

Abstract
Macrophages pre-exposed to a sub-lethal dose of anthrax lethal toxin (LeTx) are refractory to subsequent high cytolytic doses of LeTx, termed toxin-induced resistance (TIR). A small population of TIR cells (2–4%) retains TIR characteristics for up to 5 to 6 weeks. Through studying these long-term TIR cells, we found that a high level of histone deacetylase (HDAC)8 expression was crucial for TIR. Knocking down or inhibition of HDAC8 by siRNAs or the HDAC8-specific inhibitor PCI-34051, respectively, induced expression of the mitochondrial death genes Bcl2 Adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), BNIP3-like (BNIP3L) and Metastatic Lymph Node (MLN)64, and re-sensitized TIR cells to LeTx. Among multiple histone acetylations, histone H3 lysine 27 acetylation (H3K27Ac) was most significantly decreased in TIR cells in an HDAC8-dependent manner, and the association of H3K27Ac with the genomic regions of BNIP3 and MLN64, where HDAC8 was recruited to, was diminished in TIR cells. Furthermore, over-expression of HDAC8 or knocking down the histone acetyltransferase CREB-binding protein (CBP)/p300, known to target H3K27, rendered wild-type cells resistant to LeTx. As in RAW264.7 cells, primary bone marrow-derived macrophages exposed to a sub-lethal dose of LeTx were resistance to LeTx in an HDAC8-dependent manner. Collectively, this study demonstrates that epigenetic reprogramming mediated by HDAC8 plays a key role in determining the susceptibility of LeTx-induced pyroptosis in macrophages.
Keywords: anthrax lethal toxin, pyroptosis, histone H3, histone deacetylases
Introduction
Pyroptosis is a rapid programmed cell death that concurs with the release of potent inflammatory cytokines such as interleukin (IL)-1β and IL-18 (1). The canonical pathway of pyroptosis is initiated by the nucleotide oligomerization domain-like receptors (NLRs), such as NLRP1b, NLRP3, NLRC4 and NAIP, in response to various microbial components as well as foreign or endogenous inflammatory molecules. Activated NLRs form a multi-protein complex together with the adaptor protein ASC and caspase-1, known as the “inflammasome”. Inflammasome is a platform for activating caspase-1 which produces biologically active IL-1β/-18 and executes pyroptosis (2–4). Production of IL-1β/18 plays a crucial role in host immune responses against invading pathogens (5–11); whereas, the role and mechanism of pyroptosis are not yet clear. Pyroptosis can be beneficial to the hosts by removing intracellular niches of pathogens (8, 12–14) and exploited by pathogens for immune evasion and dissemination (15–18). Aberrant pyroptosis can also be detrimental to the host, since it causes depletion of immune cells (17) and breakdown of splenic architecture (14).
Anthrax lethal toxin (LeTx) is a cytotoxic bacterial toxin released by Bacillus anthracis. LeTx induces pyroptosis in certain murine myeloid cells such as macrophages and dendritic cells through activating NLRP1b (19). We and others have shown that macrophages pre-exposed to a sub-lethal dose of LeTx are refractory to subsequent high cytolytic doses of LeTx (20–22), termed “toxin-induced resistance (TIR)”. A small portion of TIR cells (2–4 %) further retains TIR characteristics for up to 5–6 weeks (21). These cells are normal in cleaving the mitogen-activated protein kinase kinases (MEKs) and activating caspase-1, but resistant to LeTx-induced mitochondrial dysfunction, including hyper-polarization and ROS generation, through down-regulating the mitochondrial death genes, Bcl2/adenovirus E1B-interacting proteins (BNIP)3, BNIP3-like (BNIP3L; also known as NIX) (21) and MLN64 (also known as the steroidogenic acute regulatory-related lipid transfer protein 3) (23). To date, how these death genes are down-regulated throughout multiple cell cycles in TIR cells is unknown.
Epigenetic reprogramming is a cellular mechanism that conveys phenotype to daughter cells without altering genomic DNA sequences. It includes DNA methylation, histone modifications (methylation, acetylation, sumoylation and ubiquitination) and production of micro RNAs, which involves many aspects of cell function, differentiation, proliferation and tumorigenesis (24–26). Based on the non-permanent, but inheritable characteristics of TIR, we examined the possible involvement of epigenetic reprogramming in TIR. Here, we show that the histone deacetylase (HDAC)8 plays a crucial role in TIR, in part through deacetylating histones, such as H3K27Ac, and subsequently inhibiting expression of the mitochondrial death genes.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Protective antigen (PA) and lethal factor (LF) were purified as previously described (27, 28). Antibodies of H3Ac, H3K4Ac, H3K9Ac, H3K14Ac, H3K18Ac, H3K23Ac, H3K27Ac, H3K36Ac, H3K56Ac and H3K79Ac were purchased from Active Motif (CA, USA), and antibodies for pan H3 and β-actin were purchased from Bio Vision (CA, USA) and Rockland Inc. (PA, USA), respectively. Antibodies against EGFP, MEK-1-NT, p38 and caspase-1 were purchased from Life Technologies (ON, Canada), Stressgen Bioreagents (BC, Canada), Santa Cruz Biotechnology (TX, USA) and Cell Signaling (MA, USA), respectively. DiOC6 (3-3′-dihexyloxacarbocyanine iodide) and Mito-SOX-red were purchased from Calbiochem (EMD Biosciences, CA, USA) and Life Technologies (Molecular Probes®, ON, Canada). The following is the list of epigenetic chemical inhibitors used in this study: aza-2-deoxycytidine (azacitidine; Sigma, ON, Canada), panobinostat (LBH-589; Selleck, TX, USA), tozasertib (LC Lab, MA, USA), PCI-34051 (Cayman; CA, USA), CAY10603 (Cayman) and anacardic acid (Bio vision, CA, USA). Pre-designed small interfering RNA (siRNA) oligonucleotides directed against HDAC8 (No.SI1063902) were purchased from Qiagen. Pre-designed siRNAs directed against CREB binding protein (CBP; No.4390771), E1A binding protein p300 (No.AM16708), mouse PCAF (No. AM16708), and mouse caspase-1 (No. 4390771) were purchased from Life Technologies (Ambion®). Full-length mouse HDAC8 cDNA (NM 027382) was amplified from cDNA library by PCR using primers (forward; 5′-TTGCGAATCTGATGGAGATGCCAGAGGAACCC-3′, reverse; 5′-TTGCGGATCCCGGACCACATGCTTCAGATTCCC-3′) and cloned into the pEGFP-N1 vector using EcoR I and BamH I restriction enzymes.
Cell culture
RAW 264.7 murine macrophages were cultured in Dulbecco’s modified Eagle’s medium containing 8% heated-inactivated fetal bovine serum (Sigma-Aldrich, Canada), 10 mM MEM non-essential amino acids solution, 100 units/ml penicillin G sodium, 100 μg/ml streptomycin sulfate, and 1 mM sodium pyruvate. Cells were grown at 37°C in 5% CO2. TIR cells were generated as previously reported (21). Briefly, RAW 264.7 macrophages were treated with LeTx (500 ng/ml LF and 1 μg/ml PA) for 5 h, and surviving cells were plated in a fresh cell culture media. Two weeks later surviving clones were individually picked and plated on a 96-well plate. Each clone was propagated and tested for LeTx sensitivity. TIR clones were selected individually or pooled for further cell culture and were confirmed for their sensitivity to LeTx. Primary bone marrow-derived macrophages (BMDMs) were prepared from 129/S1/Svlmj mice as previously described (29). BMDMs were treated with a sub-lethal dose of LeTx (100 ng/ml LF and 100ng/ml PA) for 24 h and surviving cells were plated onto new culture dishes with fresh media. These cells were used as primary BMDM TIR cells.
