibition of Hst3p leads to a loss of virulence in mice.
Figure 6.
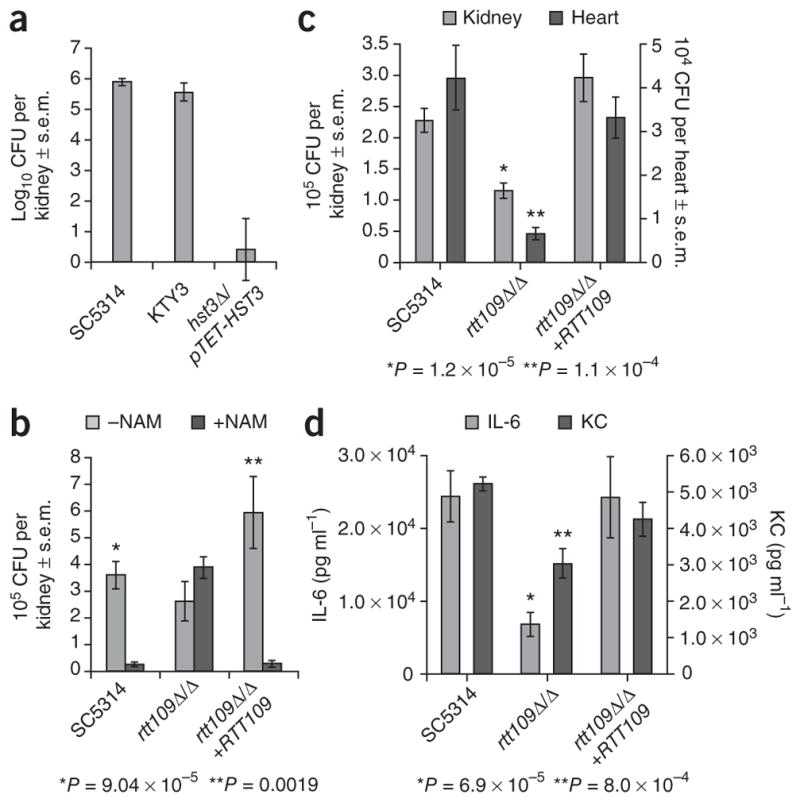
Modulation of H3K56ac levels reduces virulence in a mouse model of C. albicans infection. (a) Kidney fungal loads (CFU, colony-forming units) of A/J mice injected with 5 × 104 cells of each strain. Mice were given tetracycline in their drinking water to repress the pTET-HST3 allele. SC5314 and KTY3 are two distinct wild-type strains. (b) Kidney fungal loads of A/J mice injected with 3 × 105 cells of each strain. Mice were injected with nicotinamide 30 min before and 8 h after C. albicans infection. P values for two-tailed Student’s t-test are indicated for comparison of fungal loads in NAM-treated and untreated mice. (c) Kidney and heart fungal loads of A/J mice injected with 3 × 105 cells of each strain. P values for two-tailed Student’s t test are indicated for comparison of fungal loads in mice injected with rtt109Δ/Δ versus SC5314 WT cells. (d) Cytokine levels in the blood of the mice described in c. P values for two-tailed Student’s t test are indicated for comparison of cytokine levels in mice injected with rtt109Δ/Δ versus SC5314 WT cells. IL-6, interleukin-6. KC, keratinocyte-derived chemokine.
We also investigated the effect of the RTT109 deletion on virulence. We injected A/J mice intravenously with wild-type, rtt109Δ/Δ or rtt109Δ/Δ+RTT109 revertant cells. At the time of killing (24 h after infection), only mice injected with wild-type and revertant cells showed clinical manifestations of advanced candidiasis (data not shown). This prompted us to investigate the effect of RTT109 deletion on other parameters of C. albicans virulence. A/J mice suffer from cardiac dysfunction and accumulate high fungal loads in kidneys and the heart32. We found reduced fungal loads in the heart and kidneys of mice infected with rtt109Δ/Δ as compared with wild-type or revertant cells (Fig. 6c). We also observed that, compared with wild-type and revertant strains, the rtt109Δ/Δ mutant elicited a weaker inflammatory response in A/J mice, as evidenced by significantly lower amounts of interleukin-6, a key proinflammatory cytokine, and keratinocyte-derived chemokine, which is crucial for granulocyte recruitment (Fig. 6d). These results show that loss of Rtt109p reduces the virulence of C. albicans in A/J mice.
