

Abstract
Activation of vascular endothelial growth factor receptor-2 (VEGFR-2), an endothelial cell receptor tyrosine kinase, promotes tumor angiogenesis and ocular neovascularization. We report the methylation of VEGFR-2 at multiple Lys and Arg residues, including Lys1041, a residue that is proximal to the activation loop of the kinase domain. Methylation of VEGFR-2 was independent of ligand binding and was not regulated by ligand stimulation. Methylation of Lys1041 enhanced tyrosine phosphorylation and kinase activity in response to ligands. Additionally, interfering with the methylation of VEGFR-2 by pharmacological inhibition or by site-directed mutagenesis revealed that methylation of Lys1041 was required for VEGFR-2–mediated angiogenesis in zebrafish and tumor growth in mice. We propose that methylation of Lys1041 promotes the activation of VEGFR-2 and that similar posttranslational modification could also regulate the activity of other receptor tyrosine kinases.
INTRODUCTION
The molecular mechanisms underlying the activation of receptor tyrosine kinases (RTKs) and their downstream signaling mechanisms are highly conserved. Activation of RTKs results in an increase in the intrinsic catalytic activity and creates binding sites on the RTKs that recruit cytoplasmic signaling proteins (1). The autophosphorylation sites either are involved in the regulation of kinase activity of the receptor itself or can serve as binding sites for SH2 (Src homology 2) domain– and PTB (protein tyrosine binding) domain–containing proteins (2). The interplay between posttranslational modifications is essential for appropriate regulation of RTK signaling, protein-protein interactions, degradation, and trafficking (3). An example of crosstalk between posttranslational modifications of RTKs is that between phosphorylation and ubiquitination events. Phosphorylation of RTKs such as epidermal growth factor receptor (EGFR) (4) and platelet-derived growth factor receptor (PDGFR) (5) and serine phosphorylation of vascular endothelial growth factor receptor-2 (VEGFR-2) (6, 7) recruit ubiquitin E3 ligases to these RTKs, which ultimately triggers their ubiquitination and targets them for proteasomal degradation.
Methylation is a posttranslational modification that occurs on the side chains of Lys and Arg residues of various proteins such as histone proteins (8), the tumor suppressor p53 (9, 10), forkhead box O (FOXO) transcription factors (11), and RTKs such as EGFR (12) and VEGFR-1 (also known as FLT-1) (13). Similar to protein phosphorylation and ubiquitination, methylation can regulate protein-protein interactions and other key protein functions. The transfer of methyl groups from S-adenosylmethionine (SAM), a universal methyl donor, to the side-chain nitrogen atoms of Arg and Lys residues is catalyzed by protein arginine methyltransferases and lysine methyltransferases, respectively (14).
VEGFR-2 is a key RTK in endothelial cells and plays a major role in embryonic development and pathological angiogenesis during cancer and ocular neovascularization (15). VEGFR-2 consists of an extracellular region that contains a ligand binding site, a transmembrane domain, and a cytoplasmic region, which has intrinsic tyrosine kinase activity. In the inactive state, the activation loop occupies the active site, preventing substrate access and adenosine 5′-triphosphate (ATP) binding (16, 17). Activation of VEGFR-2 stimulates various signal transduction pathways including phosphatidylinositol 3-kinase, which is involved in endothelial cell survival and proliferation (18), phospholipase C–γ1 (PLCγ1), which stimulates tubulogenesis in endothelial cells (19), Src family kinases (20, 21), and IQGAP1 (20). Here, we provide evidence that VEGFR-2 undergoes methylation at multiple Arg and Lys residues and that methylation of Lys1041 plays a critical role in the tyrosine phosphorylation of VEGFR-2 and VEGFR-2–mediated angiogenesis.
RESULTS
VEGFR-2 is methylated on Lys and Arg residues in cells
We investigated methylation of VEGFR-2 in human primary umbilical vein endothelial cells (HUVECs). VEGFR-2 was methylated on both Lys and Arg residues in HUVECs (Fig. 1A), as detected by anti–methyl-lysine and arginine antibodies (fig. S1, A and B). We used a chimeric VEGFR-2 called CKR [in which the extracellular domain of VEGFR-2 was replaced with the extracellular domain of human colony-stimulating factor 1 (CSF-1) receptor (16)] to establish that the cytoplasmic domain of VEGFR-2 is directly methylated and also to avoid possible crosstalk and dimerization between VEGFR-2 and other VEGFR isoforms. Metabolic labeling of human embryonic kidney (HEK) 293 or porcine aortic endothelial (PAE) cells expressing CKR with the methyl donor SAM showed that VEGFR-2 was methylated in cells (Fig. 1B). Both immature and mature (fully glycosylated and cell surface–localized) CKRs were detected, suggesting that methylation of VEGFR-2 occurs before ligand-mediated tyrosine phosphorylation of VEGFR-2 (Fig. 1B). Methylation of CKR was not altered by stimulation with CSF-1 and was not detected in control vector-transfected PAE cells (Fig. 1C).
Fig. 1. VEGFR-2 is methylated on lysine and arginine residues.

