

Abstract
Objective
To report the auditory and vestibular phenotypes of patients with GATA3 mutation.
Study design
Case series of 6 patients
Setting
Tertiary referral center
Patients
All patients had the classic triad of GATA3 deficiency: hypoparathyroidism, hearing loss, and renal dysplasia. Patients (29-60 years old, mean age 42.5 years, 3M/3F) were confirmed to have heterozygous mutations involving GATA3 by Sanger sequencing.
Interventions
Behavioral audiometry, distortion product otoacoustic emissions (DPOAEs) and auditory brainstem responses (ABRs) were used to assess hearing. Rotational vestibular testing was used to assess vestibular function.
Results
All patients with GATA3 mutation presented with hearing loss during childhood. The mean three frequency (.5/1/2/ kHz) pure tone average was 67 dB HL (range 50-83 dB HL, SD 9.3). The average speech discrimination score was 73% (range 36-100%, SD 15.9). DPOAEs were absent in all patients. ABRs were remarkably robust and provided no evidence of retrocochlear dysfunction. Some patients complained of dizziness, but rotary chair testing was normal across participants for whom testing occurred.
Conclusions
Patients with GATA3 mutation present with early onset sensorineural hearing loss (SNHL). DPOAEs were absent, supporting outer hair cell dysfunction, while ABRs were present and robust. Rotational vestibular testing revealed no evidence of abnormal horizontal semicircular canal function.
Introduction
Genetic hearing loss is one of the most common inherited sensory disorders, affecting 1 in every 1000 births. Non-syndromic hearing loss accounts for ~70% of hereditary congenital deafness, whereas syndromic hearing loss accounts for the other 30%. Currently, more than 150 genetic loci have been linked to genetic hearing loss, and more than 70 genes have been identified [1]. GATA3 belongs to the GATA family of zinc finger transcription factors, which are named according to their DNA binding sequence (GATA). GATA3 plays an important role in inner ear development [2-4], and mutations affecting GATA3 are associated with HDR syndrome, which stands for hypoparathyroidism, deafness, and renal dysplasia [5-22]. It has been reported that patients with GATA3 mutations present with early-onset sensorineural hearing loss. However, the vestibular phenotype has never been reported in these patients. In this study, we describe auditory and vestibular findings in 6 patients with GATA3 mutations.
Materials and Methods
Six patients with heterozygous GATA3 mutations were included in this case series. The study was approved by the National Institutes of Health Institutional Review Board (NCT00404560, NCT01222741). Clinical examination was performed in all patients. Conventional behavioral audiometry and distortion product otoacoustic emissions (DPOAEs) were used to assess peripheral auditory status. Auditory brainstem responses (ABRs) were conducted in 4/6 patients. Rotational testing using sinusoidal harmonic acceleration (SHA) was used to assess vestibular function in 4/6 patients.
Case series (Table 1)
Case #1
31 year-old man was diagnosed with sensorineural hearing loss (SNHL) at the age of 18 months, and was fit with bilateral hearing aids at 22 months of age. The patient reported that his hearing has been stable. He denied any vertigo and disequilibrium. His otoscopic examination was normal bilaterally. His audiogram showed bilateral downsloping moderate to severe/profound (right/left, respectively) SNHL (Figure 1), with word recognition scores of 82% and 74% for the right and left ears, respectively. DPOAEs were absent bilaterally, and ABRs showed morphologically normal waveforms with normal absolute and interpeak latencies. Vestibulo-ocular reflex (VOR) gain, phase and symmetry were normal on SHA testing.
Figure 1.