DISCUSSION
Two major classes of chemicals are commonly used to treat candidiasis: azoles and echinocandins. However, the emergence of strains that are resistant to these agents requires the development of new approaches to clinical intervention. In this report, we have identified two enzymes as attractive new therapeutic targets to treat C. albicans infections and possibly those caused by other pathogenic fungi. These enzymes, Rtt109p and Hst3/Hst4p, respectively acetylate and deacetylate histone H3K56 in C. albicans, S. cerevisiae and S. pombe1,11–15,29,30, and potential orthologs exist in several other pathogenic fungi. In C. albicans, disruption of the RTT109 gene conferred acute sensitivity to two distinct echinocandin family members, and this sensitivity was suppressed by antioxidants. Given that RTT109 mutations render C. albicans and other yeasts sensitive to genotoxic agents that impede DNA replication11–13,29,30, it seems likely that the marked echinocandin sensitivity of rtt109 mutants arises from oxidative damage to DNA35. Deletion of RTT109 also leads to pseudohyphae formation and reduces the growth rate of C. albicans in rich medium. Concordant with this, we found that rtt109Δ/Δ mutants are less virulent than wild-type C. albicans cells in the A/J mouse model. Given these results, Rtt109p inhibitors would be expected to enhance the cytotoxicity of echinocandins and reduce the virulence of C. albicans. CREB-binding protein (CBP) and p300 acetylate H3K56 and other substrates in human cells7,23. Despite similarities in their polypeptide backbones suggesting an ancestral relationship, the catalytic cores, catalytic mechanisms and substrate specificity of yeast Rtt109p and human p300/CBP are vastly different23. Therefore, it is likely that chemical library screens could identify specific inhibitors of Rtt109p that do not affect human p300/CBP.
We found that repression of the C. albicans HST3 gene is lethal even in the absence of antifungal agent. In A/J mice, Hst3p inhibition by gene repression or nicotinamide treatment considerably reduced C. albicans proliferation and clinical manifestations of candidiasis. Nicotinamide is a vitamin that serves as a precursor of NAD+. It is well tolerated and used systemically to treat pellagra and skin lesions36,37. Despite its beneficial effects, complex immunomodulatory effects of nicotinamide have been reported in mammalian cells34. This suggests that the ability of nicotinamide to curtail C. albicans infection might be due to an enhanced immune response of the host. However, three lines of evidence firmly establish that the main therapeutic effect of nicotinamide is exerted through inhibition of Hst3p, thus blocking H3K56 deacetylation. First, nicotinamide is not cytotoxic to C. albicans cells lacking Rtt109p and H3K56ac. Second, the antifungal property of nicotinamide in A/J mice depends on the presence of the RTT109 gene. Third, as judged by the absence of various inflammation markers, short-term nicotinamide treatment has no obvious immunomodulatory effects under our experimental conditions.
There are sound biochemical arguments to indicate that selective inhibitors of Hst3p may not have major side effects in humans. Despite the fact that sirtuin-1 (SIRT1) and SIRT2 have been implicated in H3K56 deacetylation in human cells7,10, there are sequence motifs conserved among fungal Hst3p family members that are absent from human sirtuins24. For instance, differences occur in the peptide binding channel of sirtuins38. Unlike human SIRT1 and SIRT2 (refs. 39–41), Hst3p seems to be highly substrate specific. In S. cerevisiae1 or C. albicans (Supplementary Table 1), acetylation of several sites in the N-terminal tails of H3 or H4 does not increase in the absence of Hst3 or Hst4p. Hence, it is conceivable that small molecules that specifically block the deacetylase activity of fungal Hst3p orthologs, without affecting human sirtuins, might be identified. Indeed, small molecules that inhibit even closely related sirtuins with high selectivity have been identified by screening chemical libraries42.
We determined the sensitivity to nicotinamide of a broad spectrum of fungal species. All of the C. albicans clinical isolates tested, including azole- and echinocandin-resistant strains, were very sensitive to nicotinamide. This is clinically important because, among the numerous species that cause candidiasis, C. albicans is responsible for the majority of cases43 and frequently develops resistance to azoles and echinocandins21,22. The non-albicans Candida strains that we tested showed various degrees of sensitivity. The strong sensitivity of C. krusei to nicotinamide is of particular interest, because C. krusei is intrinsically resistant to many antifungal drugs44. We also found that two different Aspergillus species, A. fumigatus and A. nidulans, were very sensitive to nicotinamide. This is noteworthy because A. fumigatus causes the second most common invasive mycoses in humans, after candidiasis45. Our data thus support the notion that nicotinamide or future fungal-specific sirtuin inhibitors may prove useful to treat various fungal infections and provide a solid physiological and molecular basis for the development of new antifungal agents that inhibit the enzymes that regulate H3K56ac. Although further in vivo studies will be necessary to assess the true therapeutic potential of nicotinamide and related chemicals, our results constitute a proof of principle that this strategy represents a useful therapeutic avenue.
