

Abstract
The glycoprotein milk fat globule-EGF factor 8 (MFG-E8) is expressed in several tissues and mediates diverse homeostatic functions. However, whether MFG-E8 plays a role in bone homeostasis has not been established. Here we show for the first time that osteoclasts express and are regulated by MFG-E8. Bone marrow-derived osteoclast precursors (OCPs) from MFG-E8–deficient (Mfge8−/−) mice underwent increased RANKL-induced osteoclastogenesis leading to enhanced resorption pit formation as compared with wild-type controls. Consistently, exogenously added MFG-E8 inhibited RANKL-induced osteoclastogenesis from mouse or human OCPs. Upon induction of experimental periodontitis, an oral inflammatory disease characterized by loss of bone support of the dentition, Mfge8−/− mice exhibited higher numbers of osteoclasts and more bone loss than wild-type controls. Accordingly, local microinjection of anti-MFG-E8 mAb exacerbated periodontal bone loss in wild-type mice. Conversely, microinjection of MFG-E8 inhibited bone loss in experimental mouse periodontitis. In comparison to wild-type controls, Mfge8−/− mice also experienced >60% more naturally occurring chronic periodontal bone loss. In conclusion, MFG-E8 is a novel homeostatic regulator of osteoclasts and could be exploited therapeutically to treat periodontitis and perhaps other immunological disorders associated with inflammatory bone loss.
Introduction
Originally identified as a milk protein, milk fat globule-epidermal growth factor (EGF)-factor 8 (MFG-E8; also termed lactadherin) is now known to be expressed in a range of tissues where it performs diverse homeostatic functions (1). The molecule comprises two N-terminal EGF-like domains and two C-terminal discoidin-like domains with sequence similarity to blood coagulation factors V and VIII. A signal peptide at the N-terminus of the nascent MFG-E8 protein mediates secretion (1).
MFG-E8 is expressed by macrophages, fibroblasts, dendritic and epithelial cells in various organs and tissues, including the mammary glands, spleen, lungs, liver, kidneys, and intestine (2). Numerous studies in mouse models of physiology and disease have shown that MFG-E8 mediates apoptotic cell clearance (3), maintenance and repair of intestinal epithelia (4), anti-inflammatory action in neutrophils and macrophages (5, 6), and regulation of physiological (or pathological) angiogenesis (7, 8). In both humans and animal models, the expression of MFG-E8 declines considerably in inflammatory conditions, including sepsis, colitis, acute lung injury, ischemia/reperfusion injury, atherosclerosis, and Alzheimer’s disease (2, 9, 10). Importantly, experimental exogenous administration of MFG-E8 mitigates inflammation and tissue damage in several disease models (11).
Although MFG-E8 has been investigated extensively, a possible role in bone homeostasis and disease has not been established for this functionally versatile molecule. Here we show for the first time that MFG-E8 is expressed by and regulates osteoclasts (OCLs), giant multinucleated cells (MNCs) that resorb bone during normal bone remodeling and also under pathologic inflammatory conditions that potentiate their resorptive activity (e.g., rheumatoid arthritis and periodontitis) (12, 13). OCLs differentiate from bone marrow (BM) precursors in the monocyte/macrophage lineage. In this process, macrophage colony-stimulating factor (M-CSF) promotes the survival and proliferation of osteoclast precursors which are induced to express receptor activator of NF-κB (RANK), thereby becoming competent to respond to RANK ligand (RANKL), a key cytokine for OCL differentiation and activation (12). Our present findings indicate that MFG-E8 is a novel regulator that restrains RANKL-induced osteoclast differentiation and function and can be used therapeutically to inhibit inflammatory bone loss.
Materials and Methods
Mice
Mfge8−/− mice were generated as previously described and were speed backcrossed to the C57BL/6NCr genotype to generate mice that were ≈99% identical to C57BL/6NCr mice (14). Colonies of Mfge8−/− and C57BL/6NCr WT controls (Charles River Laboratories) were established at the University of Pennsylvania. Mice were housed in a pathogen-free environment and used when they were 8–10 wk-old except in experiments of aging lasting up to the age of 13 mo. All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Periodontitis models
(a) Ligature-induced periodontitis
The placement of ligatures accelerates bacteria-mediated inflammation and bone loss (15). To induce bone loss, a 5-0 silk ligature was tied around the maxillary left second molar, as previously described (16). The contralateral molar tooth in each mouse was left unligated to serve as baseline control for bone loss measurements. The ligatures remained in place in all mice throughout the experimental period. The mice were euthanized at various timepoints (0 to 10d) after placement of the ligatures and defleshed maxillae were used to measure bone heights (i.e., the distances from the cementoenamel junction [CEJ] to the alveolar bone crest [ABC]) under a Nikon SMZ800 microscope using a 40 × objective. Images of the maxillae were captured using a Nicon Digital Sight DS-U3 camera controller and CEJ-ABC distances were measured at 6 predetermined points on the ligated molar and adjacent regions using NIS-Elements software (Nikon Instruments Inc.) (16). To calculate bone loss, the 6-site total CEJ-ABC distance for the ligated side of each mouse was subtracted from the 6-site total CEJ-ABC distance of the contralateral unligated side. The results were presented in mm and negative values indicated bone loss relative to the baseline (unligated control). In intervention experiments, anti-MFG-E8 mAb (B1F10 (8)) or rMFG-E8 (R&D Systems), or corresponding controls (IgG2a and BSA, respectively), were microinjected into the palatal gingiva of the ligated second maxillary molar, as previously described (17, 18).
