

Abstract
The physiological functions of members of the tumor-necrosis factor (TNF) receptor (TNFR)–associated factor (TRAF) family in T cell immunity are not well understood. We found that in the presence of interleukin 6 (IL-6), naive TRAF5-deficient CD4+ T cells showed an enhanced ability to differentiate into the TH17 subset of helper T cells. Accordingly, TH17 cell–associated experimental autoimmune encephalomyelitis (EAE) was greatly exaggerated in Traf5−/− mice. Although it is normally linked with TNFR signaling pathways, TRAF5 constitutively associated with a cytoplasmic region in the signal-transducing receptor gp130 that overlaps with the binding site for the transcription activator STAT3 and suppressed the recruitment and activation of STAT3 in response to IL-6. Our results identify TRAF5 as a negative regulator of the IL-6 receptor signaling pathway that limits the induction of proinflammatory CD4+ T cells that require IL-6 for their development.
The activation and differentiation of CD4+ T cells are regulated by three key signaling components from the T cell antigen receptor (TCR) (signal 1), costimulatory molecules (signal 2) and cytokine receptors (signal 3). The signals from cytokine receptors on CD4+ T cells serve an essential role in the lineage ‘decision’ of helper T cell subsets1,2. Binding of the proinflammatory cytokine IL-6 with a complex of the receptor for interleukin 6 (IL-6R) and the signal-transducing receptor gp130 results in the recruitment and activation of the transcription activator STAT3 (refs. 3–5), and that event serves a dominant role in the differentiation of naive CD4+ T cells into IL-17-producing helper T cells (TH17 cells) by inducing the lineage-specific transcription factor RORγt6–9. Signaling via IL-6 can also contribute to the development of T helper type 2 (TH2) cells in many situations10–12. How IL-6-driven differentiation of CD4+ T cells is modulated and regulated is incompletely understood.
The six members of the tumor-necrosis factor (TNF) receptor–associated factor (TRAF) family function as adaptors for members of the TNF receptor superfamily (TNFRSF) by associating with the intracellular domains of those receptors to mediate downstream signaling events13–15. The carboxy-terminal domain of TRAF proteins, which is composed of a coiled-coil leucine-zipper domain followed by a TRAF domain (TRAF-C), contributes to self-oligomerization of the TRAF proteins and their recognition of a variety of cytoplasmic molecules, including members of the TNFRSF. In addition to regulating signaling from members of the TNFRSF16–18, TRAF proteins are also recruited by the Toll-like receptor family and the RIG-I-like receptor family15,19, and other data suggest they also serve important functions in adaptive immunity controlling T cell signaling from the TCR20,21.
Some reports suggest that TRAF proteins can be modulatory in both a positive manner and a negative manner. The regulatory activity of TRAF proteins in T cell signaling has been demonstrated for TRAF1 in the transcription factor NF-κB1 pathway22, TRAF2 and TRAF3 in modulating activation of NF-κB2 (refs. 21,23), and TRAF6 in signaling via the kinases PI(3)K and PKB24. TRAF5 is suggested to be both a positive regulator and a negative regulator of T cells13,25–28. Although TRAF5 is structurally most homologous to TRAF3 (ref. 29), its molecular function is most similar to that of TRAF2 in the context of TNF-induced activation of NF-κB30. In contrast to mice deficient in TRAF2 or TRAF3, which become runted and die prematurely, TRAF5-deficient mice are viable and show no obvious abnormalities26. A pronounced TH2 response is induced in Traf5−/− CD4+ T cells, and allergic lung inflammation is more exaggerated in Traf5−/− mice than in wild-type mice27, which indicates that TRAF5 limits T cell–mediated inflammatory diseases such as asthma and suggests that TRAF5 has a function that has not been delineated in detail in T cells. However, it is unclear how TRAF5 negatively regulates the signaling and inflammatory responses mediated by CD4+ T cells.
In this study, we found that after receiving signal 1 and signal 2 in the presence of signal 3 from IL-6, naive Traf5−/− CD4+ T cells produced more IL-17 than did their wild-type counterparts and developed a pronounced TH17 phenotype both in vitro and in vivo. In accordance with that, Traf5−/− mice showed enhanced clinical signs of experimental autoimmune encephalomyelitis (EAE), and Traf5−/− CD4+ T cells induced exaggerated EAE in TRAF5-sufficient recipient mice. We also found that TRAF5 constitutively associated with gp130 and negatively controlled IL-6–STAT3 signaling. Our data demonstrate that TRAF5 regulates signal 3 in CD4+ T cells and works as an anti-inflammatory factor to limit immune responses mediated by effector CD4+ T cells that had been primed with IL-6.
RESULTS
TRAF5 deficiency facilitates IL-6-driven TH17 differentiation
To investigate how TRAF5 regulates the differentiation of CD4+ T cells, we cultured highly purified naive T cells from wild-type or Traf5−/− C57BL/6 (B6) mice in polarizing cytokine conditions (Fig. 1). After culturing naive T cells for 3–5 d in plates coated with antibody to the invariant signaling protein CD3 (anti-CD3) and antibody to the coreceptor CD28 (anti-CD28), we stained the cells for intracellular interferon-γ (IFN-γ), IL-4, IL-17A and the transcription factor Foxp3 to assess development of the TH1, TH2, TH17 and regulatory T cell (Treg cell) subsets, respectively. Notably, when we added IL-6 to the cultures, the proportion of Traf5−/− CD4+ T cells that expressed IL-17A was significantly higher than that of wild-type CD4+ T cells (Fig. 1a). TH17 cell–associated genes, such as Rorc, Il17a, Il17f and Il23r, were also significantly upregulated in Traf5−/− T cells (Fig. 1b), and IL-17A and IL-21 were much more abundant in Traf5−/− cultures than in wild-type cultures, but IFN-γ was not (Fig. 1c). Naive Traf5−/− CD4+ T cells expressed the same amount of IL-6R-gp130 as wild-type naive CD4+ T cells did (Supplementary Fig. 1a). In these polarized conditions, we found no significant difference between wild-type and Traf5−/− T cells in their expression of IFN-γ, IL-4 or Foxp3 protein (Fig. 1a) or Tbx21, Gata3 or Foxp3 mRNA (Supplementary Fig. 1b). Expression of Traf5 mRNA in wild-type CD4+ T cells was significantly downregulated after stimulation with anti-CD3 and anti-CD28, and this was not affected much by the presence of exogenous cytokines (Supplementary Fig. 1c). Wild-type and Traf5−/− CD4+ T cells proliferated and produced IL-2 in response to anti-CD3 and anti-CD28 in an equivalent manner27 (data not shown). These results demonstrated that TRAF5 in CD4+ T cells negatively regulated the IL-6-mediated development of TH17 cells.
Figure 1.