ONLINE METHODS
Strain construction
Detailed methodologies for genetic manipulations, Southern blot and northern blot analyses are presented in the Supplementary Methods. The C. albicans strains constructed in this study are listed in Supplementary Table 2, and the primers used for their construction are shown Supplementary Table 3. The molecular characterization of the mutant strains is described in detail in the Supplementary Results.
Immunoblots
We prepared whole-cell extracts for SDS-PAGE by an alkaline method46 or a glass bead method30. Antibodies against H3K56ac (AV105), the H3 N-terminal domain (AV71/72) and recombinant S. cerevisiae histone H4 (AV 94) have been previously described2,30,47.
Identification of H3 K56 acetylation by mass spectrometry
We isolated histones from C. albicans as previously described for S. cerevisiae48. Acid-extracted histones were further purified by reverse phase HPLC49. Nano liquid chromatography–tandem mass spectrometry analyses of H3 tryptic peptides with multiple reaction monitoring were performed on an AB/MDS Analytical Technologies 4000 Q-Trap mass spectrometer equipped with a Nanospray II interface. We injected samples in triplicate, using multiple reaction monitoring for relative quantification.
RT-PCR
We extracted total RNA from 2 × 107 cells by the hot phenol method50. One microgram of total RNA was reverse-transcribed to cDNA with the SuperscriptIII reverse transcriptase enzyme (Invitrogen). Quantitative PCR was performed in triplicate with a SYBR green master mix containing Jumpstart Taq DNA polymerase (Sigma) and SYBR green nucleic acid stain (Invitrogen). We performed quantitative PCR on a Step One qPCR instrument (Applied Biosystems). Results were normalized to the ACT1 gene signal. The Supplementary Methods contain primer sequences.
Drug susceptibility assays
We performed spot and liquid microtiter plate assays as described previously51. We monitored cell growth after 48 h of incubation at 30 °C, unless otherwise specified. We purchased genotoxic and antifungal drugs from Sigma unless otherwise stated. Caspofungin was a gift from Merck Frosst Canada and micafungin was purchased from Astellas Pharma Canada.
Microscopy
We performed DIC and epifluorescence microscopy with a Zeiss Axio-Imager Z1 microscope. Image analysis was carried out with the Zeiss AxioVision 4.8 software. DAPI staining of DNA, CFW staining of cell walls and septa and propidium iodide staining of dead cells were performed as previously described52.
In vivo disseminated candidiasis model
Eight to twelve week old A/J mice were purchased from The Jackson Laboratories. Housing and all experimental procedures were approved by the Biotechnology Research Institute Animal Care Committee, which operates under the guidelines of the Canadian Council of Animal Care. To turn off the expression of the HST3 gene (Fig. 6a), 5% sucrose and 2 mg ml−1 tetracycline were added to the drinking water of the mice 3 d before infection with C. albicans. Inoculums of the C. albicans strains were grown overnight in YPD medium. Mice were injected via the tail vein with 3 × 105 blastospores, except for the conditional repression of HST3 with tetracycline (Fig. 6a), where a lower dose was used (5 × 104 blastospores) to allow time for tetracycline to act. Mice were closely monitored for clinical symptoms of candidiasis such as lethargy, hunched back and ruffled fur. Mice showing extreme lethargy were deemed moribund and were killed by exsanguination. This was typically performed at 24 h and 48 h after injection for 3 × 105 and 5 × 104 C. albicans blastospores, respectively. For determination of fungal load, kidneys and hearts were homogenized in PBS, and appropriate dilutions were plated on Sabouraud broth-agar plates. For the in vivo nicotinamide treatments (Fig. 6b), mice (six per treatment) were injected intraperitoneally with PBS containing nicotinamide (500 mg per kg body weight) 30 min before the C. albicans infection, as it takes 20–30 min for the nicotinamide concentration to peak in the blood stream53. A second dose of nicotinamide was injected 8 h after C. albicans infection to maintain high nicotinamide concentrations in circulation.
Cytokine analysis
We collected mouse blood in 1.1 ml Z-gel microtubes (Sarstedt), and samples were processed as recommended by the manufacturer. To determine the levels of cytokines in circulation, we analyzed sera (12.5 μl) with the BD CBA Flex sets (BD Biosciences) according to the manufacturer’s instructions. Fluorescence levels were recorded with the BD LSRII and data analysis was carried out with the FCAP Array software (BD BioSciences).
Statistical analysis
Statistical comparisons used two-tailed Student’s t test (kidney and heart fungal load estimations and cytokines analyses). Error bars are expressed as the s.e.m.