(A) Anti–methyl-arginine and anti–methyl-lysine immunoprecipitates (IP) from HUVECs were blotted for VEGFR-2. Whole-cell lysates (WCL) were blotted for total VEGFR-2 and PLCγ1 as a loading control. Data are representative of three independent experiments. (B) Autoradiography of VEGFR-2 immunoprecipitates from PAE and HEK-293 cells expressing chimeric VEGFR-2 (CKR) and labeled with SAM. Whole-cell lysates were blotted for total VEGFR-2 and PLCγ1. Data are representative of three independent experiments. (C) Serum-starved PAE cells expressing empty vector (pMSCV) or CKR were stimulated with CSF-1. Anti–methyl-lysine immunoprecipitates were blotted for VEGFR-2. Whole-cell lysates were also blotted for total VEGFR-2 and PLCγ1. Blots are representative of three independent experiments. (D) Serum-starved PAE cells expressing wild-type VEGFR-2 or K1041R–VEGFR-2 were either not stimulated (–) or stimulated with VEGF (+), and cell lysates were blotted for phospho-Tyr1052-VEGFR-2, total VEGFR-2, and PLCγ1 as a loading control. Results are presented as induction of VEGFR-2 phosphorylation at Tyr1052 in response to VEGF stimulation, normalized to total VEGFR-2. Median of three independent experiments is shown. Error bars indicate range. *P= 0.05. (E) Immunoprecipitated wild-type VEGFR-2 and K1041R mutant from serum-starved PAE cells were subjected to in vitro kinase assays. The same membrane was reblotted for total VEGFR-2. Results are presented as induction of tyrosine phosphorylation of VEGFR-2 in response to ATP stimulation, normalized to total VEGFR-2. Median of three independent experiments is shown. Error bars indicate range. *P = 0.05. AU, arbitrary units.
To identify the methylated Arg and Lys residues on VEGFR-2, we analyzed VEGFR-2 immunoprecipitated from PAE cells expressing VEGFR-2 by liquid chromatography–tandem mass spectrometry (LC-MS/MS). The MS analysis identified five methylated residues on VEGFR-2: Arg817, Lys856, Lys861, Lys1041, and Arg1115 (fig. S2A). Lys856, Lys861, Lys1041, and Arg1115 are located in the kinase domain of VEGFR-2 (22) and are surface-exposed, suggesting that they could be posttranslationally modified (fig. S2B). Lys856 and Lys861 are located in the C-loop of the kinase domain (22) upstream of the ATP binding site Lys866 (fig. S2, B and C). Lys1041 is located in the unstructured and flexible linker region between the two β sheets of the kinase domain, and Arg1115 is located at the N-terminal end of the kinase domain, distal to the key tyrosine autophosphorylation sites (Tyr1052 and Tyr1057) and Lys866 in the ATP binding site (fig. S2B). In vitro methylation assays of synthesized peptides corresponding to Lys856, Lys861, Lys1041, and Arg1115 showed that Lys856, Lys861, Lys1041, and Arg1115 were methylated, thus demonstrating that VEGFR-2 can be directly methylated (fig. S2D).
Further MS analysis showed that the stoichiometry of methylation of Lys1041 was similar to that of tyrosine phosphorylation of VEGFR-2 (fig. S3), suggesting that methylation of Lys1041 could be as prevalent as phosphorylation of Tyr1057, a phosphorylation event that is required to stimulate the kinase activity of VEGFR-2 (20).
Methylation of Lys1041 is required for the tyrosine phosphorylation and kinase activation of VEGFR-2
To ascertain the role of methylation of Lys856, Lys861, Lys1041, and Arg1115 in kinase activation and tyrosine phosphorylation of VEGFR-2, we individually mutated these residues in the CKR chimera to either a different basic residue (Lys to Arg or Arg to Lys) or a hydrophobic residue (Ala) (16) to generate methylation mutant VEGFR-2 constructs and avoid complications of endogenous VEGF receptors in endothelial cells.
We assessed the phosphorylation status of Tyr1052 (which corresponds to Tyr1054 in human VEGFR-2) and Tyr1173 (which corresponds to Tyr1175 in human VEGFR-2) in the CKR mutants with phospho-VEGFR-2–specific antibodies. The ligand-dependent tyrosine phosphorylation of the CKR chimera was significantly inhibited by the K1041R mutation but not by the R817K, K856R, or R1115K mutation (fig. S4A). The K1041A mutation also inhibited the phosphorylation of VEGFR-2 in response to ligand stimulation (fig. S4B). Unexpectedly, the K1041A mutation inhibited tyrosine phosphorylation to a greater extent than did the K1041R mutation (fig. S4B); in addition, the K1041F mutation also produced a similar effect (fig. S5, A to C), suggesting that Phe and Ala substitutions at Lys1041 may induce structural changes in VEGFR-2 that prevent phosphorylation of the autocatalytic tyrosine residues. Substitution of Arg to Phe in some cases mimics the effect of Arg methylation (23), but a similar mutation in VEGFR-2 did not act as a Lys methylation mimetic mutation. Substitution of Arg to Gln mimics the effect of Arg acetylation through neutralization of the positive charges of lysine (24, 25). Unlike the Arg→Phe or Arg→Ala mutation, the K1041Q mutation partially preserved ligand-induced tyrosine phosphorylation of VEGFR-2 (fig. S5, D to F).
In full-length mouse VEGFR-2, the K1041R mutation reduced VEGF-stimulated tyrosine phosphorylation and in vitro kinase activation (Fig. 1, D and E). Because ligand-induced tyrosine phosphorylation of RTKs requires ligand binding to RTKs followed by RTK dimerization (1), we assessed whether K1041R mutation affected ligand binding and ligand-induced VEGFR-2 dimerization. The K1041R VEGFR-2 mutant bound similar amounts of VEGF as wild-type VEGFR-2 (fig. S6A). In addition, VEGF-induced dimerization of the K1041RVEGFR-2 mutant was similar to that of the wild-type VEGFR-2 (fig. S6B), indicating that reduced methylation of Lys1041 in VEGFR-2 was not associated with impaired ligand binding or dimerization. Together, these data demonstrate that Lys1041 is methylated and that mutation of Lys1041 inhibits phosphorylation of VEGFR-2 without altering ligand binding and dimerization properties.