(b) Naturally-occurring periodontitis
Mfge8−/− mice and WT controls were reared in parallel from the age of 10 wk until 13 mo. Defleshed maxillae from euthanized mice were used for bone measurements. CEJ-ABC distances were measured on 14 predetermined points on maxillary molars (19). To calculate bone loss, the 14-site total CEJ-ABC distance for each mouse was subtracted from the mean CEJ-ABC distance of sham-infected mice.
Osteoclastogenesis
RANKL-induced osteoclastogenesis was performed according to standard protocols using mouse BM-derived monocyte/macrophage precursor cells (20), RAW264.7 precursor cells (21), or human CD14+ monocytes (22).
(a) BM-derived precursors
BM cells were flushed from femurs and tibias of mice. After lysis of erythrocytes using RBC lysis buffer (eBioscience), BM cells were cultured on petri dishes with recombinant murine M-CSF (5 ng/ml; R&D Systems) for 16h. The nonadherent cell population was recovered and further cultured in α-MEM media/10%FBS with 100 ng/ml M-CSF for 3d. Floating cells were removed and attached cells were used as BM-derived monocyte/macrophage precursor cells (“osteoclast precursors”; OCPs). OCPs (1×105 per well in a 96-well plate) were cultured for 3d in the presence of 50 ng/ml soluble recombinant RANKL (R&D Systems) and 100 ng/ml M-CSF to generate OCLs. The cells were fixed and stained for TRAP using an acid phosphatase leukocyte diagnostic kit (Sigma-Aldrich) and TRAP+ multinucleated (≥ 3 nuclei) cells were counted (20).
(b) RAW264.7 cells
To induce osteoclastogenesis from RAW264.7 cells, the cells were plated at a density of 2×103 cells per well into a 96-well plate and cultured with α-MEM media/10%FBS in the presence of 20 ng/ml RANKL for 4d (M-CSF was not added as this cytokine is produced by RAW264.7 cells). Cultures were re-fed and re-treated with RANKL at d3 and TRAP+ multinucleated cells were counted the following day (21).
(c) Human monocytes
To generate human OCLs (22), CD14+ monocytes were isolated from human peripheral blood mononuclear cells (obtained from the University of Pennsylvania Human Immunology Core) using anti-CD14 magnetic beads as instructed by the manufacturer (StemCell Technologies). After incubation with M-CSF (20 ng/ml) for 24h, the generated OCPs were added to 96-well plates at a seeding density of 5×104 cells per well and incubated in α-MEM media/10%FBS supplemented with M-CSF (20 ng/ml) and RANKL (40 ng/ml) for 5d. Media and cytokines were replenished on d3. TRAP+ multinucleated cells were counted on d5.
In all experiments, TRAP+ MNCs were imaged using a Nikon Eclipse NiE automated upright fluorescent microscope and met the criteria of authentic OCLs, manifested by expression of OCL differentiation markers and bone-resorbing activity on Ca3(PO4)2-coated wells (see Results). In experiments of mouse or human osteoclastogenesis designed to determine the effects of MFG-E8, mouse or human rMFG-E8 (R&D Systems) was added together with RANKL.
Resorption pit formation
OCL resorption activity was determined using Osteo Assay Surface plates following the protocol of the manufacturer (Corning). Briefly, mouse OCPs from BM were plated at a density of 1×105cells/well in a 96-well plate coated with inorganic bone biomaterial, crystalline Ca3(PO4)2. The cells were cultured in the presence of M-CSF (100 ng/ml) with or without RANKL (50 ng/ml) for 4d. Similarly, human OCPs prepared as above (see Osteoclastogenesis) were plated at a density of 5×104 cells/well in a 96-well plate coated with Ca3(PO4)2 and cultured with 20 ng/ml M-CSF, with or without 40 ng/ml RANKL, for 5d. At the end of the incubation period, both mouse and human OCLs were removed by 5-min treatment with 10% bleach and resorptive areas were visualized by light microscopy. The total resorbed area was measured using Photoshop CS6.
Histological TRAP staining
Maxillae with intact surrounding tissue were fixed in 4% paraformaldehyde, decalcified in Immunocal solution (Decal Chemical) for 14d, and embedded in OCT compound. TRAP staining was performed on coronal sections (6-μm thick) using the leukocyte acid phosphatase kit (Sigma-Aldrich). Slides were viewed using a Nikon Eclipse NiE microscope and TRAP+ MNCs were considered to be OCLs.