Naive polyclonal Traf5−/− CD4+ T cells display an enhanced TH17 phenotype in vitro. (a) Expression of IFN-γ, IL-4, Foxp3 and IL-17A in activated CD4+ T cells generated from naive wild-type (WT) or Traf5−/− (KO) B6 CD4+ T cells cultured with anti-CD3 and anti-CD28 in various polarizing conditions (left margin; α-, anti-) and restimulated for 5 h with the phorbol ester PMA and ionomycin. Numbers adjacent to outlined areas (left) indicate percent IFN-γ+, IL-4+, Foxp3+ or IL-17A+ (top to bottom) CD4+ T cells. (b) Quantitative RT-PCR analysis of the expression of TH17-associated genes (horizontal axis) in activated CD4+ T cells generated from naive wild-type or Traf5−/− B6 CD4+ T cells cultured for 24 h with anti-CD3 and anti-CD28 in TH17-polarizing conditions (right margin), presented relative to the expression of the gene encoding β-actin. ND, not detected. (c) Enzyme-linked immunosorbent assay of primary IL-17A, IL-21 and IFN-γ in supernatants of cells as in a (polarizing conditions, below graphs), assessed at day 2 (IL-17A and IFN-γ) or at day 3 (IL-21) after initial activation of naive CD4+ T cells. (d) Expression of IFN-γ, IL-4, IL-17A and Foxp3 in activated CD4+ T cells generated from naive wild-type or Traf5−/− B6 CD4+ T cells cultured with anti-CD3 plus wild-type B6 splenic APCs (after depletion of T cells), in various polarizing conditions (left margin), then restimulated for 5 h with PMA and ionomycin. Numbers adjacent to outlined areas indicate percent IFN-γ+IL-4− cells (top left) or IFN-γ−IL-4+ cells (bottom right) (top two rows), or IL-17A+Foxp3− cells (top left) or IL-17A−Foxp3+ cells (bottom right) (bottom two rows). NS, not significant; *P < 0.05 and **P < 0.01 (Student’s t-test). Data are from one experiment representative of at least two independent experiments with similar results (average and s.d. of triplicate wells).
In agreement with results reported above, enhanced TH17 development was also induced by stimulation of Traf5−/− CD4+ T cells with soluble anti-CD3 and splenic antigen-presenting cells (APCs) from wild-type B6 mice (Fig. 1d). Moreover, in the same culture, the development of Foxp3-expressing T cells was suppressed in the absence of TRAF5 (Fig. 1d). This supported the findings that IL-17+ CD4+ T cells and Foxp3+ CD4+ T cells were reciprocally regulated during differentiation and that signaling via IL-6 controls the down-regulation of Foxp3 expression31.
Next, to evaluate more precisely the role of TRAF5 in TH17 differentiation, we obtained naive CD4+ T cells from wild-type or Traf5−/− OT-II mice (which have transgenic expression of an oval-bumin (OVA)-specific TCR) and cultured the cells with a low dose of OVA peptide (amino acids 323–339) and splenic APCs from wild-type B6 mice in the presence or absence of IL-6. The primary production of IL-17A and IL-21 was significantly higher in Traf5−/− OT-II cultures than in wild-type cultures (Fig. 2a). Recall IL-17A expression in primed Traf5−/− OT-II cells was additionally about twofold higher than that in wild-type cells (Fig. 2b). Proliferative responses to antigen and IL-6 (Supplementary Fig. 2a,b), endogenous IL-6 production in the T cell culture (Supplementary Fig. 2c) and susceptibility to growth arrest mediated by transforming growth factor-β (TGF-β) (Supplementary Fig. 2d) were similar in the two groups. Thus, these results substantiated the results presented above showing enhanced TH17 differentiation of polyclonal T cells in the absence of TRAF5 and suggested that TRAF5 regulated IL-6 signaling but not cell proliferation, IL-6 production or TGF-β signaling.
Figure 2.
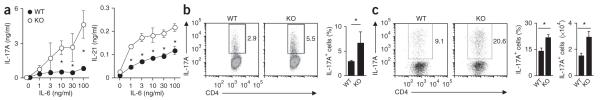
Traf5−/− OT-II CD4+ T cells exhibit enhanced TH17 development in vitro and in vivo. (a) Enzyme-linked immunosorbent assay of primary IL-17A and IL-21 in supernatants of activated CD4+ T cells generated from naive wild-type or Traf5−/− OT-II CD4+ T cells stimulated for 3 d with 0.1 μM OVA peptide (amino acids 323–339) and wild-type B6 splenic APCs (after depletion of T cells) in the presence of various concentrations of IL-6 (horizontal axes). (b) Recall IL-17A production in activated CD4+ T cells generated from naive wild-type or Traf5−/− OT-II CD4+ T cells stimulated for 5 d with OVA and APCs as in a in the presence of 10 ng/ml of IL-6, then restimulated for 5 h with PMA and ionomycin. Numbers adjacent to outlined areas (left) indicate percent IL-17A+CD4+ cells. (c) Quantification of IL-17A+ OT-II (CD4+CD45.2+) cells obtained from the draining lymph nodes of CD45.1+ B6.SJL hosts given naive wild-type or Traf5−/− OT-II (CD45.2+) donor cells on day 0, followed by immunization of the host with OVA in CFA on day 1 and node harvest on day 8, then restimulation of the cells in vitro for 5 h with PMA and ionomycin (numbers adjacent to outlined areas (left), as in b). *P < 0.05 (Student’s t-test). Data are from one experiment representative of three independent experiments with similar results (average and s.d. of triplicate wells in a; average and s.e.m. of three replicates in b; average and s.e.m. of four mice per group in c).
To determine whether TRAF5 serves a similar function in vivo, we adoptively transferred naive wild-type or Traf5−/− OT-II T cells into congenic B6 mice, followed by subcutaneous immunization of the recipient mice with OVA protein in complete Freund’s adjuvant (CFA). We then assessed recall IL-17 responses in donor OT-II cells in the draining lymph nodes 7 d after immunization (Fig. 2c). IL-17A+ Traf5−/− OT-II cells were about two times more abundant than IL-17A+ wild-type OT-II cells, in both frequency and absolute number (Fig. 2c). Collectively, these results demonstrated that TRAF5 antagonized the differentiation of TH17 cells both in vitro and in vivo.
TRAF5 acts as a negative regulator of IL-6R signaling
To evaluate how TRAF5 controls CD4+ T cell responses mediated by IL-6, we purified polyclonal splenic CD4+ T cells from wild-type or Traf5−/− B6 mice and stimulated the cells with a complex of IL-6 and IL-6R (IL-6–IL-6R) in isolation, without triggering the antigen receptor, then measured the phosphorylation of STAT3 at Tyr705. CD4+ T cells of both genotypes had equivalent expression of gp130 on the surface (Fig. 3a). IL-6–IL-6R promoted the phosphorylation of STAT3 in a dose-dependent way (Fig. 3b), and the response peaked at 10 min, then decreased (Fig. 3c). Notably, under these conditions, Traf5−/− CD4+ T cells displayed significantly larger amounts of phosphorylated STAT3 than did their wild-type counterparts (Fig. 3b–d). Traf5−/− CD8+ T cells also showed enhanced phosphorylation of STAT3 mediated by IL-6 (Supplementary Fig. 3a), which indicated that this TRAF5 function was not specific to CD4+ T cells. However, expression of Traf5 mRNA or Il6st mRNA (which encodes gp130) was significantly lower in other cell populations, such as B cells, natural killer T cells, natural killer cells and macrophages, than in T cells (Supplementary Fig. 3b,c), and IL-6-mediated phosphorylation of STAT3 in Traf5−/− macrophages was similar to that in wild-type macrophages (Supplementary Fig. 3d). Thus, TRAF5 may not be important for signaling via IL-6 in cells with lower expression of TRAF5 or gp130. The phosphorylation of STAT3 mediated by IL-10 or IL-21 was not affected by TRAF5 deficiency (Supplementary Fig. 3e,f), which confirmed the specificity of these results. Collectively, these results suggested that TRAF5 antagonized an early signaling activity downstream of gp130 in T cells.
Figure 3.