Additional methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgments
We are grateful to T. Roemer (Merck Research Laboratory) for providing strain CaSS1 and plasmids pHIS3 and pSAT-Tet, G. St-Germain (Laboratoire de santé publique du Québec) for strains MY067497, MY067743, MY070362, MY070589, MY069520, MY070954, MY070968 and MY070627, C. Restieri and M. Laverdière (Hôpital Maisonneuve-Rosemont) for strain 372.73374.1, C. Bachewich (Concordia University) for strain R153 and T. Edlind (Drexel University College of Medicine) for strain 20464. We are indebted to T. Roemer and M. Whiteway for critical reading of the manuscript. We thank F. Malenfant, J. Leroy, M. Mercier and P. Liscourt for excellent assistance in mouse handling. We are also grateful for the availability of the Candida Genome Database. This work was supported by research grants from the Canadian Institutes of Health Research to M.R. (CTP-79843, III-94587), A.M. (CTP-79843) and A.V. (CTP-79392) and from the National Science and Engineering Research Council of Canada to P.T. (STGP-322143-05). This is National Research Council of Canada publication number 52756. The Institute for Research in Immunology and Cancer is supported by funds from the Canadian Center of Excellence in Commercialization and Research, the Canadian Foundation for Innovation, and the Fonds de la Recherche en Santé du Québec.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
AUTHOR CONTRIBUTIONS
H.W. and S.T. designed and performed most experiments; G.L. constructed the yeast strains used in this study; J.T. and S.T. performed the fungal load and cytokine analyses; A.M. and M.R. designed and supervised the mouse studies; P.D., P.T. and A.V. designed and performed mass spectrometry experiments; E.-H.L. provided technical support; all authors participated in manuscript preparation; M.R. and A.V. supervised the study.
References
- 1.Celic I, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–1289. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle–regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 3.Hyland EM, et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:10060–10070. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozdemir A, et al. Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae. J Biol Chem. 2005;280:25949–25952. doi: 10.1074/jbc.C500181200. [DOI] [PubMed] [Google Scholar]
- 5.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Zhou H, Madden BJ, Muddiman DC, Zhang Z. Chromatin assembly factor 1 interacts with histone H3 methylated at lysine 79 in the processes of epigenetic silencing and DNA repair. Biochemistry. 2006;45:2852–2861. doi: 10.1021/bi0521083. [DOI] [PubMed] [Google Scholar]
- 7.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage–responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:1878–1889. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie W, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–427. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J, Pu M, Zhang Z, Lou Z. Histone H3–K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009;8:1747–1753. doi: 10.4161/cc.8.11.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–652. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 13.Tsubota T, et al. Histone H3–K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–712. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–119. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Thaminy S, et al. Hst3 is regulated by Mec1-dependent proteolysis and controls the S phase checkpoint and sister chromatid cohesion by deacetylating histone H3 at lysine 56. J Biol Chem. 2007;282:37805–37814. doi: 10.1074/jbc.M706384200. [DOI] [PubMed] [Google Scholar]
- 16.Berman J, Sudbery PE. Candida albicans: a molecular revolution built on lessons from budding yeast. Nat Rev Genet. 2002;3:918–930. doi: 10.1038/nrg948. [DOI] [PubMed] [Google Scholar]
- 17.Cowen LE, Anderson JB, Kohn LM. Evolution of drug resistance in Candida albicans. Annu Rev Microbiol. 2002;56:139–165. doi: 10.1146/annurev.micro.56.012302.160907. [DOI] [PubMed] [Google Scholar]
- 18.Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wisplinghoff H, et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 20.Sims CR, Ostrosky-Zeichner L, Rex JH. Invasive candidiasis in immunocompromised hospitalized patients. Arch Med Res. 2005;36:660–671. doi: 10.1016/j.arcmed.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 21.Akins RA. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med Mycol. 2005;43:285–318. doi: 10.1080/13693780500138971. [DOI] [PubMed] [Google Scholar]
- 22.Perlin DS. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat. 2007;10:121–130. doi: 10.1016/j.drup.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/ CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 25.Kelly J, Rowan R, McCann M, Kavanagh K. Exposure to caspofungin activates Cap and Hog pathways in Candida albicans. Med Mycol. 2009;47:697–706. doi: 10.3109/13693780802552606. [DOI] [PubMed] [Google Scholar]