Methylation inhibitors reduce the activity of VEGFR-2
To further investigate the importance of methylation of VEGFR-2, we also used various pharmacological approaches. Treatment of PAE cells with adenosine, periodate oxidized (adenosine-2′,3′-dialdehyde, or “Adox”) or 3-deazaneplanocin A·HCl (DZNep) (26–28), both of which are global inhibitors of methylation, reduced the methylation of the CKR chimera (fig. S7A). The in vitro kinase activity of hypomethylated CKR was significantly inhibited (Fig. 2A). Similarly, phosphorylation of VEGFR-2 in response to ligand stimulation was also inhibited by treating cells with DZNep and Adox (Fig. 2B). The inhibitory effect of Adox on the tyrosine phosphorylation of VEGFR-2 is unlikely due to a direct effect on the kinase activity of VEGFR-2, because Adox did not inhibit the in vitro kinase activity of VEGFR-2 (Fig. 2C). Furthermore, the inhibitory effect of Adox on the phosphorylation of VEGFR-2 was reversed after Adox was washed off the cells (Fig. 2D). The tyrosine phosphorylation of VEGFR-2 after washout was similar to that of VEGFR-2 in nontreated cells (Fig. 2D), indicating that the methylation of newly synthesized VEGFR-2 was not inhibited. In addition, Adox treatment did not inhibit the ligand-stimulated tyrosine phosphorylation of the K1041Q CKR mutant (fig. S7B), indicating that the inhibitory effect of Adox on the tyrosine phosphorylation of VEGFR-2 requires an intact Lys1041residue.
Fig. 2. Adox and DZNep inhibit tyrosine phosphorylation of VEGFR-2.
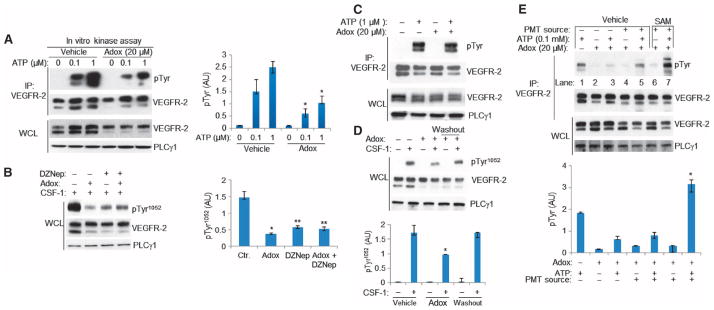
(A) Serum-starved PAE cells expressing CKR were treated with vehicle, Adox, or DZNep. VEGFR-2 immunoprecipitates from these cell lysates were subjected to in vitro kinase assays. Whole-cell lysates were blotted for total CKR and PLCγ1 as a protein loading control. Results are presented as induction of phosphorylation of CKR normalized to total CKR protein. Median of three independent experiments is shown. Error bars indicate range.*P= 0.049.(B) CKR-expressing PAE cells were pretreated with Adox and DZNep and stimulated with CSF-1. Cell lysates were blotted for anti-pTyr1052 (top panel) and reblotted for total CKR and PLCγ1. Results are presented as induction of phosphorylation of Tyr1052-VEGFR-2 normalized to total VEGFR-2. Median of three independent experiments is shown. Error bars indicate range. *P = 0.046; **P = 0.049 (C) CKR immunoprecipitates from serum-starved PAE cells were subjected to in vitro kinase assay in the presence or absence of Adox and immunoblotted with anti-pTyr antibody. The same membranes were reblotted for total CKR. Whole-cell lysates were blotted for CKR and PLCγ1. Data are representative of three independent experiments. (D) Cell lysates from PAE cells expressing CKR, which were untreated, treated with Adox, or treated with Adox and washed off, were blotted for anti-pTyr1052 and reblotted for VEGFR-2 and PLCγ1. Data are represented as phospho-Tyr1052 normalized to total CKR. Median of three independent experiments is shown. Error bars indicate range. *P = 0.05 for treatment with Adox compared to control; P = 0.05 for Adox treatment compared to washout; P = 0.827 for washout compared to no treatment. (E) PAE cells expressing VEGFR-2 were treated with Adox to generate hypomethylated VEGFR-2. The hypomethylated VEGFR-2 was immunoprecipitated and subjected to in vitro methylation assay followed by in vitro kinase assay. Results are presented as induction of tyrosine phosphorylation of VEGFR-2 normalized to total VEGFR-2 protein. Median of three independent experiments is shown. Error bars indicate range. *P = 0.049 for lane 7 compared to lane 5.
We next attempted to rescue the effects of methylation inhibitors on tyrosine phosphorylation of VEGFR-2 by in vitro remethylation (Fig. 2E). Hypomethylated CKR was immunoprecipitated from Adox-treated PAE cells and subjected to in vitro methylation assays containing SAM and cell homogenates as a source of protein methyltransferases (fig. S8A). We first confirmed that Adox inhibited the methylation of VEGFR-2 in PAE cells expressing VEGFR-2 (fig. S8B). Adox treatment inhibited the in vitro kinase activity of CKR (Fig. 2E, lane 5 compared to lane 1), an inhibition that was reversed upon subjecting CKR to in vitro methylation (Fig. 2E, lane 7 compared to lane 5). Furthermore, the addition of exogenous SAM and cell homogenate to the in vitro reactions increased kinase activity over that in the condition, suggesting that increased methylation correlated to increased kinase activity (Fig. 2E, lane 7 compared to lane 1). Addition of cell homogenate alone did not affect the tyrosine phosphorylation of VEGFR-2 (Fig. 2E, lane 4). Addition of exogenous SAM and cell homogenate did not increase the basal kinase activity of the K1041R VEGFR-2 mutant (fig. S8C, lane 1 compared to lane 7). These data demonstrate that hypomethylated VEGFR-2 with reduced kinase activity can be remethylated, and the kinase activity of remethylated VEGFR-2 is comparable to that of wild-type VEGFR-2.