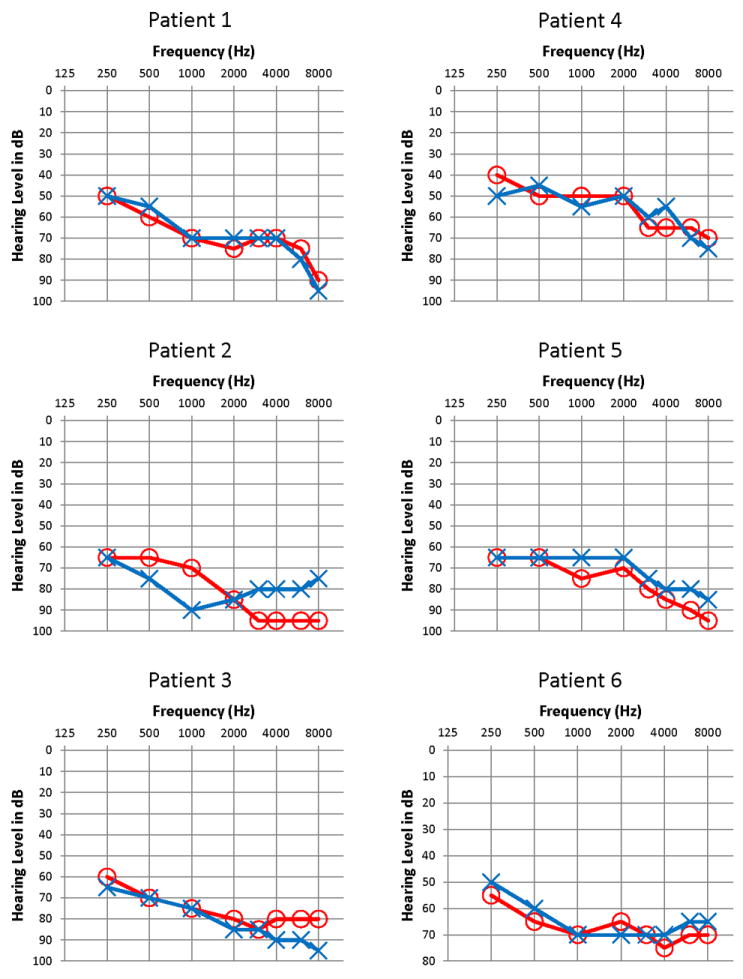
Pure tone audiograms in patients with GATA3 mutation. The right ear thresholds are shown in red (circles), and the left ear thresholds are shown in blue (crosses). There were no clinically significant air-bone gaps (bone conduction thresholds not shown).
Case #2
43 year-old man was diagnosed with SNHL in early childhood and was fit with bilateral hearing aids at 5 years of age. The patient reported that his hearing has been stable. He denied any vertigo and disequilibrium. His otoscopic examination was normal bilaterally. His audiogram showed downsloping moderately-severe to severe SNHL for the left ear and moderately-severe to profound predominantly SNHL for the right ear (Figure 1), with word recognition scores of 62% and 68% for the right and left ears respectively. DPOAEs were absent and the ABRs (Figure 2) were within normal limits for absolute and interpeak latencies bilaterally.
Figure 2.
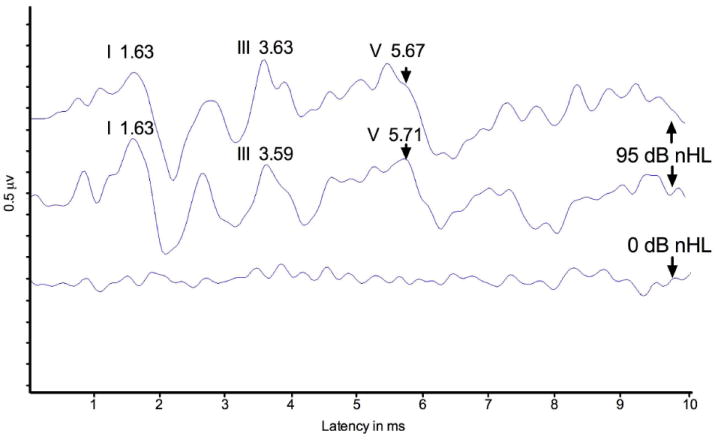
ABR recordings from the left ear of patient 2 at 95dB and 0dBnHL. The 95dBnHL ABR was tested twice for confirmation. ABR was stimulated using broadband clicks.