Immunofluorescence histochemistry
Maxillary sections prepared as above were stained with antibodies to MFG-E8 (18A2-G10, MBL) or cathepsin K (polyclonal; Abcam) followed by secondary reagents (AlexaFluor594–conjugated goat anti-hamster IgG or AlexaFluor488-conjugated goat anti-rabbit IgG; Life Technologies). The specificity of staining was confirmed using appropriate isotype control or non-immune IgG. Images were captured using a Nikon Eclipse NiE automated fluorescent microscope.
Quantitative real-time PCR (qPCR)
Total RNA was extracted from excised gingival tissue or cultured cells using TRIzol (InVitrogen) or RNeasy Mini Kit (Qiagen) and quantified by spectrometry at 260 and 280 nm. The RNA was reverse-transcribed using the High Capacity RNA-to-cDNA Kit (Life Technologies) and qPCR with cDNA was performed using the Applied Biosystems 7500 Fast Real-Time PCR System according to the manufacturer’s protocol (Life Technologies). Data were analyzed using the comparative (ΔΔCt) method. TaqMan probes, sense primers, and antisense primers for detection and quantification of genes investigated in this paper were purchased from Life Technologies.
Determination of bacterial counts
In ligature-induced periodontitis, the ligatures were recovered from euthanized mice and gently washed with PBS to remove food residue and other debris. Subsequently, the sutures were placed in Eppendorf tubes with 1 ml PBS and the bacteria were extracted by vortexing for 2 min at 3000 rpm. Serial dilutions of the bacterial suspensions were plated onto blood agar plates and CFU were enumerated following anaerobic growth at 37°C for 7d. Results were normalized by dividing CFU by the length (mm) of the corresponding suture. To assess the oral microbial burden in naturally-occurring periodontitis, the murine oral cavity was sampled for 1 min using sterile polyester tipped applicators (Puritan Medical Products Co.) held against the gumlines and the extracts were processed as above for CFU enumeration.
Antimicrobial activity
The disk inhibition zone assay was used to determine possible antimicrobial activity of MFG-E8 using Imipenem and PBS as positive and negative control, respectively. The assay was performed according to the Performance Standards for Antimicrobial Susceptibility Testing (Twenty First Informational Supplement, M10S21). A total of 15 anaerobic bacterial isolates were randomly selected from ligature-induced periodontal lesions of 5 mice (3 isolates per mouse). Sterile filter paper discs (7-mm diameter; 185-μm thickness) were impregnated with various amounts of the test and control compounds and placed on Gifu anaerobic medium (GAM)-based blood agar plates (Nissui Pharmaceutical), which had been previously spread with 100 μl of inocula, each containing bacterial suspension equivalent to 0.5 McFarland standard. The plates were incubated at 37°C for 7d and the diameter of the growth inhibition zones around the discs was measured using a vernier caliper.
Immunoprecipitation and immunoblotting
Cell lysates were prepared using the RIPA Lysis Buffer System (Santa Cruz Biotechnology) and protein content concentrations were determined by Nano Drop 2000C spectrophotomer (Thermo Scientific). Immunoprecipitation was carried out using goat polyclonal anti-MFG-E8 antibody or non-immune IgG control (R&D Systems) and protein G-coupled magnetic beads according to the manufacture’s protocol (Life Technologies). Proteins were separated by standard SDS-PAGE on 10% acrylamide gels (Life Technologies) and transferred to polyvinylidene difluoride membrane (Bio-Rad) by electroblotting. The membranes were incubated in blocking buffer (5% nonfat dried milk, 10 mM Tris [pH 7.5], 100 mM NaCl, and 0.05% Tween 20) followed by probing with goat polyclonal anti-MFG-E8 antibody (R&D Systems) or anti-MFG-E8 mAb (18A2-G10; MBL) and visualization with horseradish peroxidase-conjugated secondary antibody and chemiluminescence using the Amersham Biosciences ECL system. Images were captured using a FluorChem M imaging system (ProteinSimple).
Statistical analysis
Data were evaluated by ANOVA and the Dunnett multiple-comparison test using the InStat program (GraphPad). Where appropriate (comparison of two groups only), two-tailed unpaired t tests were performed. All experiments were performed two or more times for verification. A p value <0.05 was taken as the level of significance.
Results
Expression of MFG-E8 in the periodontal tissue and by in vitro generated OCLs
In view of the anti-inflammatory potential of MFG-E8 (1, 2), we investigated its role in periodontitis, a microbiota-induced inflammatory disease causing loss of bone support of the dentition (23). Using the ligature-induced periodontitis model in mice (15, 16), we first monitored the expression of MFG-E8 mRNA in the periodontal tissue. Consistent with MFG-E8 downregulation in various models of inflammation (2), periodontal MFG-E8 mRNA levels weresignificantly decreased within 24h (from d0 to d1) upon placement of the ligatures (Fig. 1A). Subsequently, and unexpectedly, MFG-E8 mRNA expression gradually increased until d8 (Fig. 1A). The resurgence of MFG-E8 expression correlated with the appearance of OCLs, the numbers of which increased to d8 but dropped on d10 (Fig. 1B and Supplemental Fig. 1), when MFG-E8 expression also appeared to decline (Fig. 1A). Being a cumulative process, bone loss continued to rise to d10 (Fig. 1B, right Y axis). The correlation of MFG-E8 re-expression with osteoclastogenesis suggested that MFG-E8 might derive from OCLs in the course of periodontitis. Consistent with this notion, MFG-E8 was detected in regions of cathepsin K expression, at the interface of connective tissue (periodontal ligament) and bone (Fig. 1C). Moreover, the sites of MFG-E8 and cathepsin K expression coincided with TRAP+ cells (Fig. 1C; bottom row).