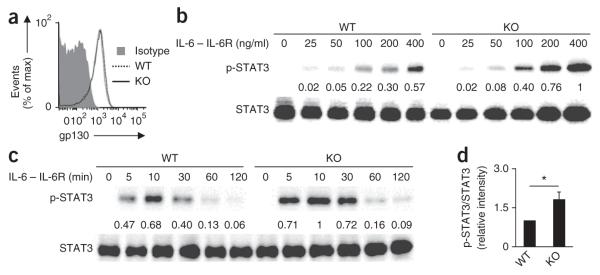
IL-6-mediated phosphorylation of STAT3 is enhanced in Traf5−/− CD4+ T cells. (a) Expression of gp130 on purified splenic wild-type and Traf5−/− polyclonal CD4+ T cells. Isotype, isotype-matched control antibody. (b,c) Immunoblot analysis of STAT3 phosphorylated at Tyr705 (p-STAT3) and total STAT3 in wild-type and Traf5−/− CD4+ T cells stimulated for 10 min with various concentrations (above lanes) of IL-6–IL-6R (b) or stimulated for various times (above lanes) with 200 ng/ml of IL-6–IL-6R (c). Numbers between blots indicate densitometry (p-STAT3/STAT3), presented relative to the highest ratio, set as 1. (d) Ratio of phosphorylated STAT3 to total STAT3 in wild-type and Traf5−/−CD4+ T cells stimulated for 10 min with 200 ng/ml of IL-6–IL-6R (presented as in b,c). *P < 0.05 (Student’s t-test). Data are from one experiment representative of three independent experiments with similar results (average and s.e.m. of three replicates in d).
Binding of TRAF5 to gp130 antagonizes recruitment of STAT3
To explore the molecular mechanism by which TRAF5 inhibited the activation of STAT3, we transfected HEK293T human embryonic kidney cells to express both c-Myc-tagged gp130 and V5-tagged TRAF5 and immunoprecipitated proteins from cell lysates with anti-c-Myc or anti-V5. TRAF5 immunoprecipitated together with gp130, and a reverse immunoprecipitation produced the same result (Fig. 4a). Notably, we detected a constitutive association between TRAF5 and gp130 in primary CD4+ T cells (Fig. 4b), which showed that the binding of TRAF5 to gp130 was physiologically relevant. The TRAF-C domain is responsible for binding to the cytoplasmic tails of members of the TNFRSF; thus, this domain of TRAF5 might be critical for its association with gp130. To assess this, we transfected HEK cells to express gp130 together with either the amino terminus of TRAF5 (amino acids 1–241), which contains RING and zinc-finger domains (TRAF5(1–241)), or the carboxyl terminus of TRAF5 (amino acids 242–558), which contains leucine-zipper and TRAF-C domains (TRAF5(242–558)). As expected, TRAF5(242–558) immunoprecipitated together with gp130 (Fig. 4c). The finding that TRAF5 did not need its RING or zinc-finger domains to interact with gp130 indicated that TRAF5(242–558) might antagonize the IL-6-mediated recruitment of STAT3 to gp130. To investigate this, we transfected HEK cells to express gp130 and STAT3 in the presence or absence of TRAF5(242–558) and evaluated the binding of STAT3 to gp130 by immunoprecipitation. After stimulation with IL-6–IL-6R, STAT3 was recruited to gp130, and that recruitment was strongly suppressed by the expression of TRAF5(242–558) (Fig. 4d). In addition, the amount of TRAF5 associated with gp130 increased further after stimulation with IL-6–IL-6R (Fig. 4d). Collectively, these results demonstrated that constitutive binding of TRAF5 to gp130 was antagonistic for IL-6-driven recruitment of STAT3 to gp130.
Figure 4.

Constitutive binding of TRAF5 to gp130 inhibits the IL-6-mediated recruitment of STAT3 to gp130. (a) Immunoassay of HEK293T cells transiently transfected with plasmid vectors encoding V5-tagged TRAF5 and c-Myc-tagged gp130, followed by immunoprecipitation (IP) of proteins from lysates with anti-V5, anti-c-Myc or control immunoglobulin G (IgG) and immunoblot analysis with anti-c-Myc and anti-V5. (b) Immunoprecipitation of endogenous gp130 from lysates of splenic wild-type or Traf5−/− B6 CD4+ T cells (2.5 × 108 per sample) with monoclonal antibody to gp130, followed by immunoblot analysis with anti-gp130 or anti-TRAF5. Input (bottom), immunoblot analysis of lysates without immunoprecipitation. *, heavy chain. (c) Immunoprecipitation of gp130 from lysates of HEK cells transiently transfected with plasmid vectors encoding c-Myc-tagged gp130 together with V5-tagged TRAF5(1–558), TRAF5(1–241) or TRAF5(242–558) (above lanes; identified at top), followed by immunoblot analysis with anti-V5 or anti-c-Myc. *, heavy chain. (d) Immunoassay of HEK cells transduced to express c-Myc-tagged gp130 and STAT3 together with V5-tagged TRAF5(242–558) (lanes 2, 4 and 5) or empty vector (lanes 1 and 3), then left unstimulated (US) or stimulated for 15 min with IL-6–IL-6R (500 ng/ml), followed by immunoprecipitation of proteins from lysates with anti-c-Myc or control IgG and immunoblot analysis with anti-c-Myc, anti-STAT3 or anti-V5. (e–g) Immunoassay of HEK cells transduced with plasmids encoding gp130 mutants (above lanes) with various deletions in the cytoplasmic region in positions 774–848 (above blot, e) or with substitution of alanine for other amino acids in TRAF-binding sites 1 and 2 (above blots, f,g) and cotransfected to express V5-TRAF5(242–558), followed by immunoprecipitation of proteins from lysates with control IgG or anti-c-Myc and immunoblot analysis with anti-V5 or anti-c-Myc. Data are from one experiment representative of at least two independent experiments with similar results.
To map the cytoplasmic region of gp130 necessary for binding TRAF5, we prepared five deletion mutants of gp130 (mutants 2–6) that differed in the length of their cytoplasmic tail (Supplementary Fig. 4a). The ability of TRAF5(242–558) to bind gp130 was much lower for gp130 mutants 3, 4, 5 and 6 (Supplementary Fig. 4a), which indicated that amino acid residues 774–848 of gp130 were critical for binding. To investigate this further, we prepared four more mutants of gp130 (mutants 7–10) with various deletions in the cytoplasmic region from residue 774 to residue 848 (Fig. 4e). The gp130 mutants 7 and 8 displayed lower binding to TRAF5(242–558) (Fig. 4e), which indicated that amino acids 774–798 of gp130 were critical for this binding. That region in gp130 contains a ‘Ser-X-X-Glu’ motif (where ‘X’ indicates any amino acid; site 1, Ser-Arg-Ser-Glu) and two diacidic amino acids (site 2, Glu788-Glu789 and Glu792-Asp793) (Fig. 4f), which are recognition elements for the TRAF-C domain32–34. To investigate whether those motifs were critical for binding to TRAF5, we prepared mutants of gp130 with substitution of alanine for other amino acids (Fig. 4f,g). The gp130 mutant with substitutions in site 1 (AAAA, positions 746–779) or in site 2 (AARPAA, positions 788–793) showed considerably diminished binding to TRAF5(242–558) relative to that of wild-type gp130 (Fig. 4f). In contrast, the binding of gp130 mutants 12 (AARPED, positions 788–793) and 13 (EERPAA, positions 788–793) was similar to that of wild-type gp130 (Fig. 4g), which indicated that either of the two diacidic motifs in gp130 site 2 was necessary for the association of gp130 with TRAF5. Collectively, we concluded that both Ser-Arg-Ser-Glu (positions 746–779) and Glu-Glu-Arg-Pro-Glu-Asp (positions 788–793) in the cytoplasmic tail of gp130 were essential for its recognition by TRAF5. We found that those regions in gp130 are highly conserved across various species (Supplementary Fig. 4b).