Transfection of cells
Transfection of RAW 264.7 cells with small interference (si)RNAs and the pEGFP-HDAC8 plasmid was performed using Lipofectamine RNAi Max kit and Lipofectamine 2000 (Invitrogen™), respectively, according to the manufacturer’s instruction. Briefly, cells plated on 6-well plates were transfected with 100 pmol (for RAW364.7 cells) or 150 pmol (for BMDMs) of siRNAs in RNAi Max reagent for 16 h and fresh media were added for the next 24 h, unless otherwise indicated in the figure legends. Total RNAs were harvested and levels of mRNA expression were analyzed by reverse transcription real-time quantitative polymerase chain reaction (RT-qPCR). Primers used for qPCR are listed in Supplemental Table 1S. For stable transfection, RAW264.7 cells were transfected with pEGFP or pEGFP-HDAC8 using Lipofectamine 2000 (Invitrogen) and stably transfected cells were selected in the presence of G418 (1 mg/ml) antibiotics for 1 week and surviving cells were pooled.
Cell treatments, lysate preparation and Western blot analysis
After cells were cultured for the indicated times in the presence or absence of LeTx, chemicals or siRNAs, cells were collected and lysed in ice-cold cell lysis buffer (20 mM MOPS, 2 mM EGTA, 5 mM EDTA, 1 mM Na3VO4, 40 mM β-glycerophosphate, 30 mM sodium fluoride, and 20 mM sodium pyrophosphate, pH 7.2) containing 1% Triton X-100, and protease inhibitor mixtures (Roche Applied Science). Cell lysates were incubated on ice for 10 min and sonicated for 5 minutes. These extracts were run on 12.5% SDS-PAGE gels, followed by transfer onto nitrocellulose membranes. Membranes were subsequently blocked at room temperature for 1 h with 5% (w/v) skim milk. Various anti H3 antibodies, β-actin, p38 and GFP were used at dilutions ranging from 1:500–1:1500 and incubated overnight at room temperature. After extensive washing with TTBS buffer (Tris-buffered saline containing 0.05% Tween 20), the secondary antibody was applied at 1:5000 dilution and incubated for 60 min. The membranes were then washed with TTBS and developed using the Enhanced Chemiluminescent Detection reagent (Thermo scientific), followed by densitometry analysis using Image J program (NIH).
Measurement of cell Viability
A microtiter tetrazolium (MTT) assay or propidium iodide (PI) staining, followed by fluorescent activated cell sorting (FACS) analysis, was used to measure cytotoxicity. RAW264.7 macrophages were cultured in the presence or absence of LeTx in 96 well plates, and MTT was then added at a final concentration of 1mg/ml. After an additional 2 h incubation at 37 °C, culture media were carefully aspirated and 100 μl of dimethysulfoxide (DMSO) was added to dissolve crystals. Optical densities of the wells were analyzed using an automatic ELISA plate reader (Bio-Rad) at a wavelength of 570 nm. The ratio of cell death was estimated based on the optical density of wells by comparing with those from non-treated cells as 100% survival. For PI and FACS analysis, RAW 264.7 cells were cultured in the presence or absence of LeTx in 12 or 6 well plates for the time indicated, and cells were then harvested. After washing twice, cells were resuspended in PBS containing 2 μg/ml PI at a concentration of 1 × 106 cells/ml and PI positive cells were analyzed by FACS Calibur (Beckton-Dickson Biosciences) using CellQuest Pro software (Beckton-Dickson Biosciences).
Mitochondrial ROS measurements
Mitochondrial ROS production was assessed by Mito-SOX-red (Invitrogen, Molecular Probes) as previously described (23). Briefly, RAW264.7 cells were stained with 2.5uM Mito-SOX-Red for 15min at 37°C and washed twice with fresh media. After collecting, cells were then resuspended in PBS and analyzed by FACS Calibur and CellQuest Pro software (Beckton-Dickson Biosciences).
Mitochondrial membrane polarization measurements
Mitochondrial membrane polarization was measured as previously described (23) using 3-3′-dihexyloxacarbocyanine iodide (DiOC6). Briefly, cells were treated with LeTx and washed twice with pre-warmed fresh media. Cells were further incubated with fresh media containing DiOC6 (2 μg/ml ) at 37°C for 10 min. After two washes with fresh media, cells were then harvested and analyzed by FACS.
Quantitative Real-time PCR
Briefly, total cellular RNA was isolated using TRIzol (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized from 2 μg of total RNA using Moloney murine leukemia virus (M-MuLV) reverse transcriptase (New England Biotechnology) and oligo (dT) primers. qPCR analyses were performed with a Rotor-Gene RG3000 quantitative multiplex PCR instrument (Montreal Biotech) using Express SYBR Green ER qPCR Super Master Mix (Invitrogen). The data were normalized by expression of the GAPDH housekeeping gene.
Chromatin immunoprecipitation (ChIP) analysis
ChIP analysis was conducted as described previously (30), using the H3K27Ac (Active motif, California) or the RNA polymerase II (Pol-II; phospho S5; Abcam, Ontario, Canada) antibodies and primers for BNIP3, BNIP3L, and MLN64, as shown in Supplemental Table S1. Briefly, RAW 264.7 cells were cross-linked with 1% formaldehyde, and DNAs were extracted in lysis buffer (50 mM Tris at pH 8.0, 5 mM EDTA and 1% sodium dodecyl sulfate, supplemented with protease inhibitors; Thermo Scientific) and diluted with dilution buffer (50 mM Tris at pH 8.0, 0.5% Triton X-100, 0.1 M NaCl, and 2 mM EDTA). Chromatin from isolated nuclei was sheared by sonication (Bioruptor UCD-200 ultrasound sonicator, Diagenode) for 20 min resulting in DNA fragments between 150 and 200 bp in size. After preclearing the protein G-conjugated Dynabeads (Life Technologies) for 1 h, immunoprecipiation was conducted with a mixture of G-conjugated Dynabeads with 4–6 μg of H3K27Ac (Active Motif, CA), Pol-II (phosphor S5; Abcam Inc., ON, Canada), HDAC3 (Epigentek, NY), HDAC8 (Epigentek) or rabbit IgG (Sigma) antibodies overnight at 4 °C. The immunoprecipitates were washed extensively and heated overnight at 65 °C to reverse cross-linking. DNA fragments were then purified using QIAquick Spin columns (Qiagen) and subjected to quantitative real-time PCR (qPCR) analysis using SYBR Green ER qPCR Super Master Mix (Life Technologies) for H3K27Ac, HDAC3 and HDAC8 analysis. DNAs from Pol-II immunoprecipitation were subjected to qPCR using SYBR Green ER qPCR system for MLN64 or TaqMan® qPCR analysis for BNIP3 and BNIP3L, using the ZEN™ quencher system (Integrated DNA Technologies, Iowa) containing the qPCR mixture (50 units/ml of Taq DNA polymerase, 1X ThermoPol reaction buffer, 200 uM dNTPs, 900nM forward and reverse primers, and 250 nM double-quenched probe). For H3K27Ac, HDAC3, HDAC8 and Pol-II ChIP assays, primers were designed to target the genomic sites known to be associated with H3K27Ac based on the ENCODE data base (http://encodeproject.org/ENCODE/): BNIP3 (expanding ~ 1 kb in front of exon 1), BNIP3L (expanding ~3 kb within intron 3) and MLN64 (~4 kb in front of exon 1). Primers targeting the promoter regions of BNIP3, BNIP3L, MLN64 and glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) were designed to encompass the transcription start sites, based on the Transcriptional Regulatory Element Database (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home). Data are presented as enrichment of the precipitated target sequence as compared to input DNA.