Figure 1. Expression of MFG-E8 in the periodontal tissue and by in vitro generated OCLs.

(A) Timecourse of MFG-E8 mRNA expression in the periodontal tissue after ligature-induced periodontitis. Results (means ± SD; n = 5 mice) were normalized to GAPDH mRNA and presented relative to those at d0, set as 1. (B) Mice were treated as above and TRAP+ MNCs were enumerated and averaged (with SD) from 60 random coronal sections (20 from each of three mice per group) of ligated teeth with surrounding periodontal tissue. Dashed line with open circles shows timecourse of bone loss in similarly treated mice (means ± SD; n = 5). (C) Tissue sections from ligature-induced periodontitis at d5 were stained as indicated. Bottom row images involve the same section processed for immunofluorescence and TRAP staining. B, bone; D, dentin; DIC, differential interference contrast; PL, periodontal ligament. Scale bars; 50μm (white), 500μm (black). (D) RANKL-induced OCLs from progenitor RAW264.7 cells (+RANKL) were assayed for mRNA expression of indicated molecules. Results (means ± SD; n = 3) were normalized to GAPDH mRNA and presented relative to those of undifferentiated RAW264.7 cells (−RANKL), set as 1. (E) Light microscopy of undifferentiated (−RANKL) and RANKL-differentiated RAW264.7 cells (scale bar, 50μm) and enumeration of TRAP+ MNCs in the cultures (means ± SD; n = 3). (F) DIC and fluorescent images of RANKL-differentiated OCL stained for MFG-E8 and nuclei (DAPI) (scale bar, 50μm). (G) Anti-MFG-E8 immunoblotting of cell lysates from undifferentiated RAW264.7 cells (−RANKL) and differentiated OCLs (+RANKL). (H) Immunoprecipitation of MFG-E8 from culture supernatants of RANKL-differentiated OCLs using goat anti-MFG-E8 IgG antibody (1, OCLs differentiated from RAW264.7 cells; 2, OCLs differentiated from mouse BM-derived precursors) followed by immunoblotting with the same antibody. *P < 0.01. NS, not significant.
RAW264.7 cells are widely used to model OCL differentiation, as RANKL-induced RAW264.7 gene expression and developmental and functional characteristics are similar to those of OCLs in vivo or OCLs generated in vitro from primary precursor cells (21). Consistent with the concept that OCLs may constitute a source of MFG-E8, we showed that RANKL-differentiated RAW264.7 OCLs express MFG-E8 mRNA (six-fold upregulation as compared to undifferentiated RAW264.7 cells), in addition to established activation and functional markers, such as NFATc1, the heterodimeric αvβ3 integrin (CD51/CD61), and cathepsin K (Fig. 1D). NFATc1, the master transcription factor for OCL differentiation (12), was also upregulated at the protein level (Supplemental Fig. 2A). The generated OCLs (identity further confirmed morphologically after TRAP staining; Fig. 1E) also expressed MFG-E8 protein, as shown by cell immunofluorescence (Fig. 1F), immunoblotting of cell lysates (Fig. 1G), and immunoprecipitation from culture supernatants (Fig. 1H). MFG-E8 protein expression, determined by whole-cell lysate immunoblotting, was also shown for primary OCLs generated from mouse BM-derived precursors or from human CD14+ monocytes (Supplemental Fig. 2B). These data show for the first time that OCLs express MFG-E8.
MFG-E8 regulates osteoclast differentiation and function
To characterize the role of MFG-E8 in osteoclastogenesis, we examined the differentiation of RANKL-stimulated RAW264.7 cells in the absence or presence of exogenously added rMFG-E8. rMFG-E8 inhibited the expression of OCL differentiation and functional markers, namely, NFATc1, β3 integrin, and cathepsin K (Fig. 2). To obtain conclusive evidence that MFG-E8 is involved in homeostatic regulation of OCLs, we generated OCLs from WT or Mfge8−/− osteoclast precursors (OCP) from BM (20). Mfge8−/− OCPs underwent more efficient osteoclastogenesis (higher numbers of TRAP+ MNCs) than WT OCPs (Fig. 3A), consistent with higher expression of OCL markers (cathepsin K, TRAP, and integrin β3) (Fig. 3B). Moreover, Mfge8−/− OCLs caused enhanced resorption pit formation compared with their WT counterparts (Fig. 3C). Importantly, addition of rMFG-E8 dose-dependently inhibited osteoclastogenesis from Mfge8−/− OCPs (Fig. 3D). The degree of Mfge8−/− osteoclastogenesis in the presence of 1–2 μg/ml rMFG-E8 was comparable to WT osteoclastogenesis (Fig. 3D). These data implicate MFG-E8 as a novel negative regulator of osteoclastogenesis, at least in mice. MFG-E8 may have a similar function in humans. Indeed, in a system of osteoclastogenesis from human CD14+ monocytes, human rMFG-E8 inhibited RANKL-induced expression of OCL differentiation and functional markers (Fig. 4A), osteoclastogenesis (Fig. 4B), and resorption pit formation (Fig. 4C).