TRAF5-gp130 binding negatively controls TH17 differentiation
To determine whether the carboxy-terminal domain of TRAF5 is inhibitory for TH17 differentiation, we cultured naive Traf5−/− CD4+ T cells in polarizing conditions with IL-6, transduced differentiating T cells with a retroviral vector encoding green fluorescent protein (GFP) and TRAF5(1–558), TRAF5(1–241) or TRAF5(242–558), and assessed the frequency of IL-17A+ cells in the GFP+CD4+ gated population. TRAF5(1–558) and TRAF5(242–558) significantly suppressed the development of IL-17A-expressing T cells, but TRAF5(1–241) did not (Fig. 5a). Thus, the carboxy-terminal domain of TRAF5 served an inhibitory role in IL-6-mediated TH17 development.
Figure 5.

The TRAF5-gp130 interaction limits the development of TH17 cells. (a) Flow cytometry of naive Traf5−/− CD4+ T cells activated for 12 h with anti-CD3 and anti-CD28 plus IL-2 in the presence of blocking antibodies to IFN-γ and IL-4 and then transduced with retroviral vector expressing GFP alone (empty vector (EV)) or GFP and TRAF5 (1–558) or TRAF5(1–241) or TRAF5(242–558) (above plots (left) and below graph (right)), followed 24 h later by the induction of TH17 differentiation for an additional 3 d through the addition of IL-6–IL-6R and TGF-β, then restimulation for 5 h with PMA and ionomycin. Numbers adjacent to outlined areas (left) indicate percent IL-17A+ cells among total GFP+CD4+ cells. (b) Flow cytometry of naive wild-type or Traf5−/− CD4+ T cells activated as in a and then transduced with retroviral vector encoding GFP alone (EV) or GFP-tagged gp130(769–800) with wild-type sequence (WT) or the alanine substitutions specified in Results (Ala-mut). NS, not significant; *P < 0.05 (Student’s t-test). Data are from one experiment representative of at least two independent experiments with similar results (average and s.d. of triplicate wells).
The finding that both site 1 and site 2 in gp130 (Fig. 4f) were responsible for its binding to TRAF5 raised the possibility that expression of a peptide containing those amino acid residues might inhibit the endogenous interaction between TRAF5 and gp130. To investigate this, we transfected HEK cells to express TRAF5(242–558), gp130 and a GFP fusion protein containing both site 1 and site 2 in gp130 (GFP-gp130(769–800)) (Supplementary Fig. 4c,d). As expected, GFP-gp130(769–800) immunoprecipitated together with TRAF5(242–558), but mutant GFP-gp130(769–800) with substitution of alanine for various amino acids in gp130 did not (Supplementary Fig. 4c), and expression of GFP-gp130(769–800) inhibited the binding of gp130 to TRAF5(242–558) (Supplementary Fig. 4d). Thus, these results demonstrated that GFP-gp130(769–800) competitively inhibited the binding of TRAF5 to gp130.
To elucidate how endogenous binding of TRAF5-gp130 affected TH17 differentiation, we transduced differentiating wild-type or Traf5−/− CD4+ T cells with retroviral vector encoding GFP-gp130(769–800) with wild-type sequence (VPSVQVFSRSESTQPLL DSEERPEDLQLVDSV; underlining indicates sequence altered in the mutant) or the mutant GFP-gp130(769–800) described above with substitution to alanine (VPSVQVFAAAASTQPLLDSAARPAALQL VDSV). GFP-gp130(769–800) with wild-type sequence significantly enhanced the generation of IL-17A-producing wild-type CD4+ T cells relative to that elicited by control vector or the mutant GFP-gp130(769–800) (Fig. 5b). We did not detect such activity in Traf5−/− CD4+ T cells (Fig. 5b). These results indicated that intracellular expression of a peptide containing both site 1 and site 2 in gp130 augmented IL-6-driven TH17 differentiation through the sequestration of endogenous TRAF5 away from gp130.
TRAF5 deficiency in CD4+ T cells exacerbates EAE
Finally, to investigate TRAF5 function in an IL-6-dependent disease model35, we immunized groups of Traf5−/− B6 mice and their wild-type littermates with a peptide of amino acids 35–55 myelin oligodendrocyte glycoprotein (MOG) emulsified in CFA on day 0, to induce EAE. We collected draining lymph nodes from wild-type and Traf5−/− mice on day 8 and restimulated those nodes with the MOG peptide to measure recall cytokine responses. IFN-γ and IL-17 responses in Traf5−/− cells from draining lymph nodes were higher than those in their wild-type counterparts (Fig. 6a); IL-4 and IL-10 were below the limit of detection (data not shown). Clinical signs of disease were evident on day 11 and peaked between days 14 and 18 (Fig. 6b). Traf5−/− mice developed EAE with kinetics similar to that of wild-type mice but with a considerably higher clinical score (Fig. 6b) and more body-weight loss (Supplementary Fig. 5a) than that of wild-type mice. The accumulation of IFN-γ+ CD4+ T cells and IL-17+ CD4+ T cells in the central nervous system on day 23 was also much greater in Traf5−/− mice (Fig. 6c). Although not only TH17 responses but also TH1 responses were enhanced in Traf5−/− mice (Fig. 6c), these results demonstrated that TRAF5 limited the generation of pathogenic MOG-specific T cells responsible for the induction of EAE. IFN-γ-producing cells may develop from IL-17-producing cells (‘ex-TH17 cells’) after receipt of other inflammatory signals35,36.
Figure 6.

TRAF5 serves an inhibitory role in EAE. (a) IL-17A and IFN-γ recall responses (right) in cells of draining lymph nodes obtained from wild-type and Traf5−/− mice 8 d after subcutaneous immunization (at the base of the tail) with MOG peptide (MOG(35–55)) in CFA, followed by culture (at a density of 3 × 106 cells per ml) for 36 h in vitro with various concentrations (horizontal axes) of MOG peptide. Left, quantification of total and CD4+ cells. (b) Clinical signs of EAE in mice as in a, monitored over 22 d. (c) Quantification of IL-17A+ or IFN-γ+ CD4+ lymphocytes isolated from central nervous system of mice as in a at day 23 after immunization, then restimulated in vitro for 5 h with PMA and ionomycin. (d) Clinical signs of EAE in irradiated B6.SJL (CD45.1+) recipient mice given no T cells or adoptive transfer of wild-type or Traf5−/− (CD45.2+) CD4+ T cells (5 × 106 cells per host), followed by immunization with MOG peptide in CFA, monitored over 25 d. †, mouse death. *P < 0.05 and **P < 0.01 (Student’s t-test). Data are from one experiment representative of two experiments (a) or are from three (b) or two (c) pooled experiments (average and s.e.m. of three (a), ten (b) or six (c) mice per genotype) or are from one experiment (d; average and s.e.m. of three mice (no cell transfer) or six mice (adoptive transfer).