Statistical analysis
All data were analyzed using GraphPad Prism 4.0 (GraphPad Software, Inc. California). The results are presented as the mean of three independent replicates. Data were analyzed by Student’s t-test or two-way ANOVA test, followed by Tukey’s test. Statistical significance was defined at P < 0.05 (*) and P < 0.01 (**).
RESULTS
The broad-spectrum HDAC inhibitor panobinostat sensitizes TIR cells to LeTx-induced pyroptosis
To examine the involvement of epigenetic reprogramming in TIR, we first examined the effects of various chemical inhibitors in different concentrations (up to 200-fold higher than known IC50), targeting enzymes involved in epigenetic reprogramming, including tozasertib (broad spectrum aurora kinase inhibitor), azacitidine (broad spectrum DNA methyltransferase inhibitor), panobionstat (LBH-589; broad spectrum HDAC inhibitor), mocetinostat (HDAC inhibitor specific for HDAC 1, 2, & 3)(31), CAY10603 (HDAC6-specific inhibitor) and anacardic acid (broad spectrum histone acetyltransferase inhibitor). TIR cells pre-treated with these inhibitors in various concentrations for 24 h were exposed to LeTx for 5 h and analyzed for cell death using MTT assays. Among these inhibitors, azacitidine and panobinostat (PN) re-sensitized TIR cells to LeTx-induced cell death in a dose-dependent manner (Fig. 1A and Supplemental Fig. S1). Here, we further examined the involvement of HDACs in re-sensitization of TIR cells to LeTx by PN. We first confirmed that cell death of PN-treated TIR cells by LeTx was pyroptosis, since knocking down caspase-1 by si-RNAs almost completely prevented cell death (Fig. 1B, left panel). si-RNAs targeting caspase-1 decreased more than 75% of caspase-1 m-RNA and protein levels in these cells (Fig. 1B, right panel). TIR cells are resistant to LeTx-induced mitochondrial dysfunction, including generation of reactive oxygen species (ROS) and hyperpolarization of transmembrane electrical gradients, through down-regulating the expression of the mitochondrial death genes BNIP3, BNIP3L and MLN64 (21, 23). PN induced the expression of the mitochondrial death genes in TIR cells (Fig. 1C), and PN-treated TIR cells were susceptible to LeTx-induced mitochondrial ROS generation (Fig. 1D).
Figure 1. The HDAC inhibitor panobinostat induces expression of the mitochondrial death genes, and sensitizes to LeTx-induced mitochondrial dysfunction and pyroptosis in TIR RAW264.7 cells.

(A) TIR RAW264.7 cells were cultured in the presence or absence of different concentrations of panobinostat (PN) for 24 h. Equal number of cells were re-plated on a 96-well plate with fresh media and treated with LeTx (500 ng/ml LF and 1μg/ml PA) for 5 h. Cell death was measured using MTT assays as described in Experimental Procedures. (B) TIR cells were transfected with scrambled (si-scramble) or caspase-1 (si-Casp-1) siRNAs for 24 h and then treated with PN (50 nM) or none for 16 h. These cells were then exposed to LeTx (500ng/ml LF and 1 μg/ml PA) for 5 h. Cell death, and caspase-1 mRNA and protein levels were measured using MTT assays (left panel), RT-qPCR (right upper panel) and Western bot (right lower panel) analysis, respectively, as described in Experimental Procedures. (C) Similarly, mRNA levels of the mitochondrial cell death genes BNIP3, BNIP3L and MLN64, were measured in TIR cells non-treated or treated with PN (50 nM) for 16 h. Levels of mRNAs were expressed as fold of change relative to those of wild-type non-treated RAW264.7 cells (dotted line). (D) Wild-type and TIR cells were pre-exposed to si-scramble or si-Casp-1 for 24 h. Cell were then treated with none or PN (50 nM) for 16 h, followed by LeTx (500ng/ml LF and 1 μg/ml PA) for 3 h. Mitochondrial ROS production was measured using Mito-SOX-red. Data were acquired by flow cytometry using Cellquest software and data shown are representative results of 3 independent experiments. (A–C) Data are expressed as means ± SD (n=3), * P < 0.05 (Student’s t-test); ** P < 0.01.
Up-regulation of HDAC8 is correlated with TIR, which is involved in the suppression of mitochondrial death genes expression and resistance to LeTx-induced mitochondrial transmembrane hyper-polarization and pyroptosis
To unravel HDACs involved in re-sensitizing TIR cells, we examined whether expression of any specific HDACs was specifically correlated with TIR, using RT-qPCR analysis. Among the various HDACs examined (Supplemental Fig. S2), only the level of HDAC8 mRNA was closely correlated with the extent of TIR (R2=0.8; Fig. 2A). No apparent correlation between HDAC1/2 expression and TIR, and no effect of the HDAC 1, 2 & 3-specific inhibitor mocetinostat on cell death (Supplemental Fig. S1) consistently rule out the involvement of HDAC1 and 2 in TIR. To examine the role of HDAC8 in TIR, the HDAC8-specific inhibitor PCI-34051 with >200-fold selectivity over the other HDAC isoforms (32) was exposed to TIR cells for 24 h and then treated with LeTx. As shown in Fig. 2B, PCI-34051 at 100 nM was able to sensitize TIR cells to LeTx. To confirm the role of HDAC8 in TIR, HDAC8 and/or caspase-1 were specifically knocked down in TIR cells, and sensitivities to LeTx-induced mitochondrial dysfunction and cell death were examined. Knocking down HDAC8 by siRNAs significantly re-sensitized TIR cells to LeTx-induced cell death, which was abolished by knocking down caspase-1 (Fig. 2C). As shown in Fig. 2D, TIR cells expressed higher levels of HDAC8 proteins than those of wild-type cells, which was diminished by treating si-RNAs against HDAC8. Furthermore, knocking down HDAC8 increased expression of BNIP3 (~400% of wild-type cells), BNIP3L (~300%) and MLN64 (~75%) (Fig. 2E), and rendered TIR cells sensitive to LeTx-induced mitochondrial transmembrane hyper-polarization (Fig. 2F). Collectively, these results suggest a key role of HDAC8 in TIR.
Figure 2. High levels of HDAC8 expression are correlated with the extent of TIR, and involved in suppression of the mitochondrial death genes expression and in resistance to LeTx-induced mitochondrial dysfunction/pyroptosis in TIR cells.