Figure 2. MFG-E8 inhibits RANKL-induced differentiation of RAW264.7 OCLs.
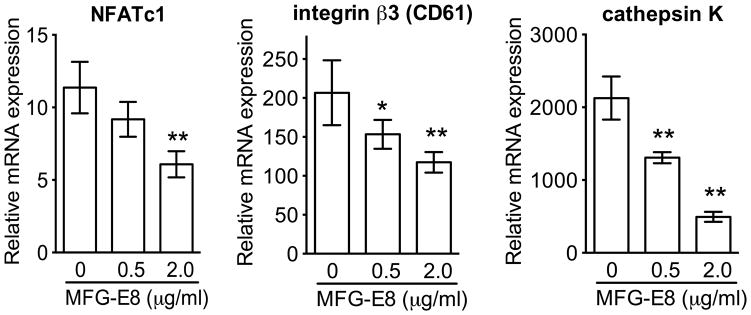
RANKL-induced osteoclastogenesis from progenitor RAW264.7 cells was performed in the absence or presence of the indicated concentrations of MFG-E8 and mRNA expression of the indicated molecules was assayed by qPCR. Results (means ± SD; n = 4) were normalized to GAPDH mRNA and presented relative to those of undifferentiated RAW264.7 cells, set as 1. *P < 0.05; **P < 0.01.
Figure 3. MFG-E8 regulates osteoclast differentiation and function.

(A) Enumeration of TRAP+ MNCs in RANKL-stimulated cultures of WT and Mfge8−/− OCPs and representative photomicrographs (scale bar, 100μm). (B) RANKL-induced OCLs were generated from WT or Mfge8−/− OCPs and after 3d were assayed for mRNA expression of the indicated molecules. Results were normalized to those of GAPDH mRNA and presented relative to those of undifferentiated OCPs, set as 1. (C) WT and Mfge8−/− OCPs were cultured under osteoclastogenic conditions for 4d on Ca3(PO4)2-coated wells and the total resorption area (dark spots) in each culture was measured and expressed relative to the WT group, set as 1. (D) RANKL-induced osteoclastogenesis from Mfge8−/− OCPs in the presence of increasing MFG-E8 concentrations. TRAP+ MNCs were counted (left). Dashed line marks the number of OCLs formed from WT OCPs (no exogenous MFG-E8 added). (D) Data are means ± SD (A, B, and D, n = 3; C, n = 6). *P < 0.05, **P < 0.01 compared to WT or untreated control.
Figure 4. Regulation of human osteoclastogenesis and resorption pit formation by MFG-E8.

Human CD14+ monocytes underwent RANKL-induced osteoclastogenesis, as detailed in Methods, in the presence of the indicated increasing concentrations of MFG-E8. (A) The mRNA expression of the indicated molecules was assayed by qPCR. Results were normalized to GAPDH mRNA and presented relative to those of undifferentiated monocytes (not treated with RANKL), assigned an average value of 1. (B) Cells were stained for TRAP expression and TRAP+ MNCs were counted (left). −RANKL indicates control monocytes not subjected to osteoclastogenesis. (C) CD14+ monocytes on Ca3(PO4)2-coated wells were cultured under osteoclastogenic conditions for 5d and resorptive areas (dark spots) were visualized by light microscopy. The total resorbed area in each culture was measured and expressed relative to the WT group, set as 1 (left). Data are means ± SD (A and B, n = 3; C, n = 8). *P < 0.05, **P < 0.01 compared to untreated control.
MFG-E8 deficiency is associated with increased osteoclastogenesis and bone loss in vivo
To test the relevance of MFG-E8 in in vivo osteoclastogenesis, we subjected WT and Mfge8−/− mice to ligature-induced periodontitis. Mfge8−/− mice exhibited more bone loss (Fig. 5A) and higher numbers of OCLs in the periodontal tissue (Fig. 5B) than WT controls. Whereas Mfge8−/− mice displayed higher expression of certain proinflammatory and bone-resorption-promoting molecules (e.g., IL-17A and osteopontin [Spp1]) compared with WT mice, the two groups had comparable expression of RANKL (Tnfsf11) and its natural inhibitor, osteoprotegerin (Tnfrsf11b) (Fig. 5C), suggesting that the anti-osteoclastogenic effect of endogenous MFG-E8 may not involve alterations in RANKL expression. Local gingival microinjection of an anti-MFG-E8 mAb (but not isotype control) enhanced ligature-induced bone loss as compared to control mice (Fig. 5D), further supporting the importance of endogenous MFG-E8 in bone homeostasis. To determine the role of MFG-E8 in naturally-occurring chronic periodontitis (17, 24), we raised Mfge8−/− mice in parallel with WT controls. Although 10-wk-old Mfge8−/− and WT mice had comparable bone heights, by the age of 13 mo Mfge8−/− mice experienced >60% more bone loss than age-matched WT mice (Fig. 5E). Taken together, these data conclusively implicate MFG-E8 as a negative regulator of bone loss in periodontitis.