Case #3
60 year-old woman was seen for otologic and audiologic evaluation. This patient is the mother of Case #2. The patient reported that she was diagnosed with SNHL at the age of 5 years, and was fit with bilateral hearing aids at 12 years of age. The patient reported that her hearing has been stable. She complained of occasional tinnitus, but denied any vertigo and disequilibrium. Her otoscopic examination was normal bilaterally. Her audiogram showed downsloping moderately-severe to severe SNHL in the right ear and moderately-severe to profound SNHL in the left ear (Figure 1), with word recognition scores of 72% for the right and 70% for the left ear. DPOAEs were absent bilaterally, and ABR testing was inconclusive due to patient restlessness. A VOR was observed on SHA testing, but excessive blink artifact precluded reliable interpretation.
Case #4
34 year-old woman reported longstanding SNHL and she was fit with bilateral hearing aids since childhood. The patient reported that her hearing has been stable. Otoscopic examination was normal bilaterally. Her audiogram showed a mild-to-moderately-severe downsloping SNHL in the right ear and a moderate-to-severe SNHL in the left ear (Figure 1), with word recognition scores of 100% and 90% for the right and left ears, respectively. DPOAEs were absent bilaterally. ABRs and vestibular testing were not performed.
Case #5
58 year-old woman was seen for otologic and audiologic evaluation. This patient is the mother of Case #4. The patient reported that she was diagnosed with left ear hearing loss in 2nd grade, and right ear hearing loss in 7th grade and that her hearing had gradually worsened over time. She was fit with bilateral hearing aids. She complained of occasional dizziness but no vertigo. Her otoscopic examination was normal bilaterally. Her audiogram showed a downsloping moderately-severe to profound SNHL in the right ear and a moderately-severe to severe SNHL in the left ear (Figure 1), with word recognition scores of 36% and 68% for the right and left ears, respectively. DPOAEs were absent bilaterally. ABRs and vestibular testing were not performed.
Case #6
29 year-old man reported that he was diagnosed with SNHL during childhood, and was fit with bilateral hearing aids at 15 years of age. He reported that his hearing has been stable and denied any vertigo and disequilibrium. His audiogram showed a downsloping moderate to moderately-severe SNHL in the left ear and a moderate-to-severe SNHL in the right ear (Figure 1), with word recognition scores of 84% and 70% for the right and left ears, respectively. DPOAEs were absent bilaterally, and ABRs showed normal absolute and interpeak latencies for waves III and V bilaterally. The VOR gain, phase and symmetry were normal on SHA testing.
Discussion
In this study, we report the auditory and vestibular phenotypes in 6 patients with GATA3 mutations. GATA3 is a zinc finger transcription factor located on chromosome 10p15. It is critical for the development of multiple organ systems, including the parathyroid glands, kidneys, inner ear, thymus, and the central nervous system. Haploinsufficieny of the GATA3 gene is associated with the triad of hypoparathyroidism, hearing loss, and renal dysplasia, or HDR syndrome, which was first described by Bilous et al. in 1992 [23]. HDR syndrome is a rare congenital disorder, with no reported incidence in the literature. Several studies have reported on the auditory phenotype of patients with HDR syndrome and GATA3 mutation. Fukami and colleagues reported on a series of 6 patients with HDR syndrome [8], and found that 5 out of 6 patients presented with SNHL. However, details of auditory phenotype were not described. Nakamura and colleagues reported on 5 patients with HDR syndrome and found that all patients had SNHL [9]. They reported that the auditory thresholds were between 40-100 dB HL, but no additional details were provided. van Looij and colleagues reported on the auditory phenotype in two patients with HDR syndrome [24]. They found that these patients had moderate to severe SNHL with absent DPOAEs and normal ABR interpeak intervals.