(A) Total RNAs were purified from wild-type cells (open dots) and five TIR clones (closed dots) with different degrees of LeTx sensitivities. Expression of HDAC8 was analyzed by RT-qPCR and plotted against the extent of LeTx susceptabily (500 ng/ml LF and 1 μg/ml PA for 5 h). Correlation of coefficiency (R2) was calculated using the GraphPad Prism® program. Results are expressed as means and ± SD (n=3). (B) TIR cells were treated with various concentrations of the HDAC8-specific inhibitor PCI-34051 for 24 h. Wild-type and PCI-34051-treated cells exposed to LeTx (500ng/ml LF and 1 μg/ml PA) for 5h and cell death was measured by MTT assays. (C) TIR cells were transfected with scrambled (si-scramble), caspase-1 (si-Casp-1), HDAC8 targeting (si-HDAC8) or both caspase-1 and HDAC8 siRNAs for 40 h, followed by LeTx (500ng/ml LF and 1 μg/ml PA) treatments for 5 h. Cell death was measured using MTT assays as described in Experimental Procedures. Knock down efficiencies of si-Casp-1, si-HDAC8 in TIR cells were examined by RT-qPCR analysis. Statistical analysis was performed using two-way ANOVA test, followed by Tukey’s test (left panel) or Student’s t-test (middle and right panels). Data are expressed as means ± SD (n=3). (D) Wild-type and TIR RAW264.7 cells were transfected with si-scramble or si-HDAC8 for 40 h and expression of HDAC8 was measured by Western blots. (E–F) Similarly, TIR cells were transfected with si-scramble or si-Casp-1 for 40 h. (E) mRNA levels of BNIP3, BNIP3L and MLN64 were measured by RT-qPCR analysis and expressed as fold of change relative to those of wild-type si-scramble-treated RAW264.7 cells (dotted line). (F) Cells were then treated with LeTx (500ng/ml LF and 1 μg/ml PA) for 3 h and mitochondrial transmembrane hyperpolarization was analyzed by measuring mitochondrial uptake of DiOC6 dye. Data were acquired by flow cytometry using Cellquest software and data shown are representative results of 3 independent experiments. (E) Data are expressed as means ± SD (n=3), * P < 0.05 (Student’s t-test).
Overexpression of HDAC8 prevents LeTx-induced pyroptosis
To further verify the role of HDAC8 in regulating susceptibility to LeTx-induced pyroptosis, we examined whether overexpression of HDAC8 protected wild-type RAW264.7 cells from LeTx-induced pyroptosis. Since transfection efficiency is typically low in macrophages, RAW264.7 cells were stably transfected with the EGFP or EGFP-conjugated HDAC8 vectors as describe in Experimental Procedures. In cells transfected with pEGFP, LeTx caused more than 65% of cell death (PI-positive); whereas, cells transfected with pEFGP-HDAC8 were resistant to LeTx (Fig. 3).
Figure 3. Overexpression of HDAC8 renders resistance to LeTx-induced pyroptosis in wild-type RAW264.7 cells.
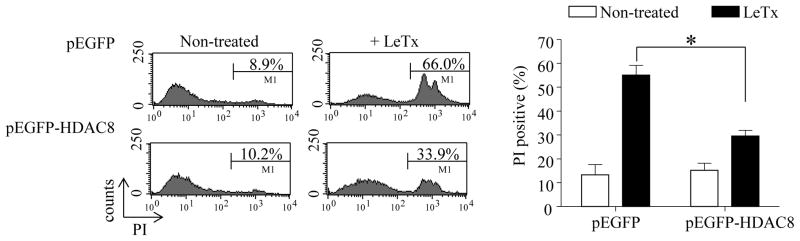
RAW264.7 cells stably transfected with pEGFP or pEGFP-HDAC8 were exposed to LeTx (500 ng/ml LF and 1 μg/ml PA) for 5 h and extents of cell death were measured by flow cytometry using propidium iodide (PI) staining. Data presented are representative histograms from 3 independent experiments (left panel) and statistical analysis of cell death is presented in the bar graph (right panel). Data are expressed as means ± SD (n=3), * P< 0.05 (Student’s t-test).
Overexpression of HDAC8 suppresses BNIP3, BNIP3L and MLN64 expression at the level of gene transcription
Since HDAC8 played a key role in down-regulating expression of BNIP3, BNIP3L and MLN64 in TIR cells (Fig. 2E), it is possible that HDAC8 suppresses expression of these genes through limiting access of transcription machineries to the promoter areas. To this end, we examined the accessibility of the RNA polymerase II (Pol-II) at the promoter regions of BNIP3, BNIP3L and MLN64 in TIR cells using ChIP-qPCR analysis. Indeed, binding of Pol-II to the promoter regions of these genes was significantly lower in TIR cells than in wild-type cells (Fig. 4A). Also, over-expression of HDAC8 in wild-type cells significantly limited the access of Pol-II to the promoter regions of these genes (Fig. 4B).
Figure 4. TIR RAW264.7 cells and HDAC8-overexpressing wild-type RAW264.7 cells have limited accessibility of the RNA polymerase II (Pol-II) to the promoter regions of BNIP3, BNIP3L and MLN64.
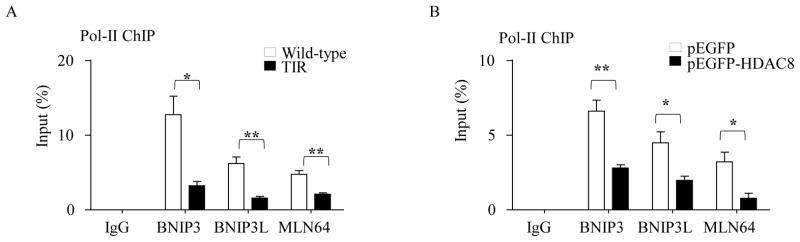
Accessibility of Pol-II to the promoter regions of BNIP3, BNIP3L and MLN64 was analyzed by ChIP-qPCR assays. DNAs prepared from RAW 264.7 wild-type cells and TIR cells (A), and RAW 264.7 wild-type cells stably overexpressing pEGFP or pEGFP-HDAC8 (B) were sonicated and immunoprecipitated using Pol-II antibodies or rabbit IgG. Power SYBER Green qPCR (for MLN64) and TaqMan® qPCR analysis using the ZEN™ quencher system (for BNIP3 and BNIP3L) were used to quantify the amounts of immunoprecipitated DNAs using primers and an internal probe, as described in the Experimental Procedures. ChIP efficiency is represented as % of input DNA recovered by immunoprecipitation. Rabbit IgG serum was used as an immunoprecipitation control. Data are expressed as means ± SD (n=3), * P< 0.05; ** P < 0.01 (Student’s t-test).
HDAC8-dependent decrease of H3K27Ac levels in TIR RAW264.7 cells is correlated with increase of HDAC8 recruitments to the H3K24Ac-associated genomic regions of BNIP3 and MLN64
Although substrates for HDAC8 are not well defined, acetylated histones are main targets of HDAC8 (33). Thus, we examined the levels of histone acetylation in wild-type and TIR cells using Western blots. We detected no overall changes in histone H4 acetylation (data not shown). However, the levels of overall histone H3 acetylation were significantly lower in TIR cells than wild-type cells (Fig. 5A). Further examination on specific histone H3 lysine residues, which undergo acetylation, showed that acetylations at the lysine residues 9, 14 and 27 were significantly lower in TIR cells than those in wild-type cells. Among them, histone H3 acetylation at lysine 27 (H3K27Ac) was most significantly down-regulated (~80%) in TIR cells. Also, the levels of H3K27Ac in TIR cells were increased by PN or si-HDAC8 treatments (Fig. 5B), suggesting that HDAC8 was involved in deacetylation of H3K27Ac in TIR cells.
Figure 5. HDAC8-dependent decrease of H3K27Ac levels in TIR RAW264.7 cells is correlated with increase of HDAC8 recruitments to the H3K24Ac-associated genomic regions of BNIP3 and MLN64.