Figure 5. MFG-E8 regulates osteoclastogenesis and bone loss in vivo.

(A) Periodontal bone loss was induced for 5d in WT or Mfge8−/− mice by ligating a maxillary second molar and leaving the contralateral tooth unligated (baseline control). (B) WT and Mfge8−/− mice were treated as above and TRAP+ MNCs were enumerated from 20 random coronal sections of the ligated molar from each of three mice (left) and averaged with SD from the total 60 sections per group (middle). Arrows in sections stained with TRAP, hematoxylin, orange-G, and aniline blue indicate OCLs adjacent to bone (right). Scale bars; 100μm (white), 500μm (black). (C) Dissected gingiva from mice used in A were processed for qPCR to determine mRNA expression of indicated molecules. Results were normalized to GAPDH mRNA and presented as fold change in the transcript levels in ligated sites relative to those of unligated sites (assigned an average value of 1). (D) Periodontal bone loss in mice locally microinjected with 5 μg anti-MFG-E8 mAb or IgG2a control 1d before placing the ligature and every day thereafter until the day before sacrifice (d5). (E) Naturally-occurring bone loss in 13-month-old Mfge8−/− mice and age-matched WT controls relative to bone measurements in 10-wk-old WT mice (0 baseline). Data are means ± SD (n = 5–8 mice per group, except for B; n = 3). *P < 0.05, **P < 0.01 compared with control or between indicated groups.
Intriguingly, MFG-E8 deficiency was associated with increased periodontal bacterial burden and, accordingly, treatment of WT mice with rMFG-E8 significantly decreased the bacterial load (Fig. 6A–C). In disk inhibition zone assays with numerous bacterial isolates from murine periodontal tissue, rMFG-E8 failed to exert direct killing activity, in contrast to the antibiotic imipenem (Fig. 6D). Therefore, the suppressive effect of MFG-E8 on the microbiota is likely mediated by its capacity to inhibit inflammation and thereby to limit growth of periodontal bacteria that utilize tissue breakdown products (25).
Figure 6. MFG-E8 reduces the periodontal bacterial burden without exerting direct antimicrobial activity.
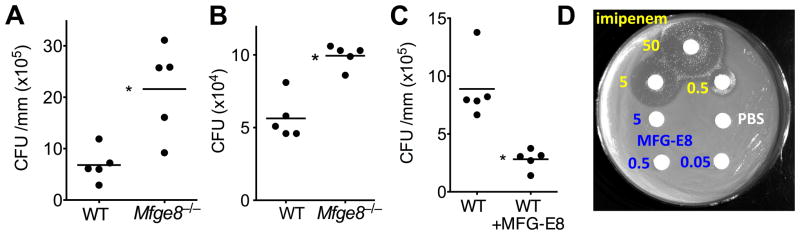
Periodontal microbiota counts were determined in WT or Mfge8−/− mice subjected to ligature-induced periodontitis (A) or to naturally-occurring periodontitis until the age of 13 months (B), as well as in WT mice in which ligature-induced periodontitis was performed with or without local treatment with 2.5 μg MFG-E8 (C). In A and C, bacteria were extracted from recovered ligatures and serial dilutions of bacterial suspensions were plated onto blood agar plates for anaerobic growth and CFU enumeration. In B, oral swabs held against the gumlines were taken and bacteria were cultured anaerobically for CFU enumeration as above. Each symbol represents an individual mouse and small horizontal lines indicate the mean. *P < 0.01. (D) Possible antimicrobial activity of MFG-E8 against mouse periodontal bacteria was determined by the disk inhibition zone method, using PBS and imipenem as negative and positive control, respectively. The experiment shown is representative of a total of 15 bacterial isolates and MFG-E8 consistently failed to inhibit bacterial growth. Numbers shown refer to μg of compound used.
Local administration of MFG-E8 protects against inflammatory bone loss
We next determined whether rMFG-E8 could suppress bone loss upon ligature-induced periodontitis in WT mice. Local microinjection of rMFG-E8 (but not BSA control) into the gingiva inhibited bone loss as compared to untreated control mice (Fig. 7A). Mice treated with rMFG-E8 also exhibited decreased expression of mRNA encoding several pro-inflammatory mediators (Fig. 7B, top) as well as adhesion molecules and innate immune receptors (Fig. 7B, bottom) relative to BSA-treated mice. Similar to its effect in WT mice, rMFG-E8 also protected Mfge8−/− mice against ligature-induced bone loss (Fig. 7C). These data suggest that MFG-E8 can be exploited therapeutically to inhibit inflammatory bone loss in periodontitis.
Figure 7. rMFG-E8 inhibits ligature-induced periodontal bone loss in vivo.