Lys1041 is important for VEGFR-2–dependent angiogenesis and tumor growth
Angiogenesis involves capillary tube formation and proliferation of endothelial cells (29). Treatment of PAE cells expressing CKR with CSF-1 stimulated capillary tube formation, but CSF-1–stimulated PAE cells expressing the K1041 mutant CKR showed no capillary tube formation (Fig. 3A). Increasing the ligand concentration enhanced the proliferation of PAE cells expressing CKR, but not that of cells expressing the K1041R CKR mutant (Fig. 3B). In addition, treatment with Adox and DZNep significantly inhibited capillary tube formation by HUVECs (Fig. 3C). In addition to its proangiogenic activities, VEGFR-2 is also expressed in solid tumors and promotes tumor growth (7, 30, 31), leading us to assess the importance of Lys1041 in VEGFR-2–mediated tumor growth. In mice, the growth of xenograft tumors formed from B16F mouse melanoma cells was enhanced by expression of wild-type VEGFR-2, but not that of the K1041R VEGFR-2 mutant (Fig. 3D).
Fig. 3. Lys1041 is required for VEGFR-2–mediated angiogenesis and tumor growth.

(A) Lys1041 mutation inhibits capillary tube formation by PAE cells. PAE cells expressing CKR and the Lys1041 mutant CKR were assessed for formation of tubular structure by microscopy. Median of three independent experiments is shown. Error bars indicate range. *P < 0.045. (B) Mutation of Lys1041 inhibits VEGFR-2–dependent proliferation of PAE cells. Cells were treated with different concentrations of CSF-1 as indicated, and proliferation was measured by MTT assay. Median of three independent experiments is shown. *P < 0.015, **P < 0.037. (C) Adox or DZNep inhibits angiogenesis of HUVECs. Data are presented as median of three independent experiments. Error bars indicate range. *P < 0.028, **P < 0.034. (D) The K1041R mutation inhibits VEGFR-2–dependent tumor growth. B16F melanoma cells expressing empty vector, VEGFR-2, or Lys1401 mutant VEGFR-2 were used in mouse tumor xenograft assays. Median of two independent experiments consisting of two animals per condition is shown. Error bars indicate range. P = 0.00071 for VEGFR-2 and P = 0.674 for K1041R, compared to controls. Expression of VEGFR-2 and the Lys1041 VEGFR-2 mutant in B16F cells is shown. Western blots are representative of two independent experiments.
The zebrafish angiogenesis model is a validated in vivo angiogenesis assay and has been used to study the molecular regulation of angiogenesis (32, 33). Zebrafish expressing wild-type VEGFR-2 showed increased caudal vein plexus formation and tail microvasculature. In contrast, zebrafish expressing the K1041R VEGFR-2 mutant exhibited significantly reduced angiogenesis compared to those expressing wild-type VEGFR-2 (Fig. 4, A and B). These data demonstrate that Lys1041 plays a key role in VEGFR-2 activation and in its ability to stimulate angiogenesis and tumor growth.
Fig. 4. Mutation of Lys1041 inhibits VEGFR-2–induced angiogenesis in zebrafish.
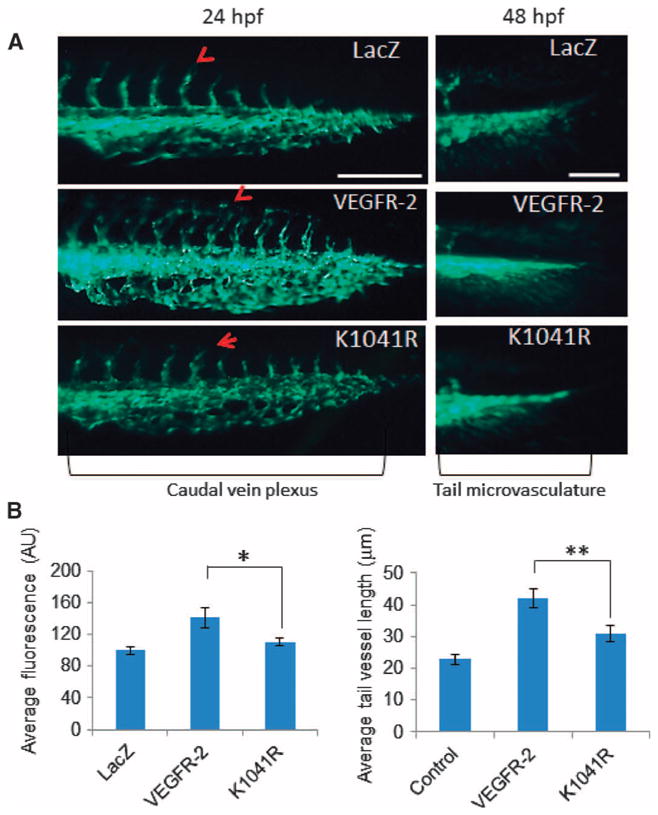
(A) mRNA encoding wild-type VEGFR-2, the Lys1041 mutant VEGFR-2, or LacZ was injected into one- or two-cell–stage embryos of Fli-eGFP transgenic zebrafish. Representative immunofluorescence images of the caudal vein plexus and tail microvasculature formation of 50 zebrafish are shown. The scale bars for caudal vein plexus and tail microvasculature images were 200 and 50 μm, respectively. hpf, hours post-fertilization. (B) Quantification of the area of the caudal vein plexus. Mean of three independent experiments is shown. Error bars represent SD.*P= 0.00016 for VEGFR-2 compared to K1041R. (C) Average tail vessel lengths. Mean vessel lengths from three independent experiments were 20.8, 42.1, and 30.4 μm for control, VEGFR-2, and K1041R, respectively. Error bars, SD. The random effects model showed P < 0.0001 for VEGFR-2 compared to control; P = 0.0322 for K1041R compared to control; and P = 0.0079 for VEGFR-2 compared to K1041R.