(A) Immunoreactivities of pan-specific acetylated histone H3 and various residue-specific histone H3 acetylation antibodies were measured in total cell lysates prepared from wild-type and TIR RAW 264.7 cells using Western blots. β-actin and histone H3 were used as loading controls. Intensities of the bands were analyzed by NIH Image J program. Immunoreactivities in TIR cells were expressed as fold of change relative to those of wild-type cells (dotted line). (B) Wild-type RAW264.7 cells, TIR RAW264.7 cells, TIR RAW264.7 cells treated with panobinostat (100nM; PN) and TIR RAW264.7 cells treated with either scrambled (si-scramble) or HDAC8 (si-HDAC8) siRNAs for 40 h were harvested, and levels of H3K27Ac were analyzed by Western blots. Data are representative images of three independent experiments. (C) Levels of H3K27Ac association with the genomic regions of the BNIP3, BNIP3 and MLN64 were evaluated by ChIP-qPCR analysis. DNAs from wild-type and TIR RAW 264.7 cells were sonicated, and immunoprecipitated using antibodies against H3K27Ac and rabbit IgG. Immunoprecipitated DNAs were analyzed by qPCR using primers targeting the genomic regions of BNIP3, BNIP3L, and MLN64, as described in Experimental Procedures. (D) Similarly, levels of HDAC8 (upper panel) and HDAC3 (lower panel) recruitments to the promoter and genomic regions of BNIP3 and MLN64 were analyzed using antibodies against HDAC8 and HDAC3, followed by qPCR targeting the genomic and promoter regions of BNIP3 and MLN64. Recruitments of HDAC8 and 3 to the promoter region of GAPDH were analyzed as controls. For ChIP efficiency, % of input DNA recovered by immunoprecipitation, was determined by qPCR. Rabbit IgG serum was used as a background control. Data are expressed as means ± SD (n=3), * P< 0.05; ** P < 0.01; N.S., not significant (Student’s t-test).
Since histone acetylation can enhance gene expression through inducing an open chromatin conformation (euchromatin), we examined whether association of H3K27Ac with the genomic regions of the mitochondrial death genes was reduced in TIR cells. Based on the ENCODE/LICR bone marrow-derived macrophage ChIP-seq database (34), H3K27Ac associates with the genomic regions of BNIP3, expanding ~ 1 kb in front of exon 1; BNIP3L, expanding ~3 kb within intron 3; MLN64, ~4 kb in front of exon 1. We performed ChIP assays using H3K27Ac antibodies, followed by qPCR targeting these gene regions (Fig. 5C). H3K27Ac association with the genomic regions of BNIP3 and MLN64 was ~12-fold and ~3-fold lower in TIR than wild-type cells, respectively. Interestingly, no differences were detected in the BNIP3L genomic region. Since H3K27Ac was associated with the gene regions of BNIP3 and MLN64, we further examined whether HDAC8 was also recruited to these regions. Indeed, HDAC8 was recruited to the H3K27Ac-associated regions of BNIP3 and MLN64, but not their promoter regions, about 2-fold higher levels in TIR than wild-type cells (Fig. 5D, upper panel). Recruitment of HDAC8 to the promoter region of GAPDH was not different between wild-type and TIR cells. Unlike HDAC8, HDAC3 was highly recruited to both promoter and H3K27Ac-associated regions of these genes; however, no significant differences were detected between wild-type and TIR cells (Fig. 5D, lower panel). These data suggest that, in TIR cells, high levels of HDAC8 recruitment to the H3K27Ac-associated genomic regions of BNIP3 and MLN64 involved in the reduction of H3K27Ac levels and suppression of BNIP3 and MLN64 expression.
Anacardic acid or knocking down histone acetyl transferases (HATs) p300 and cAMP response element-binding protein binding protein (CBP) mRNAs inhibits LeTx-induced pyroptosis
Since levels of histone acetylation are balanced by the two opposing enzymes HATs and HDACs, we suspected that inhibition of HATs could decrease levels of histone acetylation and render resistance to LeTx. As expected, the HAT inhibitor anacardic acid protected wild-type RAW264.7 cells from LeTx-induced cell death in a dose-dependent manner (Fig. 6A). Among more than 16 HATs identified in humans, p300 and CBP, but not p300-associated factor (PCAF), were shown to target H3K27 (35, 36). Indeed, knocking down p300 and/or CBP, but not PCAF, by siRNAs rendered a partial protection from LeTx-induced cell death in wild-type RAW264.7 cells (Supplemental Fig. S3A and Fig. 6B). When CBP and/or p300 were knocked down in HDAC8-overexpressing cells, their protective effects were further enhanced (Fig. 6B, solid bars). Successful stable transfection was confirmed by Western blots showing constitutive expression of EGFP or HDAC8-EGFP (Fig. 6C, left bottom panel). Knocking down p300 and/or CBP also suppressed the levels of H3K27Ac (Fig. 6C, left top and right panels) and, in HDAC8 over-expressing cells, these siRNAs caused a more dramatic decrease of H3K27Ac. RT-qPCR analysis for the specificity and efficiency of the siRNAs showed that one or a combination of siRNAs specifically knocked down ~50% of their target mRNAs (Supplemental Fig. S3B). Collectively, these results suggest that histone acetylation levels mediated by HDAC8 and p300/CBP regulated susceptibility to LeTx-induced pyroptosis in macrophages.
Figure 6. The chemical HAT inhibitor anarcardic acid or si-RNAs targeting p300 and CBP, but not PCAF, render resistance to LeTx-induced cell death and decrease H3K27 acetylation in wild-type RAW264.7 cells.

(A) RAW264.7 cells were pretreated with or without anacardic acid for 14 h and then exposed to LeTx (500 ng/ml LF and 1 μg/ml PA) for 5 h. (B) RAW264.7 cells stably expressing EGFP or HDAC8-EGFP were transfected with si-scramble, si-P300, si-CBP or both si-p300/si-CBP for 40 h. Cells were then treated with LeTx (500 ng/ml LF and 1 μg/ml PA) for 5 h. (A–B) Cell death was measured by MTT assays as described in Experimental Procedures and the extent of cell death was expressed as % of cell death relative to that of non-treated cells. Data are expressed as means ± SD (n≥3). (C) Levels of H3K27 acetylation, and stable expression of EGFP and HDAC8-EGFP were analyzed by Western blots. Data are representative images of three independent experiments. Intensities of H3K27Ac were analyzed by NIH Image J program and results are expressed as means ± SD (n=3).
Sub-lethal dose of LeTx induces TIR in bone marrow-derived primary macrophages through HDAC8
To verify that TIR is not a phenomenon limited to RAW264.7 cells, primary bone marrow-derived macrophages (BMDMs) were exposed to a sub-lethal dose of LeTx that killed ~ 20% of cells in 24 h (data not shown). The surviving cells pooled after 24 h showed clear degradation of MEK1 (Fig. 7A, top panel), indicating that these cells had no defects in incorporating LeTx into the cytoplasm, and were resistant to subsequent challenges with a lethal dose of LeTx (Fig. 7A, bottom panel). Consistent with data shown in RAW264.7 cells, BMDM TIR cells were re-sensitized to LeTx by PN, which was prevented by caspase-1 si-RNAs (Fig. 7B). Furthermore, the decrease of BNIP3 and BNIP3L mRNA expression in TIR cells were also refurnished by PN treatments (Fig. 7C). To address the involvement of HDAC8 in BMDM TIR cells, HDAC8 and/or caspase-1 were knocked down by si-RNAs and cell death was examined by MTT assays. BMDM TIR cells knocked down in HDAC8 were susceptible to LeTx-induced cell death in a caspse-1-dependent manner (Fig. 7D). Also, expression of BNIP3, BNIP3L and MLN64 mRNAs was suppressed in BMDM TIR cells, and knocking down HDAC8 refurnished the expression to above those of wild-type cells (Fig. 7E). As in RAW264.7 cells, knocking down p300 and CBP, but not PCAF, rendered BMDMs resistance to LeTx-induced cell death (Fig. 7F). Collectively, as in RAW264.7 cells, HDAC8 and p300/CBP played an opposite role in determining susceptibility to LeTx-induced pyroptosis in primary BMDMs.