(A) Periodontal bone loss in WT mice locally microinjected with 2.5 μg MFG-E8 or BSA control 1d before placing the ligature and every day thereafter until the day before sacrifice (d5). (B) Dissected gingiva from mice used in A were processed to determine mRNA expression of the indicated molecules using qPCR. Results were normalized to GAPDH mRNA and presented as fold change in the transcript levels in ligated sites relative to those of unligated sites, set as 1. (C) Periodontal bone loss in Mfge8−/− mice locally microinjected with 2.5 μg MFG-E8 or BSA control as outlined in A. Data are means ± SD (n = 5–6 mice per group). *P < 0.05, **P < 0.01 compared with control or between indicated groups.
Discussion
Homeostatic mechanisms are of paramount importance to the proper functioning of any biological system. OCLs rely on several modulators to control their function (26) and our findings indicate that MFG-E8 is one of them. This novel modulator is upregulated during osteoclastogenesis, in line with most biological systems where negative regulators are upregulated to control functional activity and prevent pathological states (27–29). The importance of MFG-E8 in restraining or fine-tuning osteoclast differentiation and function is highlighted by the effects of its absence: OCPs from Mfge8−/− mice underwent increased RANKL-induced osteoclastogenesis leading to enhanced resorption pit formation and, consistently, Mfge8−/− mice experienced more periodontal bone loss than WT controls. In both cases, administration of exogenous MFG-E8 reversed the overactive phenotype, thereby preventing excessive osteoclastic activity in vitro and ligature-induced periodontal bone loss in vivo.
It is conceivable that OCLs produce MFG-E8 at levels that would control but not abrogate their differentiation and function, whereas therapeutic doses of MFG-E8 can have stronger inhibitory effects that could effectively diminish pathologic bone resorption. This notion is consistent with the dose-dependent inhibitory effects of MFG-E8 in figure 3D: At 1–2 μg/ml, exogenously added MFG-E8 simply restrained osteoclastogenesis from Mfge8−/− OCPs, rendering it comparable to WT osteoclastogenesis (i.e., in the presence of endogenously produced MFG-E8). At a higher concentration (5 μg/ml), MFG-E8 diminished osteoclastogenesis (>75% inhibition). It thus appears that endogenously produced MFG-E8 acts homeostatically to restrain unwarranted osteoclastogenesis, although from a therapeutic standpoint higher concentrations of exogenously added MFG-E8 can inhibit further this process.
Not surprisingly, therefore, the differences between WT and Mfge8−/− osteoclastogenesis and resorption pit formation were relatively modest (Figure 3, A–C) despite being statistically and biologically significant, especially in an in vivo setting. Indeed, given adequate time, 13-month-old Mfge8−/− mice experienced >60% more periodontal bone loss than age-matched WT controls. In this regard, aging mice, like aging humans, develop naturally-occurring chronic periodontal bone loss (17, 24), and our results suggest that endogenously produced MFG-E8 is an important regulator of this process.
Our observations for decreased expression of proinflammatory cytokines and chemokines in the periodontal tissue of MFG-E8–treated mice undergoing ligature-induced periodontitis (as compared with BSA-treated controls) are consistent with the reported anti-inflammatory action of MFG-E8 (1, 5, 6). A possible anti-inflammatory mechanism of MFG-E8 involves its ability to interfere with osteopontin binding to αvβ3 integrin on macrophages, thereby preventing downstream activation of NF-κB and hence induction of inflammatory mediators (5). Another mechanism by which MFG-E8 could attenuate inflammation is through the promotion of apoptotic cell phagocytosis and thus prevention of secondary necrosis. In this regard, MFG-E8 acts as an opsonin that forms a molecular bridge between the αvβ3 integrin on phagocytes (bound by an RGD motif in the N-terminal region of MFG-E8) and phosphatidylserine on apoptotic cells (bound by the C-terminal discoidin-like domains of MFG-E8) (11). We and others have shown that inflammatory mediators including IL-1β, IL-6, and IL-17 play important roles in periodontal inflammation and bone loss (reviewed in refs. (23, 30)). The ability of MFG-E8 to inhibit the expression of these proinflammatory molecules in the current study, suggests an indirect way by which MFG-E8 can down-regulate osteoclastogenesis and bone loss. Whereas the anti-inflammatory and anti-osteoclastogenic of MFG-E8 can be readily dissociated and investigated separately in vitro, the strong connection between inflammation and osteoclastogenesis (31) suggests that the therapeutic application of MFG-E8 is capable of a two-pronged attack on periodontitis and perhaps other inflammatory bone disorders (e.g., rheumatoid arthritis and ankylosing spondylitis).
The placement of ligatures induces bacteria-mediated inflammation in the periodontal tissue (15) and this may explain the observed initial downregulation of MFG-E8 expression (90% reduction from d0 to d1), in line with similar observations in other models of inflammation (2). The subsequent resurgence of MFG-E8 expression correlated temporally and spatially with osteoclastogenesis in the course of ligature-induced periodontitis. Since in vitro formed OCLs express and are regulated by MFG-E8, it is likely that the reappearance of MFG-E8 in the course of experimental periodontitis was contributed (at least in part) by the generated OCLs, ostensibly to regulate their differentiation and function. This notion is consistent with our findings that Mfge8−/− mice experience increased osteoclastogenesis and periodontal bone loss as compared to WT controls.