To substantiate the hypothesis that the enhanced EAE responses noted above were caused by TRAF5 deficiency in CD4+ T cells, we adoptively transferred wild-type or Traf5−/− CD45.2+CD4+ T cells into congenic CD45.1+ B6 mice that had been sublethally irradiated, then immunized the recipient mice with the MOG peptide noted above in CFA on day 0 (Fig. 6d). At day 7, we detected donor CD45.2+CD4+ T cells in the peripheral blood of recipient mice, and the frequency of Traf5−/− donor cells was similar to that of wild-type cells (Supplementary Fig. 5b); this indicated that both genotypes of donor T cells populated the periphery equivalently. Recipients of Traf5−/− CD4+ T cells had significantly higher EAE scores than those of recipients of wild-type CD4+ T cells (Fig. 6d). We observed no signs of EAE in mice not been given injection of donor CD4+ T cells during this experimental period (Fig. 6d). Thus, TRAF5 limited the differentiation of pathogenic T cells responsible for EAE in a CD4+ T cell–intrinsic manner. Collectively, these results demonstrated that TRAF5 served as an important regulator for IL-6 signaling to limit the differentiation of proinflammatory CD4+ T cells.
DISCUSSION
In this study we found that the adaptor TRAF5 constitutively associated with the signal-transducing receptor gp130 in CD4+ T cells, which inhibited the IL-6-mediated activation of STAT3. Thus, TRAF5 limited the differentiation of naive CD4+ T cells into inflammatory effector T cells that received ‘instructive’ IL-6 signaling during the course of their development. Our results have identified a previously unknown anti-inflammatory activity for TRAF5 that is relevant for autoimmune and inflammatory diseases that are driven by pathogenic CD4+ T cells.
The physiological function of TRAF5 has remained elusive since its discovery. Although TRAF5 can act as a proinflammatory mediator downstream of some receptors25,26,28,37,38, several studies have suggested that TRAF5 also functions as an anti-inflammatory factor. After systemic immunization with OVA in alum adjuvant, Traf5−/− mice show greater susceptibility to the development of an asthma-like phenotype when challenged via the airway with aerosolized OVA, with enhanced airway hyper-responsiveness to methacholine, eosinophilic infiltration in their lungs, IL-5 and IL-13 in their broncho-alveolar lavage fluid, and OVA-specific immunoglobulin E in their plasma27. In the present study, when we immunized mice subcutaneously with MOG peptide (amino acids 35–55) in CFA, Traf5−/− mice developed much more severe clinical signs of EAE than did their wild-type counterparts, with greater infiltration of IL-17- and IFN-γ-producing CD4+ T cells into central nervous system. In addition to that, TRAF5-sufficient wild-type B6 mice given adoptive transfer of Traf5−/− CD4+ T cells also developed much more severe clinical signs of EAE than those of their counterparts given wild-type cells. These results indicate that TRAF5 in CD4+ T cells negatively regulates the differentiation of proinflammatory helper T cells that are critical for induction of inflammatory diseases.
Although our results support the conclusion that the notably exacerbated EAE phenotype of Traf5−/− mice was due to pronounced induction of inflammatory CD4+ T cells, TRAF5 might also control responsiveness of other cells to IL-6. It has been suggested that CD8+ T cells serve some roles in EAE. Traf5−/− CD8+ T cells exhibit defective primary population expansion of T cells and memory T cell responses in a model of infection with Listeria monocytogenes and are unresponsive to the prosurvival effects of CD27 (ref. 25), which indicates that TRAF5 is a positive signaling element in CD8+ T cells. Although Traf5−/− CD8+ T cells showed more phosphorylation of STAT3 than did wild-type CD8+ T cells, the difference was smaller for CD8+ T cells than for CD4+ T cells. Thus, it is unlikely that TRAF5 negatively controls EAE through CD8+ T cell responses. TRAF5 associates with the adaptor MyD88 and the binding partner TAB2 after stimulation via Toll-like receptors and antagonizes the association of TAB2 with TRAF6, which results in inhibition of signaling via Toll-like receptors. B cells from Traf5−/− mice produce more IL-6 in response to agonists of Toll-like recetors39. Those results suggest that B cells in Traf5−/− mice produce more IL-6 after immunization with CFA, which might contribute to the enhanced IL-17 production by Traf5−/− CD4+ T cells and more severe EAE disease in Traf5−/− mice. Although B cells expressed about twofold more Traf5 mRNA than did CD4+ T cells, we did not detect substantial expression of Il6st mRNA (which encodes gp130) or gp130 protein in B cells from wild-type and Traf5−/− mice, which indicated that TRAF5 might not be critical for IL-6 signaling in B cells. Additional work is needed for full understanding of the role of TRAF5 in cells other than CD4+ T cells and the universal role of the TRAF5-gp130 interaction in the context of inflammatory diseases.
Our original hypothesis was that the inhibitory activity of TRAF5 was derived from the modulation of signaling from a member of the TNF family. Many members of the TNFR superfamily, such as the T cell–costimulatory receptor OX40 (CD134), recruit TRAF5 to their intracellular domains40,41, and there is enhanced generation of TH2 cells driven by OX40 signaling in the context of a deficiency in TRAF5, indicative of direct regulatory activity27. We have also observed that an agonistic antibody to OX40 induced more production of IL-17 and IL-21 from antigen-responding Traf5−/− CD4+ T cells, but that was additionally accompanied by more production of IL-6 (data not shown). Because IL-6 has been linked to IL-17, IL-21 and the induction of TH2 differentiation, that suggested that IL-6 signaling might be affected mainly by TRAF5 deficiency. In agreement with that, exogenous IL-6 induced more production of IL-17 and IL-21 by Traf5−/− CD4+ T cells and more phosphorylation of STAT3 in Traf5−/− CD4+ T cells. That does not rule out the possibility of a role for TRAF5 in signaling from OX40 or other proteins of the TNFR family, but an inhibitory role during IL-6R signaling would provide a logical explanation for the enhanced differentiation of Traf5−/− CD4+ T cells into the TH2 and TH17 lineages.
STAT3 is activated during TH2 differentiation, and by directly binding to TH2 cell–associated loci, STAT3 facilitates the ability of STAT6 to bind target genes, such as the gene encoding GATA-3, which supports optimal commitment to TH2 differentiation12. STAT3 also regulates the expression of various genes encoding molecules essential for TH17 differentiation, such as Rorc, Il17a, Il17f, Il21, Il6ra and Il23r6,9,42. In our study, a pronounced TH17 response was induced in the absence of TRAF5 both in vitro and in vivo. In EAE experiments, however, not only TH17 responses but also TH1 responses were enhanced in Traf5−/− mice. During the development of EAE, MOG-specific TH17 cells rapidly lose IL-17A and produce IFN-γ instead36. That might explain why TRAF5 deficiency also resulted in greater TH1 responses in this model.