Figure 7. HDAC8 and p300/CBP are involved in determining susceptibility to LeTx in BMDMs as in RAW264.7 cells.

BMDMs from 129/S1/Svlmj mice were treated with a sub-lethal dose of LeTx (100 ng/ml LF and 100 ng/ml PA) for 24 h. Surviving cells were then re-plated onto new dishes or 96-well plates with fresh media and used as TIR BMDMs. (A) Wild-type and TIR BMDMs were harvested for Western blot analysis for MEK1 and p38 (upper panel), or treated with a lethal dose of LeTx (250 ng/ml LF and 500 ng/ml PA) for 5 h. (B) TIR BMDMs were transfected with scramble (si-scramble) or Caspase-1 targeting (si-Casp-1) siRNAs for 16 h, followed by PN or none for another 9 h. These cells were then treated with LeTx (250 ng/ml LF and 500 ng/ml PA) for 5 h. (C) TIR BMDMs were treated with none or panobinostat (PN; 50 nM) for 9 h and expression of BNIP3 and BNIP3L was analyzed by real-time qPCR. (D) TIR BMDMs were treated with scramble (si-scramble) or HDAC8 targeting (si-HDAC8) with or without caspase-1 (si-Casp-1) siRNAs for 24 h. These cells or wild-type BMDMs treated with si-scrample were exposed to LeTx (250 ng/ml LF and 500 ng/ml PA) for 5 h. (E) BMDMs were pre-exposed to LeTx with a sublethal dose of LeTx (100 ng/ml LF and 100 ng/ml PA) for 24 h, and surviving TIR cells were then transfected with scramble (si-scramble) or HDAC8 targeting (si-HDAC8) siRNAs for 24 h. Expression of BNIP3, BNIP3L and MLN64 was analyzed by RT-qPCR, and levels of mRNAs were expressed as fold of change relative to those of wild-type BMDMs (dotted line). (F) Wild-type BMDMs were treated with si-scramble or p300 (si-p300), CBP (si-CBP) or PCAF (si-PCAF) targeting siRNAs for 24 h, and then exposed to LeTx (250 ng/ml LF and 500 ng/ml PA) for 5 h. Cell death was measured by MTT assays and the extent of cell death was expressed as % of cell death relative to that of non-treated (A, B) or wild-type BMDMs treated with si-scramble but without LeTx treatments (D, F). Data are expressed as means ± SD (n=3), * P< 0.05; ** P < 0.01 (Student’s t-test).
DISCUSSION
Although the TIR phenotype persists for ~5 days in the majority of RAW264.7 macrophages, a small population of TIR cells retains their resistance to LeTx for a much longer period, 5–7 weeks (21, 23). Studying these cells allowed us to identify key mitochondrial death genes required for LeTx-induced pyroptosis. Here, we further demonstrated that TIR was an epigenetic phenomenon, sensitive to both DNMT and HDAC chemical inhibitors (Supplemental Fig. S1 & Fig. 1). Consistent with our previous study (23), the DNMT inhibitor azacitidine sensitized TIR cells to LeTx-induced cell death in a dose-dependent manner (Supplemental Fig. S1). Among the main DNMTs, expression levels of DNMT1, but not DNMT3a and 3b, were correlated with the extents of TIR (Data not shown) and knocking down DNMT1 by siRNA sensitized TIR cells to LeTx (23). These results suggest that maintenance of DNA methylation by DNMT1, rather than de novo DNA methylation by DNMT3a or b, was involved in TIR. In line with this note, global DNA methylation levels in wild-type and TIR cells were both very low (~1%) and indifferent (data not shown). Several approaches including direct sequencing of several CpG islands found in the promoter regions of BNIP3, BNIP3L and MLN64, and ChIP-sequencing using the EpiMark Methylated DNA Enrichment kit (New England Biolabs, MA) and the Illumina next generation sequencing, revealed no difference in methylation levels of these genomic regions. At this moment, how DNMT1 is involved in TIR is still elusive and warrants further extensive studies.
In addition to DNA methylation, histone deacetylation often results in gene suppression. Here, we demonstrated that TIR is a phenomenon observed in both RAW264.7 cells and primary BMDMs and, at least in part, mediated by HDAC8 (Fig. 2 & 7). The physiological role of HDAC8 in regulating susceptibility to LeTx is further substantiated by rendering wild-type RAW264.7 cells resistant to LeTx by overexpression of HDAC8 (Fig. 3). HDAC8 was required for the suppression of all three mitochondrial death genes expression in TIR cells (Fig. 2 & 4). Unlike HDAC8, we ruled out the involvement of HDAC 1–3, 5, 6, 9 and 11 in TIR, based on the facts that expression levels of HDAC1, 2, 5 and 9 did not correlate with the extent of TIR (Supplemental Fig. S2), and the HDAC1, 2, 3, and 11-specific inhibitor mocetinostat (31) and the HDAC6-specific inhibitor CAY10603 (37) had no effects on TIR (Supplemental Fig. S1). However, more definitive studies are required to rule out the involvement of these HDACs in TIR.
HDAC8 is a member of the class I HDACs, which also include HDAC1, 2 and 3 (38). HDAC8 is constitutively active and widely expressed (33, 39, 40). To date, HDAC8 was shown to be involved in adenoviral E1A-12 protein mediated gene suppression of the histocompatibility complex I genes (41) and transcriptional repression by the inversion-16 fusion gene products in acute myeloid leukemia cells (42). Its role in the regulation of the mitochondrial death genes expression found in this study is a novel observation. HDAC8 was shown to deacetylate the core histones H2A/H2B, H3 and H4 in vitro (43, 44); however, its activities toward specific histones in vivo are unknown. Unlike many HDACs, which undergo various post-translational modifications including acetylation, ubiquitination and sumoylation, the only known post-translational modification of HDAC8 both in vivo and in vitro is phosphorylation at Ser39 by the protein kinase A (PKA) (43, 45). Ser39 phosphorylation of HDAC8 by PKA results in inhibition of its deacetylase activity, and cells treated with forskolin, a PKA activator, increases acetylation levels of histones H3 and H4, suggesting that HDAC8 targets histones H3 and H4 in vivo. Consistent with these observations, TIR cells expressing high levels of HDAC8 showed an overall decrease in histone H3 acetylation levels, most strikingly at H3K27 (Fig. 5A). HDAC8 also interacts with various non-histones, including PP1 phosphatase, cAMP response element-binding protein, heat shock proteins, heat shock protein 70 binding protein, cofilin and α-actin, and the human ortholog of the yeast ever-shorter telomeres 1B (46–49), which suggests non-histone-mediated roles of HDAC8. However, involvement of these interacting proteins in cellular functions and TIR remains to be investigated.
An interesting note regarding HDAC8 regulation by PKA is that anthrax edema factor is a calmodulin-activated adenylyl cyclase which potently activates PKA (50). Therefore, it will be interesting to examine whether edema toxin potentiates LeTx-induced cytotoxic and pathophysiologic effects (51, 52) through inhibiting HDAC8 and inducing the mitochondrial death genes. Enhanced expression of the mitochondrial death genes, BNIP3, BNIP3L and MLN64, can cause cytotoxicity and is involved in hypoxia-induced cardiovascular pathology (53–56), which is also observed in anthrax animal models (57, 58).