DISCUSSION
Angiogenesis contributes to the pathology of numerous human diseases, including cancer and age-related macular degeneration (34). Substantial progress has been made in delineating the features of this complex cellular event, and VEGFR-2 signaling has emerged as a key requirement for normal and pathological angiogenesis (35, 36). Here, we report a previously unknown mechanism by which the activation of VEGFR-2, a potent angiogenic RTK, is controlled by the methylation of Lys1041. Methylation of Lys1041 is important for phosphorylation of VEGFR-2, and interfering with its methylation by site-directed mutagenesis and chemical inhibitors blocks the ability of VEGFR-2 to stimulate angiogenesis and tumor growth.
Tyrosine and serine phosphorylation switches the angiogenic signaling of VEGFR-2 among different pathways by controlling both its interactions with specific cytoplasmic partner proteins and its degradation (17, 37, 38). Lys1041 is positioned proximal to Tyr1052 in the activation loop, and autophosphorylation of Tyr1057 plays a key regulatory role in the kinase activation of VEGFR-2. Our data suggest that methylation of Lys1041 positively influences kinase activation of VEGFR-2. Replacement of Lys1041 with Gln (a hydrophobic, negatively charged amino acid) partially mimicked the effect of Lys methylation and generated resistance to the suppression of kinase activity by the methylation inhibitor Adox. It is conceivable that substitution of Lys1041 with Gln or methylation of Lys1041 relieves the strong positive charge conferred by the unmodified residue and that this state enables VEGFR-2 kinase activation. Unlike Lys1041, mutation of Arg817, Lys856, Lys861, or Arg1115 had no apparent effect on the tyrosine phosphorylation of VEGFR-2.
The research directed toward understanding the basic properties of VEGFR-2 has led to the development of various therapeutics such as bevacizumab and ranibizumab, inhibitors of VEGF receptor signaling that have been approved by the U.S. Food and Drug Administration for treatment of age-related macular degeneration and various cancers, including colorectal, lung, and kidney cancers and glioblastoma (39, 40). Demonstrating that methylation of VEGFR-2 is important for its function suggests that this posttranslational modification could be exploited in the development of new antiangiogenesis strategies. Because various small chemical inhibitors of the methylation pathway such as Adox and DZNep inhibit VEGFR-2 phosphorylation and its angiogenic activity, it is reasonable to envision that targeting methylation could offer a novel therapeutic strategy to inhibit angiogenesis.
MATERIALS AND METHODS
Growth factors, pharmacological inhibitors, and antibodies
Human recombinant CSF-1 was purchased from R&D Systems. Anti–phospho-KDR/Flk-1/VEGFR-2 (Tyr1054), clone D1W, anti-phosphotyrosine 4G10 [immunoglobulin G2b (IgG2b)], and anti–VEGFR-2 were purchased from Millipore. Anti–phospho-VEGFR-2 Tyr1175 (Tyr1173 in mouse VEGFR-2) (clone 19 A10) was purchased from Cell Signaling. Anti–phospho-VEGFR-2 Tyr801 was obtained through ECM Biosciences. The following anti-methyl antibodies were purchased from Upstate: anti–dimethyl-arginine, asymmetric (ASYM24), anti–dimethyl-arginine, symmetric (SYM11), and anti–dimethyl-arginine, symmetric (SYM10). The following anti-methyl antibodies were purchased from Abcam: anti–dimethyl-arginine (21C7), anti–methylated lysine, and anti–monomethyl-arginine (16B11). Rabbit polyclonal anti-PLCγ1 antibody (pTyr783) was purchased from Biosource. The rabbit polyclonal anti–VEGFR-2 serum was raised against a GST–VEGFR-2 C-terminal fusion protein (16). Anti-PLCγ1 (sc-81) and horseradish peroxidase–conjugated goat anti-rabbit IgG (sc-2054) and goat anti-mouse IgG (sc-2055) secondary antibodies were purchased from Santa Cruz Biotechnology Inc. Adox was purchased from Sigma (A-7154). DZNep was synthesized as described (26, 41).
MS analysis
VEGFR-2 was immunoprecipitated with anti–VEGFR-2 antibody from PAE cells ectopically expressing VEGFR-2 (6). The immunoprecipitated proteins were subjected to proteolytic digestion (incubated at 37°C for 4 hours in the presence of chymotrypsin or trypsin) on a ProGest device (Genomic Solutions). Samples were analyzed by nano–LC-MS/MS on a Thermo Fisher LTQ Orbitrap XL. Thirty microliters of hydrolysate was loaded onto a 5 mm × 75 μm inside diameter (ID) C12 (Jupiter Proteo, Phenomenex) vented column at a flow rate of 10 μl/min. Gradient elution was over a 15 cm × 75 μm ID C12 column at 300 nl/min. The mass spectrometer was operated in data-dependent mode, and the six most abundant ions were selected for MS/MS. The Orbitrap MS scan was performed at 60,000 full width at half maximum resolution. MS/MS data were searched using both MASCOT and Sequest algorithms. The search results were processed by Scaffold (http://www.proteomesoftware.com) and Proteome Discoverer (Thermo Fisher Scientific, version 1.3.0.339). Selected parameters for LTQ Orbitrap XL data require a minimum of two peptides matching per protein with minimum probabilities of 90% at the protein level and 50% at the corresponding peptide level.