Providing a molecular explanation for why TRAF5 modulates TH2 and TH17 differentiation, we identified a previously unknown interaction between TRAF5 and gp130. TRAF5 needs its carboxy-terminal domain but not its amino-terminal RING or zinc-finger domain for binding with gp130. The TRAF-C domain associates with a wide variety of cytoplasmic proteins, including members of the TNFR superfamily14,32. The region of gp130 that interacts with the TRAF5-C domain has been mapped to the amino acid sequence VFSRSESTQPLLDSEERPEDLQLVD at positions 774–798. That region has been further subdivided into site 1 (Ser-Arg-Ser-Glu; positions 776–779) and site 2 (Glu-Glu-Arg-Pro-Glu-Asp; positions 788–793). Site 1 contains a ‘Ser-X-X-Glu’ motif, and site 2 contains two diacidic amino acids (Glu788-Glu789 and Glu792-Asp793), both of which are binding motifs for the TRAF-C domain32–34. In general, most TRAF proteins, including TRAF5, already exist as trimers in the cytosol before recruitment to the cytoplasmic tails of molecules of the TNFR family, and the proximity of cytoplasmic tails of those receptors is required for the binding of those receptors to TRAF proteins33. Thus, it is reasonable to conclude that the two TRAF-binding sites in close proximity in gp130 are critical for the constitutive association of TRAF5 with gp130 and that the newly formed four TRAF-binding sites in dimerized gp130 efficiently recruit an additional TRAF5 molecule to gp130. In line with the enhanced responses of Traf5−/− T cells to IL-6, we found that the interaction between the TRAF5-C domain and gp130 antagonized the interaction of gp130 with STAT3 and inhibited the development of TH17 cells. STAT3 binds to the four distal phosphorylated-tyrosine motifs of gp130: Tyr-Arg-His-Gln, Tyr-Phe-Lys-Gln, Tyr-Lys-Pro-Gln and Tyr-Met-Pro-Gln (with phosphorylated tyrosine at positions 765, 812, 904 and 914, respectively)43. The TRAF5-binding region is located between those first two motifs, which suggests that TRAF5 inhibits the recruitment of STAT3 to these two phosphorylated-tyrosine motifs through steric hindrance in gp130 or through modulation of an optimal configuration of gp130 that might be required for binding STAT3. Additional biochemical analyses are essential for understanding the structure-function relationship of the TRAF5-gp130 interaction. The dileucine internalization motif in gp130 (Lue784-Lue785) serves a role in the ligand-independent internalization of gp130 (refs. 4,44). The TRAF5-binding region identified in our study contained that motif; thus, the TRAF5-gp130 interaction might influence the internalization of gp130. However, we found that increasing or decreasing the expression of TRAF5 did not affect the amount of gp130 on the cell surface (data not shown), which suggests this is not the mechanism by which TRAF5 alters gp130 activity.
In summary, our results have revealed an unexpected molecular function for TRAF5. The interaction between TRAF5 and gp130 limited IL-6R–gp130–dependent activation of STAT3 and suppressed STAT3-dependent gene transcription, which controls the extent of effector CD4+ T cell development and can restrain the pathogenesis of autoreactive CD4+ T cells in inflammatory and autoimmune diseases. Our data showing that TRAF5 inhibited proinflammatory IL-6R signaling in CD4+ T cells have identified a previously unknown mechanism for controlling helper T cell differentiation and highlight a regulatory event in a wide range of inflammatory responses mediated by effector CD4+ T cells.
ONLINE METHODS
Mice
Traf5−/− mice on a B6 background have been described26,27. The progeny of heterozygous Traf5+/− mice backcrossed onto B6 mice more than ten times were intercrossed to generate Traf5+/+ and Traf5−/− mice. OT-II mice (a gift from W. Heath) bred onto the B6 or Traf5+/− B6 background were used as a source of Vβ5Vα2 CD45.2+ CD4+ T cells responsive to OVA peptide (amino acids 323–339) or OVA protein. B6 and B6.SJL-PtprcaPepcb/BoyJ (SJL) mice were from The Jackson Laboratory or Japan SLC. Littermates at 6–12 weeks of age were used in experiments. Sample size was chosen according to previous experience in similar experiments. Mice were randomly chosen for each experimental group, and no ‘blinding’ was used. All mice were bred and maintained under specific pathogen–free conditions, and experiments were in compliance with protocols approved by the La Jolla Institute and the Institute for Animal Experimentation, Tohoku University Graduate School of Medicine.
Peptide, chemicals, antibodies and cytokines
OVA peptide (amino acids 323–339) was synthesized by Abgent. MOG peptide (amino acids 35–55) was from Anaspec and Sigma-Aldrich. CFSE (carboxylfluorescein diacetate, succinimidyl ester; C34554), Dynabeads protein G (100-04D) and anti-V5 (R960-25) were from Life Technologies. eFluor450–anti-F4/80 (BM8; 48-4801), phycoerythrin–anti-Foxp3 (FJK-16s; 12-5773), allophycocyanin–anti-gp130 (Kgp130; 17-1302), fluorescein isothiocyanate–anti-IFN-γ (XMG1.2; 11-7311), phycoerythrin–anti-IL-6R (D7715A7; 12-1261) and phycoerythrin–anti-IL-17A (eBio17B7; 12-7177) were from Affymetrix. Fluorescein isothiocyanate–anti-CD3ε (low endotoxin, azide free; 145-2C11; 100331 or 100306), fluorescein isothiocyanate–anti-CD19 (6D5; 115505), anti-CD28 (low endotoxin, azide free; 37.51; 102112), fluorescein isothiocyanate–peridinin chlorophyll protein–anti-CD45.2 (104; 109806 or 109826), allophycocyanin–anti-IFN-γ (low endotoxin, azide free; XMG1.2; 505810 or 505827), anti-IL-4 (low endotoxin, azide free; 11B11; 504115), phycoerythrin–anti-IL-10R (1B1.3a; 112705), phycoerythrin–allophycocyanin–anti-IL-17A (TC11-18H10.1; 506905 or 506916) and phycoerythrin–anti-IL-21R (4A9; 131906) were from Biolegend. Pacific blue–peridinin chlorophyll protein–allophycocyanin–anti-CD4 (RM4-5; 558107, 553052 or 553051), phycoerythrin–anti-IFN-γ (XMG1.2; 554412), allophycocyanin–anti-IL-4 (11B11; 554436), and phycoerythrin–anti-NK1.1 (PK136; 553165) were from BD Biosciences. Anti-c-Myc (9B11, 2276), anti-STAT3 (9139), and antibody to STAT3 phosphorylated at Tyr705 (9145) were from Cell Signaling Technology. Anti-c-Myc (9E10; sc-40), anti-gp130 (M-20; sc-656) and anti-TRAF5 (C-19; sc-6195) were from Santa Cruz Biotechnology. Anti-β-actin (C4; MAB1501) was from Millipore. Anti-gp130 (HMβ1, D022-3) and anti-GFP (598) were from MBL. Allophycocyanin–anti-gp130 (125623; FAB4681A) and anti-TGF-β (1D11; MAB1835) were from R&D Systems. Recombinant mouse IL-6 (216-16), human IL-6 (200-06), soluble human IL-6R (200-06R), mouse IL-4 (214-14), IL-10 (210-10), IL-12 (210-12), IL-21 (210-21) and human TGF-β1 (100-21C) were from PeproTech.