In our cohort, all patients had confirmed heterozygous GATA3 mutations and presented with downsloping SNHL, each with a reported early onset during childhood. The mean pure-tone average was 67 dBHL (.5/1/2 kHz; range 50-83 dBHL, SD 9.3), and the average word recognition score was 73% (range 36-100%, SD 15.9). DPOAEs were consistently absent, supporting cochlear outer hair cell dysfunction as one possible site of lesion. ABRs were normal in patients who underwent testing and were surprisingly robust given the degree of peripheral hearing loss, which further implicates cochlear dysfunction that may be confined to the outer hair cells as the primary mechanism for hearing loss. The hypothesis that GATA3 mutations affect proper outer hair cell function is supported by an animal study by Van Looij et al. [17], where heterozygouse Gata3 knockout mice had significantly lower DPOAEs and a more rapid degeneration of outer hair cells in the apex and base of the cochlea compared to wild type animals. Cochlear histology confirmed increased vacuoles in the cytoplasm of the outer hair cells, which supports dysfunction specific to these cells.
All patients in this study were fit with bilateral hearing aids and subjectively reported benefit from the devices. Our data are consistent with the study by van Looij et al. [20] and expand on prior reports [7,8] of auditory function in patients with HDR syndrome and GATA3 mutation. Some patients complained of dizziness, but rotary chair testing showed no evidence of horizontal semicircular canal dysfunction. To our knowledge, these are the first case reports of vestibular function in patients with GATA3 mutation.
Conclusions
In this study, we report the auditory and vestibular phenotypes for a series of 6 patients with GATA3 mutation. All patients in our cohort present with early-onset SNHL. DPOAEs were absent in all ears whereas ABRs were normal, supporting a cochlear site of lesion. We observed no evidence of vestibular dysfunction in these patients. To our knowledge, this is the only case series of patients with GATA3 mutation that focuses on the auditory and vestibular phenotypes of this disorder.
Supplementary Material
Acknowledgments
We would like to thank the NIDCD and NIAID intramural research funds for their support.
References
- 1.Marazita ML, et al. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet. 1993;46(5):486–91. doi: 10.1002/ajmg.1320460504. [DOI] [PubMed] [Google Scholar]
- 2.Karis A, et al. Transcription factor GATA-3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. J Comp Neurol. 2001;429(4):615–30. doi: 10.1002/1096-9861(20010122)429:4<615::aid-cne8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Lawoko-Kerali G, Rivolta MN, Holley M. Expression of the transcription factors GATA3 and Pax2 during development of the mammalian inner ear. J Comp Neurol. 2002;442(4):378–91. doi: 10.1002/cne.10088. [DOI] [PubMed] [Google Scholar]
- 4.Appler JM, et al. Gata3 is a critical regulator of cochlear wiring. J Neurosci. 2013;33(8):3679–91. doi: 10.1523/JNEUROSCI.4703-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes TS, et al. HDR syndrome: a follow-up genotype-phenotype analysis of a de novo missense Thr272Ile mutation in exon 4 of GATA3. Klin Padiatr. 2012;224(7):452–4. doi: 10.1055/s-0032-1329947. [DOI] [PubMed] [Google Scholar]
- 6.Nanba K, et al. A Novel GATA3 Nonsense Mutation in a Newly Diagnosed Adult Patient of Hypothyroidism, Deafness, and Renal Dysplasia (HDR) Syndrome. Endocr Pract. 2013;19(1):17–20. doi: 10.4158/EP12186.CR. [DOI] [PubMed] [Google Scholar]