Cell culture
PAE and HEK-293 cells were grown in Dulbecco’s modified essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and penicillin and streptomycin. HUVECs were grown in HUVEC growth medium. All cells were incubated at 37°C, 5% CO2 in a humidified chamber.
Capillary tube formation/in vitro angiogenesis assay
Matrigel (purchased from BD Biosciences) was diluted with an equal volume of DMEM and used to coat 24-well plates as described before (42). Briefly, HUVECs or PAE cells expressing CKR or mutant CKR were trypsinized, resuspended in DMEM containing 2% FBS, and counted, and 2 × 104 cells were seeded on the solidified Matrigel mixture. Cells were allowed to seed for 2 hours before adding CSF-1 at 25 ng/ml. Tube formation was allowed to take place overnight, and images were obtained with a Leica inverted microscope coupled with a charge-coupled device camera.
Mouse tumor xenograft
B16F cells (1 × 107) expressing VEGFR-2 or Lys1041 mutant VEGFR-2 were mixed with Matrigel and injected under the skin of mice. After 21 days, animals were euthanized, and the tumors were dissected and measured (43).
Zebrafish angiogenesis assay
A glass capillary needle attached to a FemtoJet injector (Eppendorf) was used for injecting mRNA (10 or 5 ng/μl in about 10 μl) into one- or two-cell–stage embryos of Fli-eGFP transgenic adult male and female zebra-fish (Danio rerio). The embryos were grown at 28°C for 3 days. The embryos were examined 28 or 50 hours after fertilization with a Nikon immunofluorescence microscope. Images of fishes under the same settings were obtained for 10 fish per group in every experiment and analyzed for the length of the tail vessels using Image-Pro software and for the tail vessel plexus fluorescence intensity using ImageJ software.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
Endothelial cell proliferation was measured with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit (Promega). Cells (8 × 103) were seeded in 24-well tissue culture plates, serum-starved for 2 days, and then treated with varying concentrations of CSF for 24 hours. Cells were then incubated with an appropriate volume of MTT dye for 2.5 hours at 37°C. Insoluble formazan crystals produced during this incubation were then solubilized with the addition of “Stop Solution.” Well volumes were thoroughly mixed and transferred to a 96-well plate for spectrophotometric analysis at 570 nm. Conditions were carried out at least in triplicate, and the entire experiment was repeated three times.
Site-directed mutagenesis
Methylation mutant VEGFR-2 constructs were created using a polymerase chain reaction–based site-directed mutagenesis strategy (42). Mutations were confirmed by sequencing. Complementary DNAs were cloned in the Not I and Sal I restriction sites of the retroviral vector pLNCX2. Viral production was achieved by transfection into 293-GPG cells, and viral supernatants were collected for 5 days; viral medium was then used as described (16).
Immunoprecipitation and Western blotting
Briefly, cells were washed three times with H/S buffer [25 mM Hepes (pH 7.4) and 150 mM NaCl] and then lysed in EB lysis buffer [10 mM tris-HCl, 10% glycerol, 5 mM EDTA (pH 7.4), 50 mM NaCl, 50 mM NaF, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 2 mM sodium orthovanadate, and aprotinin (20 μg/ml)]. Where appropriate, cells were serum-starved overnight (~16 hours) before lysis, or serum-starved and stimulated with CSF-1 (15 ng/ml) or VEGF (100 ng/ml) for 10 min at 37°C. Cell lysates were heated to 95°C for 5 min in Laemmli sample buffer, resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene difluoride (PVDF) membranes, and immunoblotted for proteins of interest.
Cellular methylation assay
HEK-293 and PAE cells expressing CKR were incubated with methionine-free DMEM for 3 hours. Cells were then treated for 2 hours with methionine-free DMEM supplemented with SAM (PerkinElmer, no. NET155H001MC) at a final concentration of 16 μCi/ml. Cells were then lysed and immunoprecipitated for VEGFR-2. Immunoprecipitated proteins were subjected to SDS-PAGE and transferred onto a PVDF membrane. The membrane was treated with 30% 2,5-diphenyloxazole for 1 hour and then extensively washed with water before being exposed to autoradiography film at −80°C for 2 weeks as described (44).
In vitro methylation assay
PAE cells expressing VEGFR-2 were pretreated with 20 μM Adox for 6 hours before lysis. A homogenate of PAE cells not expressing VEGFR-2 was prepared by scraping cells in 100 μl of H/S buffer [25 mM Hepes (pH 7.4), 150 mM NaCl, and 2 mM Na3VO4], followed by sonication on ice (4 × 3-s pulses at level 4). The supernatant was collected after centrifugation. Immunoprecipitated VEGFR-2 from VEGFR-2–expressing PAE cell lysate was then added to the homogenate with or without the addition of 2 mM SAM and incubated at 37°C for 35 min. VEGFR-2 was then immunoprecipitated with anti–VEGFR-2 antibody and subjected to in vitro kinase assays. In some cases, the synthesized peptides were dot-blotted on PVDF membranes followed by in vitro methylation assay using [3H]SAM and exposed to autoradiography.
In vitro kinase assay
Equal numbers of PAE cells harboring CKR or mutant CKR were lysed, and cell lysates were then immunoprecipitated with anti–VEGFR-2 antibody. The immunoprecipitated proteins were washed twice with lysis buffer, then twice with cold PAN buffer [10 mM Pipes (pH 7.0), 100 mM NaCl]. A third PAN wash was done, and the immunoprecipitate was divided at this step if appropriate. In vitro kinase reactions were done by incubating immunoprecipitated proteins in an equal volume of 2× kinase buffer [40 mM tris-HCl (pH 7.4), 20 mM MgCl2, 2 mM dithiothreitol, and 200 mM NaCl] with or without ATP to obtain a final ATP concentration of 0.01 or 0.1 mM as described (6). Reactions were incubated for 15 min at 30°C, stopped by the addition of an equal volume of sample buffer, and heated to 95°C for 5 min before being subjected to SDS-PAGE. Membranes were probed with anti-phosphotyrosine antibodies and reprobed for total VEGFR-2 abundance.