Plasmids and transfection
A pCMV6 entry expression vector containing cDNA encoding mouse TRAF5 (TRAF5–c-Myc; MR208836) was from Origene. A TRAF5-V5 vector was generated by insertion of TRAF5-encoding cDNA into a pcDNA3.1/V5-HisA vector (Life Technologies). A c-Myc–gp130 vector generated by insertion of mouse gp130–encoding cDNA into a pEF4/Myc-HisB vector (Life Technologies) has been described45. A STAT3-pEF-Flag vector (2353)46,47 was provided by the RIKEN BRC DNA BANK through the National Bio-Resource Project of the MEXT, Japan. DNA encoding mutant constructs of TRAF5 and gp130 was generated with a PrimeSTAR Mutagenesis Basal Kit according to the manufacturer’s instructions (Takara). TRAF5 retroviral vectors were constructed by insertion of TRAF5-encoding cDNA into pMX-IRES-EGFP. For the creation of a retroviral vector expressing the GFP-gp130(769–800) fusion, sequence encoding the gp130(769–800) region was first amplified by PCR and ligated into the plasmid pEGFP-C3 (Clontech), and GFP-gp130(769–800) was secondarily ligated into the plasmid pMXs-puro48. The correct DNA sequence of the newly generated constructs was verified with a 3100 genetic analyzer and a BigDye Terminator kit (Life Technologies). FuGENE 6 (Roche) was used for transient transfection of HEK293T cells and Plat-E retroviral packaging cells. Retrovirus was produced by transfection of retrovirus vectors into Plat-E cells48. Virus-containing supernatants at days 2 and 3 were pooled and then were concentrated by centrifugation at 8,000g at 4 °C for 16 h. Supernatants containing 5 μg/ml polybrene were added to naive T cell cultures 12 h after initial activation. The cells were spun at 800g for 1 h at 32 °C and were further cultured for 8 h. Virus-containing supernatant was removed from the cultures and replaced with fresh medium, and TH17 differentiation was initiated by the addition of 30 ng/ml IL-6–IL-6R and 0.1 ng/ml TGF-β at 36 h.
T cells APCs and T cell culture
Naive (CD44loCD62Lhi) CD4+ T cells were purified from spleens of wild-type or Traf5−/− littermates with a naive CD4+ T cell isolation kit II (130-093-227; Miltenyi Biotec) and an AutoMACS Pro cell separator (Miltenyi) or a FACSAria II cell sorter (BD Biosciences). CD4 microbeads (L3T4; 130-049-201; Miltenyi) and CD8a microbeads (Ly-2; 130-049-401; Miltenyi) were used for the separation of splenic CD4+ T cells and CD8+ T cells, respectively. Splenic APCs from wild-type B6 mice were prepared by depletion of T cells through the use of CD90.2 microbeads (130-049-101; Miltenyi) and were irradiated with 30 Gy before use. Cells were cultured in RPMI medium with penicillin, streptomycin, glutamine, 2-mercaptoethanol and 7% FCS. Naive CD4+ T cells were plated at a density of 2.5 × 105 cells per ml, together with APC populations (at a density of 5 × 106 cells per ml) that had been depleted of T cells, in the presence of 0.1 μM OVA peptide (amino acids 323–339) for cultures of OT-II cells or 1 μg/ml soluble anti-CD3 for cultures of polyclonal CD4+ T cells. Naive polyclonal CD4+ T cells (2.5 × 105 cells per ml) were stimulated with 1 μg/ml plate-bound anti-CD3 (identified above) and 1 μg/ml soluble anti-CD28 (identified above).
Flow cytometry
Cells were incubated with anti-CD16/CD32 (2.4G2; produced in-house) before being stained with the appropriate antibodies to cell-surface and intracellular antigens (identified above). For staining of intracellular cytokines, cells were stimulated for 5 h with 50 ng/ml PMA (phorbol 12-myristate 13-acetate) and 1 μg/ml ionomycin (EMD Biosciences) in the presence of GolgiPlug (BD Biosciences). After staining of surface markers, cells were fixed and permeabilized with Cytofix/Cytoperm and Perm/Wash buffer (BD Biosciences) or Foxp3 staining buffer (Affymetrix) according to the manufacturer’s instructions. Data were acquired on a FACSCalibur or a FACSCanto II (BD Biosciences) and were analyzed with FlowJo software (Tree Star).
In vivo experiments
Nonirradiated syngeneic SJL (CD45.1+) recipient mice were given intravenous injection of 5 × 104 donor naive CD4+ T cells from wild-type or Traf5−/ OT-II (CD45.2+) mice. One day after cell transfer, mice were immunized by subcutaneous injection at the base of the tail of 2 mg OVA protein in CFA (Difco). The inguinal, lumbar and sacral lymph nodes were collected 7 d after immunization.
EAE was induced by subcutaneous injection of 150 μg MOG peptide (amino acids 35–55), in 0.1 ml of CFA emulsion containing 400 μg Mycobacterium tuberculosis (Difco), into wild-type or Traf5−/− B6 mice on day 0. The mice received intraperitoneal injection of 200 ng pertussis toxin (List Biological Laboratories) on days 0 and 2. Clinical signs of EAE were assessed according to the following score: 0, no sign of disease; 1, limp tail or hind limb weakness; 2, partial hind limb paralysis; 3, complete hind limb paralysis; 4, complete hind limb paralysis and partial front leg paralysis; 5, moribund or dead.
For the isolation of mononuclear cells from central nervous system, anesthetized mice were perfused with 20 ml of PBS for the removal of blood from internal organs. The spinal cords were isolated and cut into several small pieces, then were placed in 10 ml of 2 mM EDTA in PBS and incubated for 45 min at 37 °C. The cell suspension was mixed by pipetting and was passed through a 70-μm cell strainer. Samples were enriched for mononuclear cells by centrifugation through a two-layer Percoll gradient (37% and 70%) at 700g for 20 min and were washed twice before further analysis.
For evaluation of the ability of CD4+ T cells to induce EAE, irradiated syngeneic SJL recipient mice (6 Gy) were given intravenous injection of 5 × 106 donor CD4+ T cells from wild-type or Traf5−/ B6 mice. Three days after cell transfer, mice were given subcutaneous immunization of MOG peptide and CFA followed by intraperitoneal injection of pertussis toxin.
Enzyme-linked immunosorbent assay
IFN-γ in culture supernatants was assessed by a sandwich enzyme-linked immunosorbent assay protocol with R4-6A2 (551216) and biotin-XMG1.2 (554410) from BD Biosciences. Enzyme-linked immunosorbent assay kits were as follows: for IL-6 (88-7064-88), IL-17A (88-7371-88) and IL-21 (88-8210-88), Affymetrix; for IL-21 (DY594), R&D Systems; for IL-17A (432505), Biolegend.
Immunoprecipitation and immunoblot analysis
Cells were lysed for 30 min in ice-cold 1% Nonidet P-40 (NP-40) buffer (20 mM Tris-HCl, pH7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 50 mM NaF, 1 mM Na3VO4 and 10 mM N-ethylmaleimide, containing protease-inhibitor mixture (P8340; Sigma-Aldrich)) or RIPA buffer (1% NP-40 buffer containing 1% sodium deoxycholate and 0.1% sodium dodecyl sulfate). Insoluble material was removed by centrifugation at 15,000g for 10 min. Protein content was determined by bicinchoninic acid assay (Thermo Scientific). Proteins were immunoprecipitated from lysates overnight at 4 °C with primary antibodies (identified above) immobilized on Dynabeads protein G. After being washed extensively with ice-cold lysis buffer, beads were boiled for 5 min at 100 °C in 4× lithium dodecyl sulfate sample buffer (NP0007; Life Technologies). Eluted sample were further reduced for 10 min at 70 °C with DTT or 2-mercaptoethanol for immunoblot analysis. Samples were separated by SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore) and analyzed by immunoblot with the appropriate antibodies (identified above). All blots were developed with Immobilon Western HRP substrate (Millipore).