- 26.Roemer T, et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol. 2003;50:167–181. doi: 10.1046/j.1365-2958.2003.03697.x. [DOI] [PubMed] [Google Scholar]
- 27.Deere D, et al. Flow cytometry and cell sorting for yeast viability assessment and cell selection. Yeast. 1998;14:147–160. doi: 10.1002/(SICI)1097-0061(19980130)14:2<147::AID-YEA207>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 28.Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol. 2004;12:317–324. doi: 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Haldar D, Kamakaka RT. Schizosaccharomyces pombe Hst4 functions in DNA damage response by regulating histone H3 K56 acetylation. Eukaryot Cell. 2008;7:800–813. doi: 10.1128/EC.00379-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xhemalce B, et al. Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe. J Biol Chem. 2007;282:15040–15047. doi: 10.1074/jbc.M701197200. [DOI] [PubMed] [Google Scholar]
- 31.Mullick A, et al. Dysregulated inflammatory response to Candida albicans in a C5-deficient mouse strain. Infect Immun. 2004;72:5868–5876. doi: 10.1128/IAI.72.10.5868-5876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullick A, et al. Cardiac failure in C5-deficient A/J mice after Candida albicans infection. Infect Immun. 2006;74:4439–4451. doi: 10.1128/IAI.00159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuite A, Elias M, Picard S, Mullick A, Gros P. Genetic control of susceptibility to Candida albicans in susceptible A/J and resistant C57BL/6J mice. Genes Immun. 2005;6:672–682. doi: 10.1038/sj.gene.6364254. [DOI] [PubMed] [Google Scholar]
- 34.Maiese K, Chong ZZ, Hou J, Shang YC. The vitamin nicotinamide: translating nutrition into clinical care. Molecules. 2009;14:3446–3485. doi: 10.3390/molecules14093446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dizdaroglu M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat Res. 2005;591:45–59. doi: 10.1016/j.mrfmmm.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 36.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 37.Niren NM. Pharmacologic doses of nicotinamide in the treatment of inflammatory skin conditions: a review. Cutis. 2006;77:11–16. [PubMed] [Google Scholar]
- 38.Cosgrove MS, et al. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45:7511–7521. doi: 10.1021/bi0526332. [DOI] [PubMed] [Google Scholar]
- 39.Blander G, et al. SIRT1 shows no substrate specificity in vitro. J Biol Chem. 2005;280:9780–9785. doi: 10.1074/jbc.M414080200. [DOI] [PubMed] [Google Scholar]
- 40.Garske AL, Denu JM. SIRT1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry. 2006;45:94–101. doi: 10.1021/bi052015l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 42.Hirao M, et al. Identification of selective inhibitors of NAD+-dependent deacetylases using phenotypic screens in yeast. J Biol Chem. 2003;278:52773–52782. doi: 10.1074/jbc.M308966200. [DOI] [PubMed] [Google Scholar]
- 43.Pfaller MA, et al. Results from the ARTEMIS Drosoph. Inf. Serv.K global antifungal surveillance study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida species to fluconazole and voriconazole determined by CLSI standardized disk diffusion. J Clin Microbiol. 2010;48:1366–1377. doi: 10.1128/JCM.02117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pappas PG, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:503–535. doi: 10.1086/596757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev. 2009;22:447–465. doi: 10.1128/CMR.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 47.Gunjan A, Verreault AA. Rad53 kinase–dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell. 2003;115:537–549. doi: 10.1016/s0092-8674(03)00896-1. [DOI] [PubMed] [Google Scholar]
- 48.Poveda A, et al. Hif1 is a component of yeast histone acetyltransferase B, a complex mainly localized in the nucleus. J Biol Chem. 2004;279:16033–16043. doi: 10.1074/jbc.M314228200. [DOI] [PubMed] [Google Scholar]
- 49.Drogaris P, Wurtele H, Masumoto H, Verreault A, Thibault P. Comprehensive profiling of histone modifications using a label-free approach and its applications in determining structure-function relationships. Anal Chem. 2008;80:6698–6707. doi: 10.1021/ac800739d. [DOI] [PubMed] [Google Scholar]
- 50.Schmitt ME, Brown TA, Trumpower BL. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 1990;18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsao S, Rahkhoodaee F, Raymond M. Relative contributions of the Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance. Antimicrob Agents Chemother. 2009;53:1344–1352. doi: 10.1128/AAC.00926-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trunk K, et al. Depletion of the cullin Cdc53p induces morphogenetic changes in Candida albicans. Eukaryot Cell. 2009;8:756–767. doi: 10.1128/EC.00332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horsman MR, Siemann DW, Chaplin DJ, Overgaard J. Nicotinamide as a radiosensitizer in tumours and normal tissues: the importance of drug dose and timing. Radiother Oncol. 1997;45:167–174. doi: 10.1016/s0167-8140(97)00127-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.