Image quantification and statistical analysis
Images were quantified with ImageJ software and normalized to total VEGFR-2 when appropriate. Summary statistics including the mean, median, SD, and range are presented. We compared two groups using the Wilcoxon rank sum test (or Mann-Whitney U test). When multiple vessels per zebrafish were examined, we used a random effects model with a random intercept for zebrafish to compare outcomes across three groups. This model accounts for possible correlation between multiple vessels per fish included in the analysis.
Supplementary Material
Fig. S1. Validation of the anti–methyl-Lys and anti–methyl-Arg antibodies.
Fig. S2. Mass spectra of methylated VEGFR-2 peptides and their locations.
Fig. S3. Ratios of methylation of Lys1041 and tyrosine phosphorylation of VEGFR-2.
Fig. S4. Phosphorylation of Lys and Arg mutant CKRs in response to ligand stimulation.
Fig. S5. Mutation of Lys1041 to Phe inhibits, whereas mutation to Gln partially preserves, phosphorylation of VEGFR-2.
Fig. S6. The K1041R mutation does not inhibit ligand binding or dimerization of VEGFR-2.
Fig. S7. Adox treatment inhibits the methylation of VEGFR-2.
Fig. S8. Remethylation of the K1041R VEGFR-2 mutant does not increase its tyrosine phosphorylation.
Acknowledgments
We thank N. Woolf from the Rahimi laboratory for proofreading the manuscript.
Funding: This work was supported in part through a grant from the NIH/National Eye Institute (R01EY017955 to N.R.); P41 RR010888/GM104603, S10 RR020946, and NIH/National Heart, Lung, and Blood Institute contract N01 HHSN268201000031C (to C.E.C.); and K08 award (NIH/National Institute of Diabetes and Digestive and Kidney Diseases DK080946 and Department of Medicine Career Investment Award to V.C.). This work was also supported by a grant from the Department of Pathology, Boston University, and a Massachusetts Lions Foundation grant to the Department of Ophthalmology. This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (V.E.M.).
Footnotes
Author contributions: N.R., R.D.M., E.J.H., and V.C. designed the experiments and analyzed the data. N.R. and E.J.H. wrote the manuscript. I.V.Z. helped to perform and analyze the zebrafish experiments. Y.J. performed MS analysis. C.E.C. analyzed the MS data and made corrections to the manuscript. V.E.M. provided DZNep. J.W. performed statistical analyses.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository with the data set identifier PXD000520 and DOI 10.6019/PXD000520.
References
- 1.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim WA, Pawson T. Phosphotyrosine signaling: Evolving a new cellular communication system. Cell. 2010;142:661–667. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Eden ER, Huang F, Sorkin A, Futter CE. The role of EGF receptor ubiquitination in regulating its intracellular traffic. Traffic. 2012;13:329–337. doi: 10.1111/j.1600-0854.2011.01305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyake S, Mullane-Robinson KP, Lill NL, Douillard P, Band H. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A critical role for Cbl tyrosine kinase-binding domain. J Biol Chem. 1999;274:16619–16628. doi: 10.1074/jbc.274.23.16619. [DOI] [PubMed] [Google Scholar]
- 6.Meyer RD, Srinivasan S, Singh AJ, Mahoney JE, Gharahassanlou KR, Rahimi N. PEST motif serine and tyrosine phosphorylation controls vascular endothelial growth factor receptor 2 stability and downregulation. Mol Cell Biol. 2011;31:2010–2025. doi: 10.1128/MCB.01006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaik S, Nucera C, Inuzuka H, Gao D, Garnaas M, Frechette G, Harris L, Wan L, Fukushima H, Husain A, Nose V, Fadda G, Sadow PM, Goessling W, North T, Lawler J, Wei W. SCF (β-TRCP) suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J Exp Med. 2012;209:1289–1307. doi: 10.1084/jem.20112446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 9.Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 10.Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–1439. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- 11.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, Fukamizu A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008;32:221–231. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, Lee HJ, Wang YN, Liu M, Liao HW, Shi B, Lai CC, Bedford MT, Tsai CH, Hung MC. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13:174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunizaki M, Hamamoto R, Silva FP, Yamaguchi K, Nagayasu T, Shibuya M, Nakamura Y, Furukawa Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007;67:10759–10765. doi: 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- 14.Bedford MT, Clarke SG. Protein arginine methylation in mammals: Who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahimi N. Vascular endothelial growth factor receptors: Molecular mechanisms of activation and therapeutic potentials. Exp Eye Res. 2006;83:1005–1016. doi: 10.1016/j.exer.2006.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahimi N, Dayanir V, Lashkari K. Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J Biol Chem. 2000;275:16986–16992. doi: 10.1074/jbc.M000528200. [DOI] [PubMed] [Google Scholar]