Real-time RT-PCR
SYBR Premix Ex Tag (Takara Bio) and a 7500 real-time PCR system (Life Technologies) were used for quantitative RT-PCR. Total RNA was extracted with TRIzol (Life Technologies), and cDNA was then synthesized with SuperScript III Reverse Transcriptase and oligo(dT)20 (Life Technologies). Each transcript was analyzed concurrently on the same plate with the gene encoding β-actin, and results are presented relative to the abundance of transcripts encoding β-actin. Primers were as follows: Traf5 (forward primer, 5′-CCGACACCGAGTACCAGTTTG-3′; reverse primer, 5′-CGGCACCGAGTTCAATTCTC-3′); Il6st (forward primer, 5′-TACATGGTCCGAATGGCCGC-3′; reverse primer, 5′-GGCTAAGCACACAGGC ACGA-3′); Rorc (forward primer, 5′-TCGACAAGGCCTCCTAGCCA-3′; reverse primer, 5′-CTTGGACCACGATGGGGTGG-3′); Tbx21 (forward primer, 5′-GG TTGGAGGTGTCTGGGAAGC-3′; reverse primer, 5′-GCCACGGTGAAGGACAGGAAT-3′); Gata3 (forward primer, 5′-GGCAGAACCGGCCCCTTATC-3′; reverse primer, 5′-TGGTCTGACAGTTCGCGCAG-3′); Foxp3 (forward primer, 5′-CCCATCCCCAGGAGTCTTG-3′; reverse primer, 5′-ACCATGACTAGGGGCACTGTA-3′); Il17a (forward primer, 5′-TTTAACTCCCTTGGCGCAAAA-3′; reverse primer, 5′-CTTTCCCTCCGCATTGACAC-3′); Il17f (forward primer, 5′-ACCAGCATGAAGTGCACCCGT-3′; reverse primer, 5′-AGGCAGGAACCCCTGCTTTGG-3′); Il23r (forward primer, 5′-ACTCACTGCAAGGCAGCAGG-3′; reverse primer, 5′-AGCCCTGGAAATGATGGACGC-3′); Actb (forward primer, 5′-CTGCCTGACGGCCAGG-3′; reverse primer, 5′-GGAAAAGAGCCTCAGGGCAT-3′).
Statistics
Statistical significance was assessed with Student’s t-test with two-sided distributions.
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Heath (University of Melbourne) for OT-II mice; S. Nagata (Kyoto University) and S. Akira (Osaka University) for the Flag-pEF-STAT3 vector. Supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (C) (24590571 to T.S.), the Ichiro Kanehara Foundation (T.S.), the Takeda Science Foundation (T.S.), the Suzuken Memorial Foundation (T.S.) and the US National Institutes of Health (AI049453 to M.C.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS H.Nag., M.C., N.I. and T.S. designed the experiments; H.Nag., Y.O., A.A., T.K., S.Y. and T.S. did the experiments; H.Nag., Y.O., A.A., T.K., S.Y., M.C., N.I. and T.S. analyzed data; H.Nak. contributed reagents and analytical tools; M.C., N.I. and T.S. supervised the project; M.C., N.I. and T.S. wrote the paper; and M.C., N.I. and T.S. provided funding for the project.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 3.Hirano T. Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2010;86:717–730. doi: 10.2183/pjab.86.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinrich PC, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kishimoto T. Interleukin-6: from basic science to medicine—40 years in immunology. Annu. Rev. Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 6.Zhou L, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 7.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 8.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 9.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 10.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002;39:531–536. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 11.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22:83–89. doi: 10.1016/j.cytogfr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Stritesky GL, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildebrand JM, et al. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol. Rev. 2011;244:55–74. doi: 10.1111/j.1600-065X.2011.01055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ha H, Han D, Choi Y. In: Current Protocols in Immunololgy. Coligan JE, Bierer BE, Margulies DH, Shevach EM, Strober W, editors. John Wiley & Sons; 2009. Ch.11, Unit 11.9D, 11.9D.1–11.9D.19. [Google Scholar]
- 15.Häcker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011;11:457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 16.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 17.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat. Rev. Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 18.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 20.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 21.Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J. Immunol. 2011;186:143–155. doi: 10.4049/jimmunol.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsitsikov EN, et al. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 23.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 24.King CG, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat. Med. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 25.Kraus ZJ, Haring JS, Bishop GA. TNF receptor-associated factor 5 is required for optimal T cell expansion and survival in response to infection. J. Immunol. 2008;181:7800–7809. doi: 10.4049/jimmunol.181.11.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano H, et al. Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc. Natl. Acad. Sci. USA. 1999;96:9803–9808. doi: 10.1073/pnas.96.17.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.So T, Salek-Ardakani S, Nakano H, Ware CF, Croft M. TNF receptor-associated factor 5 limits the induction of Th2 immune responses. J. Immunol. 2004;172:4292–4297. doi: 10.4049/jimmunol.172.7.4292. [DOI] [PubMed] [Google Scholar]
- 28.Esparza EM, Lindsten T, Stockhausen JM, Arch RH. Tumor necrosis factor receptor (TNFR)-associated factor 5 is a critical intermediate of costimulatory signaling pathways triggered by glucocorticoid-induced TNFR in T cells. J. Biol. Chem. 2006;281:8559–8564. doi: 10.1074/jbc.M512915200. [DOI] [PubMed] [Google Scholar]
- 29.Grech A, Quinn R, Srinivasan D, Badoux X, Brink R. Complete structural characterisation of the mammalian and Drosophila TRAF genes: implications for TRAF evolution and the role of RING finger splice variants. Mol. Immunol. 2000;37:721–734. doi: 10.1016/s0161-5890(00)00098-5. [DOI] [PubMed] [Google Scholar]
- 30.Tada K, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J. Biol. Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 31.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 32.Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell. 1999;4:321–330. doi: 10.1016/s1097-2765(00)80334-2. [DOI] [PubMed] [Google Scholar]
- 33.Park YC, Burkitt V, Villa AR, Tong L, Wu H. Structural basis for self-association and receptor recognition of human TRAF2. Nature. 1999;398:533–538. doi: 10.1038/19110. [DOI] [PubMed] [Google Scholar]
- 34.Arch RH, Thompson CB. 4–1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor κB. Mol. Cell. Biol. 1998;18:558–565. doi: 10.1128/mcb.18.1.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serada S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirota K, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulek K, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat. Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun D, et al. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat. Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchta CM, Bishop GA. TRAF5 negatively regulates TLR signaling in B lymphocytes. J. Immunol. 2014;192:145–150. doi: 10.4049/jimmunol.1301901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-κB activation. J. Biol. Chem. 1998;273:5808–5814. doi: 10.1074/jbc.273.10.5808. [DOI] [PubMed] [Google Scholar]
- 41.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009;229:173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahl N, et al. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 44.Dittrich E, Haft CR, Muys L, Heinrich PC, Graeve L. A di-leucine motif and an upstream serine in the interleukin-6 (IL-6) signal transducer gp130 mediate ligand-induced endocytosis and down-regulation of the IL-6 receptor. J. Biol. Chem. 1996;271:5487–5494. doi: 10.1074/jbc.271.10.5487. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka Y, et al. c-Cbl-dependent monoubiquitination and lysosomal degradation of gp130. Mol. Cell. Biol. 2008;28:4805–4818. doi: 10.1128/MCB.01784-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minami M, et al. STAT3 activation is a critical step in gp130-mediated terminal differentiation and growth arrest of a myeloid cell line. Proc. Natl. Acad. Sci. USA. 1996;93:3963–3966. doi: 10.1073/pnas.93.9.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kitamura T, et al. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.