This study suggested that one of the downstream substrates of HDAC8 was H3K27Ac. H3K27Ac is involved in promoting gene expression (59, 60) and we demonstrated a strong reverse correlation between HDAC8 and H3K27Ac. Either inhibition or knocking down HDAC8 by chemical inhibitors or si-RNAs, respectively, enhanced H3K27 acetylation in TIR cells (Fig. 5B). Based on our ChIP-qPCR data, H3K27Ac was associated with the genomic areas of BNIP3L (~10-folds above the basal (IgG) level) and strongly with BNIP3 and MLN64 (~100-folds and 40-folds above the basal levels) in wild-type cells (Fig. 5C). In TIR cells, H3K27Ac associations with BNIP3 and MLN64 were substantially diminished; whereas, no differences were found in BNIP3L. Consistently, HDAC8 was recruited to the H3K27Ac-associated regions, but not promoter regions, of BNIP3 and MLN64 in TIR cells (Fig. 5D). It is possible that HDAC8 was recruited to the H3K27Ac-associated enhancer regions of BNIP3 and MLN64 genes, and regulated access of Pol-II complexes to the promoter regions of these genes. Therefore, low levels of H3K27Ac in TIR cells were likely involved in the suppression of BNIP3 and MLN64 expression through inducing the closed (repressed) chromatin conformation in these genomic regions (Fig. 8). Further studies such as HDAC8 ChIP-sequencing are required to reveal whether HDAC8 is recruited to genomic regions in a sequence-specific manner and, if so, by what mechanisms.
Figure 8. A diagram illustrating the proposed epigenetic reprogramming in modulating susceptibility to LeTx-induced pyroptosis.

TIR cells induce expression of HDAC8 which favors deacetylation of histones, such as H3K27, and induce closed conformation of chromatin in the genomic regions of BNIP3, BNIP3L and MLN64. Suppression of the mitochondrial death genes renders resistance to LeTx-induced pyroptosis. Reversely, HATs such as p300 and CBP favor acetylation of histones (eg. H3K27) and induce open conformation of chromatin, consequently rendering macrophages susceptible to LeTx.
Acetylation levels of histones are balanced by two counteracting enzymes: HDACs and histone acetyltransferases (HATs). Although the mechanism of H3K27 acetylation/deacetylation is less known, previous studies have shown that the HATs, CBP and p300, target H3K27; whereas, GCN5 and PCAF target H3K9 (35, 36). Consistent with these observations, we found that both CBP and p300, but not PCAF, were involved in H3K27 acetylation (Fig. 6C) and knocking down these HATs rendered wild-type RAW264.7 cells and primary BMDMs resistance to LeTx (Fig. 6B & 7F). Since no differences in expression of CBP/p300 were detected between wild-type and TIR cells (data not shown), the decrease of H3K27Ac levels in TIR cells was mainly due to up-regulation of HDAC8. However, favoring histone deacetylation through manipulating CBP/p300 and HDAC8 in wild-type cells synergistically enhanced resistance to LeTx-induced pyroptosis (Fig. 6B & 8).
H3K27 can be either acetylated or methylated, and these two modifications are mutually exclusive (36, 61, 62). H3K27 methylation causes gene suppression through forming the polycomb repressive complex 2 (63). H3K27 methylation is balanced by the two counteracting enzymes: the methyltransferase EZH2 (enhancer of zeste homolog 2) (64) and the lysine (K)-specific demethylase family 6 (the jumnonji D3 (JMJD3) and UTX) (65). H3K27 methylation by EZH2 is crucial for survival of leukemia cells (66); whereas, JMJD3 and UTX play key roles in transcriptional activation of inflammatory genes (67) and macrophage differentiation (68–70). A previous study showed that RAW264.7 cells pre-exposed to lipopolysaccharide induce JMJD3, which is maintained in LeTx-exposed surviving cells (22). Induction of JMJD3 in toxin tolerant cells likely causes H3K27 demethylation and gene expression. However, the study did not show whether JMJD3 expression was crucial for the tolerance. Also, LeTx alone does not induce JMJD3 expression, suggesting that up-regulation of JMJD3 is induced by LPS treatment. The role of JMJD3 and H3K27 methylation in LeTx-resistance in LPS exposed cells warrants further studies.
Epigenetic regulation in expression of the mitochondrial death genes BNIP3, BNIP3L and MLN64 is not well understood. In ventricular myocytes, BNIP3 expression is inhibited by HDAC1 through direct interaction with NF-κB, a non-histone mediated mechanism (71). Certain tumors suppress expression of BNIP3 and BNIP3L through promoting histone deacetylation and subsequent methylation (72–74), particularly at H3K9 (75). We also detected decreased levels of H3K9Ac in TIR cells (Fig. 5A), but its role in the down-regulation of the mitochondrial death genes expression and LeTx resistance remains to be explored. Here we showed that HDAC8 suppressed BNIP3 and BNIP3L expression at the transcription level (Fig. 4B), and H3K27 deacetylation by HDAC8 could be involved in the suppression of BNIP3 and MLN64 expression (Fig. 5B–C).
In summary, this study demonstrated that epigenetic reprogramming mediated by HDAC8 played a key role in TIR. The epigenetic reprogramming described in this study provides a new venue for manipulating susceptibility of macrophages to pyroptosis induced by LeTx and possibly by other inflammasome-induced pyroptosis.
Supplementary Material
Acknowledgments
We would like to thank Dr. R. Dekoter and Mr. Ali Abbas for their help in designing and performing ChIP assays.
Footnotes
This work was supported by Canadian Institute of Health Research Operating Grant (MOP93551) to S.O.K.