- 17.Rahimi N. VEGFR-1 and VEGFR-2: Two non-identical twins with a unique physiognomy. Front Biosci. 2006;11:818–829. doi: 10.2741/1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dayanir V, Meyer RD, Lashkari K, Rahimi N. Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol 3-kinase and cell proliferation. J Biol Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 19.Meyer RD, Latz C, Rahimi N. Recruitment and activation of phospholipase Cγ1 by vascular endothelial growth factor receptor-2 are required for tubulogenesis and differentiation of endothelial cells. J Biol Chem. 2003;278:16347–16355. doi: 10.1074/jbc.M300259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer RD, Sacks DB, Rahimi N. IQGAP1-dependent signaling pathway regulates endothelial cell proliferation and angiogenesis. PLOS One. 2008;3:e3848. doi: 10.1371/journal.pone.0003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer RD, Dayanir V, Majnoun F, Rahimi N. The presence of a single tyrosine residue at the carboxyl domain of vascular endothelial growth factor receptor-2/FLK-1 regulates its autophosphorylation and activation of signaling molecules. J Biol Chem. 2002;277:27081–27087. doi: 10.1074/jbc.M110544200. [DOI] [PubMed] [Google Scholar]
- 22.McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure. 1999;7:319–330. doi: 10.1016/s0969-2126(99)80042-2. [DOI] [PubMed] [Google Scholar]
- 23.Mostaqul Huq MD, Gupta P, Tsai NP, White R, Parker MG, Wei LN. Suppression of receptor interacting protein 140 repressive activity by protein arginine methylation. EMBO J. 2006;25:5094–5104. doi: 10.1038/sj.emboj.7601389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 25.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilz RB, Van den Berghe G, Boss GR. Adenosine dialdehyde and nitrous oxide induce HL-60 differentiation. Blood. 1987;70:1161–1164. [PubMed] [Google Scholar]
- 28.Tseng CK, Marquez VE, Fuller RW, Goldstein BM, Haines DR, McPherson H, Parsons JL, Shannon WM, Arnett G, Hollingshead M, Driscoll JS. Synthesis of 3-deazaneplanocin A, a powerful inhibitor of S-adenosylhomocysteine hydrolase with potent and selective in vitro and in vivo antiviral activities. J Med Chem. 1989;32:1442–1446. doi: 10.1021/jm00127a007. [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 30.Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, Jürgensmeier JM, Womack C. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res. 2010;16:3548–3561. doi: 10.1158/1078-0432.CCR-09-2797. [DOI] [PubMed] [Google Scholar]
- 31.Rydén L, Stendahl M, Jonsson H, Emdin S, Bengtsson NO, Landberg G. Tumor-specific VEGF-A and VEGFR2 in postmenopausal breast cancer patients with long-term follow-up. Implication of a link between VEGF pathway and tamoxifen response. Breast Cancer Res Treat. 2005;89:135–143. doi: 10.1007/s10549-004-1655-7. [DOI] [PubMed] [Google Scholar]
- 32.Serbedzija GN, Flynn E, Willett CE. Zebrafish angiogenesis: A new model for drug screening. Angiogenesis. 1999;3:353–359. doi: 10.1023/a:1026598300052. [DOI] [PubMed] [Google Scholar]
- 33.Tobia C, De Sena G, Presta M. Zebrafish embryo, a tool to study tumor angiogenesis. Int J Dev Biol. 2011;55:505–509. doi: 10.1387/ijdb.103238ct. [DOI] [PubMed] [Google Scholar]
- 34.Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002;1:219–227. doi: 10.1016/s1535-6108(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 35.Tjwa M, Luttun A, Autiero M, Carmeliet P. VEGF and PlGF: Two pleiotropic growth factors with distinct roles in development and homeostasis. Cell Tissue Res. 2003;314:5–14. doi: 10.1007/s00441-003-0776-3. [DOI] [PubMed] [Google Scholar]
- 36.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 38.Srinivasan S, Meyer RD, Lugo R, Rahimi N. Identification of PDCL3 as a novel chaperone protein involved in the generation of functional VEGFR receptor 2. J Biol Chem. 2013;288:23171–23181. doi: 10.1074/jbc.M113.473173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 40.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 41.Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, Liu F, Marquez VE, Morgan RA, Schrump DS. Inhibition of histone lysine methylation enhances cancer–testis antigen expression in lung cancer cells: Implications for adoptive immunotherapy of cancer. Cancer Res. 2011;71:4192–4204. doi: 10.1158/0008-5472.CAN-10-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh AJ, Meyer RD, Navruzbekov G, Shelke R, Duan L, Band H, Leeman SE, Rahimi N. A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCγ1 activation and angiogenesis. Proc Natl Acad Sci USA. 2007;104:5413–5418. doi: 10.1073/pnas.0700809104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer RD, Singh A, Majnoun F, Latz C, Lashkari K, Rahimi N. Substitution of C-terminus of VEGFR-2 with VEGFR-1 promotes VEGFR-1 activation and endothelial cell proliferation. Oncogene. 2004;23:5523–5531. doi: 10.1038/sj.onc.1207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh AJ, Meyer RD, Band H, Rahimi N. The carboxyl terminus of VEGFR-2 is required for PKC-mediated down-regulation. Mol Biol Cell. 2005;16:2106–2118. doi: 10.1091/mbc.E04-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Validation of the anti–methyl-Lys and anti–methyl-Arg antibodies.
Fig. S2. Mass spectra of methylated VEGFR-2 peptides and their locations.
Fig. S3. Ratios of methylation of Lys1041 and tyrosine phosphorylation of VEGFR-2.
Fig. S4. Phosphorylation of Lys and Arg mutant CKRs in response to ligand stimulation.
Fig. S5. Mutation of Lys1041 to Phe inhibits, whereas mutation to Gln partially preserves, phosphorylation of VEGFR-2.
Fig. S6. The K1041R mutation does not inhibit ligand binding or dimerization of VEGFR-2.
Fig. S7. Adox treatment inhibits the methylation of VEGFR-2.
Fig. S8. Remethylation of the K1041R VEGFR-2 mutant does not increase its tyrosine phosphorylation.