- 7.Melis D, et al. Clinical description of a patient carrying the smallest reported deletion involving 10p14 region. Am J Med Genet A. 2012;158A(4):832–5. doi: 10.1002/ajmg.a.34133. [DOI] [PubMed] [Google Scholar]
- 8.Fukami M, et al. GATA3 abnormalities in six patients with HDR syndrome. Endocr J. 2011;58(2):117–21. doi: 10.1507/endocrj.k10e-234. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura A, et al. Molecular analysis of the GATA3 gene in five Japanese patients with HDR syndrome. Endocr J. 2011;58(2):123–30. doi: 10.1507/endocrj.k10e-246. [DOI] [PubMed] [Google Scholar]
- 10.Ohta M, et al. Novel dominant-negative mutant of GATA3 in HDR syndrome. J Mol Med (Berl) 2011;89(1):43–50. doi: 10.1007/s00109-010-0702-6. [DOI] [PubMed] [Google Scholar]
- 11.Muroya K, et al. Diabetes mellitus in a Japanese girl with HDR syndrome and GATA3 mutation. Endocr J. 2010;57(2):171–4. doi: 10.1507/endocrj.k09e-313. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, et al. Germinal mosaicism of GATA3 in a family with HDR syndrome. Am J Med Genet A. 2009;149A(4):776–8. doi: 10.1002/ajmg.a.32706. [DOI] [PubMed] [Google Scholar]
- 13.Ferraris S, et al. HDR syndrome: a novel “de novo” mutation in GATA3 gene. Am J Med Genet A. 2009;149A(4):770–5. doi: 10.1002/ajmg.a.32689. [DOI] [PubMed] [Google Scholar]
- 14.Saito T, et al. A novel mutation in the GATA3 gene of a Japanese patient with PTH-deficient hypoparathyroidism. J Bone Miner Metab. 2009;27(3):386–9. doi: 10.1007/s00774-008-0015-9. [DOI] [PubMed] [Google Scholar]
- 15.Benetti E, et al. 10p12.1 deletion: HDR phenotype without DGS2 features. Exp Mol Pathol. 2009;86(1):74–6. doi: 10.1016/j.yexmp.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Adachi M, et al. A novel mutation in the GATA3 gene in a family with HDR syndrome (Hypoparathyroidism, sensorineural Deafness and Renal anomaly syndrome) J Pediatr Endocrinol Metab. 2006;19(1):87–92. doi: 10.1515/jpem.2006.19.1.87. [DOI] [PubMed] [Google Scholar]
- 17.van Looij MA, et al. GATA3 haploinsufficiency causes a rapid deterioration of distortion product otoacoustic emissions (DPOAEs) in mice. Neurobiol Dis. 2005;20(3):890–7. doi: 10.1016/j.nbd.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 18.Mino Y, et al. Identification of a novel insertion mutation in GATA3 with HDR syndrome. Clin Exp Nephrol. 2005;9(1):58–61. doi: 10.1007/s10157-004-0327-6. [DOI] [PubMed] [Google Scholar]
- 19.Zahirieh A, et al. Functional analysis of a novel GATA3 mutation in a family with the hypoparathyroidism, deafness, and renal dysplasia syndrome. J Clin Endocrinol Metab. 2005;90(4):2445–50. doi: 10.1210/jc.2004-1969. [DOI] [PubMed] [Google Scholar]
- 20.Verri A, et al. Chromosome 10p deletion in a patient with hypoparathyroidism, severe mental retardation, autism and basal ganglia calcifications. Ann Genet. 2004;47(3):281–7. doi: 10.1016/j.anngen.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 21.van der Wees J, et al. Hearing loss following Gata3 haploinsufficiency is caused by cochlear disorder. Neurobiol Dis. 2004;16(1):169–78. doi: 10.1016/j.nbd.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Muroya K, et al. GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. J Med Genet. 2001;38(6):374–80. doi: 10.1136/jmg.38.6.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bilous RW, et al. Brief report: autosomal dominant familial hypoparathyroidism, sensorineural deafness, and renal dysplasia. N Engl J Med. 1992;327(15):1069–74. doi: 10.1056/NEJM199210083271506. [DOI] [PubMed] [Google Scholar]
- 24.van Looij MA, et al. Characteristics of hearing loss in HDR(hypoparathyroidism, sensorineural deafness, renal dysplasia) syndrome. Audiol Neurootol. 2006;11(6):373–9. doi: 10.1159/000095899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.