References
- 1.Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8:372–379. doi: 10.1038/nri2296. [DOI] [PubMed] [Google Scholar]
- 2.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 3.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 4.Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol. 2008;83:13–30. doi: 10.1189/jlb.0607402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galan JE. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedra JH, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, Flavell RA, Fikrig E. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J Immunol. 2007;179:4783–4791. doi: 10.4049/jimmunol.179.7.4783. [DOI] [PubMed] [Google Scholar]
- 7.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, Flavell RA. Roles of caspase-1 in Listeria infection in mice. Int Immunol. 2004;16:335–343. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- 10.Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, Takeda K, Zychlinsky A. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 11.Fortier A, Diez E, Gros P. Naip5/Birc1e and susceptibility to Legionella pneumophila. Trends Microbiol. 2005;13:328–335. doi: 10.1016/j.tim.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog. 2012;7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aachoui Y, I, Leaf A, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao EA, I, Leaf A, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freche B, Reig N, van der Goot FG. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin Immunopathol. 2007;29:249–260. doi: 10.1007/s00281-007-0085-0. [DOI] [PubMed] [Google Scholar]
- 17.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, Alexander WS, Kile BT, Croker BA. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37:1009–1023. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soong G, Chun J, Parker D, Prince A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis. 2012;205:1571–1579. doi: 10.1093/infdis/jis244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 20.Salles, Tucker AE, Voth DE, Ballard JD. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc Natl Acad Sci U S A. 2003;100:12426–12431. doi: 10.1073/pnas.2134042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha SD, Ng D, Lamothe J, Valvano MA, Han J, Kim SO. Mitochondrial proteins Bnip3 and Bnip3L are involved in anthrax lethal toxin-induced macrophage cell death. J Biol Chem. 2007;282:26275–26283. doi: 10.1074/jbc.M703668200. [DOI] [PubMed] [Google Scholar]
- 22.Das ND, Jung KH, Chai YG. The role of NF-kappaB and H3K27me3 demethylase, Jmjd3, on the anthrax lethal toxin tolerance of RAW 264.7 cells. PLoS One. 2010;5:e9913. doi: 10.1371/journal.pone.0009913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha SD, Park S, Han CY, Nguyen ML, Kim SO. Cellular adaptation to anthrax lethal toxin-induced mitochondrial cholesterol enrichment, hyperpolarization, and reactive oxygen species generation through downregulating MLN64 in macrophages. Mol Cell Biol. 2012;32:4846–4860. doi: 10.1128/MCB.00494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gasser SM, Paro R, Stewart F, Aasland R. The genetics of epigenetics. Cell Mol Life Sci. 1998;54:1–5. doi: 10.1007/s000180050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin. 2013;6:24. doi: 10.1186/1756-8935-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saetrom P, Snove O, Jr, Rossi JJ. Epigenetics and microRNAs. Pediatr Res. 2007;61:17R–23R. doi: 10.1203/pdr.0b013e318045760e. [DOI] [PubMed] [Google Scholar]
- 27.Wesche J, Elliott JL, Falnes PO, Olsnes S, Collier RJ. Characterization of membrane translocation by anthrax protective antigen. Biochemistry. 1998;37:15737–15746. doi: 10.1021/bi981436i. [DOI] [PubMed] [Google Scholar]
- 28.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 29.Warren MK, Vogel SN. Bone marrow-derived macrophages: development and regulation of differentiation markers by colony-stimulating factor and interferons. J Immunol. 1985;134:982–989. [PubMed] [Google Scholar]
- 30.DeKoter RP, Geadah M, Khoosal S, Xu LS, Thillainadesan G, Torchia J, Chin SS, Garrett-Sinha LA. Regulation of follicular B cell differentiation by the related E26 transformation-specific transcription factors PU.1, Spi-B, and Spi-C. J Immunol. 2010;185:7374–7384. doi: 10.4049/jimmunol.1001413. [DOI] [PubMed] [Google Scholar]
- 31.Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, Macleod AR, Besterman JM, Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 32.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 33.Murko C, Lagger S, Steiner M, Seiser C, Schoefer C, Pusch O. Expression of class I histone deacetylases during chick and mouse development. Int J Dev Biol. 2010;54:1527–1537. doi: 10.1387/ijdb.092971cm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 35.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010;38:4958–4969. doi: 10.1093/nar/gkq244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Bjerling P, Silverstein RA, Thon G, Caudy A, Grewal S, Ekwall K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol Cell Biol. 2002;22:2170–2181. doi: 10.1128/MCB.22.7.2170-2181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfson NA, Ann Pitcairn C, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99:112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Ricciardi RP. E1A is the component of the MHC class I enhancer complex that mediates HDAC chromatin repression in adenovirus-12 tumorigenic cells. Virology. 2006;352:338–344. doi: 10.1016/j.virol.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 42.Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol. 2003;23:607–619. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu E, Chen Z, Fredrickson T, Zhu Y, Kirkpatrick R, Zhang GF, Johanson K, Sung CM, Liu R, Winkler J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J Biol Chem. 2000;275:15254–15264. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- 44.Buggy JJ, Sideris ML, Mak P, Lorimer DD, McIntosh B, Clark JM. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem J. 2000;350(Pt 1):199–205. [PMC free article] [PubMed] [Google Scholar]
- 45.Lee H, Rezai-Zadeh N, Seto E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol Cell Biol. 2004;24:765–773. doi: 10.1128/MCB.24.2.765-773.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson SD, Yates JR, 3rd, Montminy M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- 47.Karolczak-Bayatti M, Sweeney M, Cheng J, Edey L, Robson SC, Ulrich SM, Treumann A, Taggart MJ, Europe-Finner GN. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J Biol Chem. 2011;286:34346–34355. doi: 10.1074/jbc.M111.278549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waltregny D, Glenisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–968. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 49.Lee H, Sengupta N, Villagra A, Rezai-Zadeh N, Seto E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol Cell Biol. 2006;26:5259–5269. doi: 10.1128/MCB.01971-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tournier JN, Quesnel-Hellmann A, Cleret A, Vidal DR. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007;9:555–565. doi: 10.1111/j.1462-5822.2006.00866.x. [DOI] [PubMed] [Google Scholar]
- 52.Tournier JN, Quesnel-Hellmann A, Mathieu J, Montecucco C, Tang WJ, Mock M, Vidal DR, Goossens PL. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J Immunol. 2005;174:4934–4941. doi: 10.4049/jimmunol.174.8.4934. [DOI] [PubMed] [Google Scholar]
- 53.Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–127. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374–383. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tichauer JE, Morales MG, Amigo L, Galdames L, Klein A, Quinones V, Ferrada C, Alvarez AR, Rio MC, Miquel JF, Rigotti A, Zanlungo S. Overexpression of the cholesterol-binding protein MLN64 induces liver damage in the mouse. World J Gastroenterol. 2007;13:3071–3079. doi: 10.3748/wjg.v13.i22.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 57.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guichard A, Nizet V, Bier E. New insights into the biological effects of anthrax toxins: linking cellular to organismal responses. Microbes Infect. 2012;14:97–118. doi: 10.1016/j.micinf.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin JH, Li WR, Gao Y, Baldwin Rt, Li CJ. Genome-wide ChIP-seq mapping and analysis reveal butyrate-induced acetylation of H3K9 and H3K27 correlated with transcription activity in bovine cells. Funct Integr Genomics. 2012;12:119–130. doi: 10.1007/s10142-012-0263-6. [DOI] [PubMed] [Google Scholar]
- 60.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lan F, Shi Y. Epigenetic regulation: methylation of histone and non-histone proteins. Sci China C Life Sci. 2009;52:311–322. doi: 10.1007/s11427-009-0054-z. [DOI] [PubMed] [Google Scholar]
- 63.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 64.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 65.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 66.Fiskus W, Pranpat M, Balasis M, Herger B, Rao R, Chinnaiyan A, Atadja P, Bhalla K. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol Cancer Ther. 2006;5:3096–3104. doi: 10.1158/1535-7163.MCT-06-0418. [DOI] [PubMed] [Google Scholar]
- 67.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 68.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 69.Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, FtCarson W, Cavassani KA, Li X, Lukacs NW, Hogaboam CM, Dou Y, Kunkel SL. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res. 2011;26:2665–2671. doi: 10.1002/jbmr.464. [DOI] [PubMed] [Google Scholar]
- 71.Shaw J, Zhang T, Rzeszutek M, Yurkova N, Baetz D, Davie JR, Kirshenbaum LA. Transcriptional silencing of the death gene BNIP3 by cooperative action of NF-kappaB and histone deacetylase 1 in ventricular myocytes. Circ Res. 2006;99:1347–1354. doi: 10.1161/01.RES.0000251744.06138.50. [DOI] [PubMed] [Google Scholar]
- 72.Bacon AL, Fox S, Turley H, Harris AL. Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene. 2007;26:132–141. doi: 10.1038/sj.onc.1209761. [DOI] [PubMed] [Google Scholar]
- 73.Perez-Perarnau A, Coll-Mulet L, Rubio-Patino C, Iglesias-Serret D, Cosialls AM, Gonzalez-Girones DM, de Frias M, de Sevilla AF, de la Banda E, Pons G, Gil J. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics. 2011;6:1228–1235. doi: 10.4161/epi.6.10.17200. [DOI] [PubMed] [Google Scholar]
- 74.Knutson AK, Welsh J, Taylor T, Roy S, Wang WL, Tenniswood M. Comparative effects of histone deacetylase inhibitors on p53 target gene expression, cell cycle and apoptosis in MCF-7 breast cancer cells. Oncol Rep. 2012;27:849–853. doi: 10.3892/or.2011.1590. [DOI] [PubMed] [Google Scholar]
- 75.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109:E3367–3376. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.