Our findings on the indirect antimicrobial effects of MFG-E8 add to accumulating evidence that inhibition of periodontal inflammation exerts a negative impact on the periodontal microbiota. Indeed, local treatments with anti-inflammatory or pro-resolution agents, such as meloxicam (selective cyclooxygenase-2 inhibitor), anti-IL-17 mAb, or resolvin E1, cause a significant reduction in the total counts of periodontal bacteria in animal models of periodontitis (17, 32, 33). Consistent with these findings, a recent metagenomic study showed that the bacterial biomass of human periodontitis-associated biofilms increases with increasing periodontal inflammation (34). In this regard, inflammation generates tissue breakdown products (e.g., peptides and heme-containing compounds) that can serve critical nutritional needs of periodontal bacteria (25). Conversely, and consequently, the control of inflammation would be expected to limit bacterial growth, thereby explaining the inhibitory effects of MFG-E8 on the periodontal microbiota despite lacking intrinsic antimicrobial activity.
In summary, autocrine MFG-E8 regulates OCL homeostasis and rMFG-E8 could be a new therapeutic platform for the treatment of bone loss disorders. The anti-inflammatory action of MFG-E8 can further contribute to the control of bone loss in inflammatory conditions. Whereas diminished expression of MFG-E8 is associated with certain inflammatory diseases (2, 9, 10), MFG-E8 is expressed at high levels and is implicated in the pathogenesis of certain other pathologic conditions, such as chronic pancreatitis, obesity, and tumorigenesis (in humans and/or animal models) (8, 35, 36). Therefore, caution is required in future MFG-E8–based therapeutic strategies, although the local administration of MFG-E8 in conditions with localized bone loss (e.g., periodontitis and rheumatoid arthritis) should not involve undue risks.
Supplementary Material
Abbreviations
- BM
bone marrow
- EGF
epidermal growth factor
- MFG-E8
milk fat globule-EGF factor 8
- qPCR
quantitative real-time PCR
- OCL
osteoclast
- OCP
osteoclast precursor
- MNC
multinucleated cell
- RANK
receptor activator of NF-κB
- RANKL
RANK ligand
- TRAP
tartrate-resistant acid phosphatase
- WT
wild-type
Footnotes
Supported by grants from the NIH (DE015254, DE017138, and DE021685; GH), the Deutsche Forschungsgemeinschaft (CH279/5-1) and the European Research Council (TC), and by the NIH Intramural Research Program (MCU).
References
- 1.Raymond A, Ensslin MA, Shur BD. SED1/MFG-E8: a bi-motif protein that orchestrates diverse cellular interactions. J Cell Biochem. 2009;106:957–966. doi: 10.1002/jcb.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aziz M, Jacob A, Matsuda A, Wang P. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011;16:1077–1086. doi: 10.1007/s10495-011-0630-0. [DOI] [PubMed] [Google Scholar]
- 3.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 4.Bu HF, Zuo XL, Wang X, Ensslin MA, Koti V, Hsueh W, Raymond AS, Shur BD, Tan XD. Milk fat globule-EGF factor 8/lactadherin plays a crucial role in maintenance and repair of murine intestinal epithelium. J Clin Invest. 2007;117:3673–3683. doi: 10.1172/JCI31841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aziz MM, Ishihara S, Mishima Y, Oshima N, Moriyama I, Yuki T, Kadowaki Y, Rumi MA, Amano Y, Kinoshita Y. MFG-E8 attenuates intestinal inflammation in murine experimental colitis by modulating osteopontin-dependent alphavbeta3 integrin signaling. J Immunol. 2009;182:7222–7232. doi: 10.4049/jimmunol.0803711. [DOI] [PubMed] [Google Scholar]
- 6.Aziz M, Matsuda A, Yang WL, Jacob A, Wang P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol. 2012;189:393–402. doi: 10.4049/jimmunol.1200262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silvestre JS, Thery C, Hamard G, Boddaert J, Aguilar B, Delcayre A, Houbron C, Tamarat R, Blanc-Brude O, Heeneman S, Clergue M, Duriez M, Merval R, Levy B, Tedgui A, Amigorena S, Mallat Z. Lactadherin promotes VEGF-dependent neovascularization. Nat Med. 2005;11:499–506. doi: 10.1038/nm1233. [DOI] [PubMed] [Google Scholar]
- 8.Motegi S, Leitner WW, Lu M, Tada Y, Sardy M, Wu C, Chavakis T, Udey MC. Pericyte-derived MFG-E8 regulates pathologic angiogenesis. Arterioscler Thromb Vasc Biol. 2011;31:2024–2034. doi: 10.1161/ATVBAHA.111.232587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boddaert J, Kinugawa K, Lambert JC, Boukhtouche F, Zoll J, Merval R, Blanc-Brude O, Mann D, Berr C, Vilar J, Garabedian B, Journiac N, Charue D, Silvestre JS, Duyckaerts C, Amouyel P, Mariani J, Tedgui A, Mallat Z. Evidence of a role for lactadherin in Alzheimer’s disease. Am J Pathol. 2007;170:921–929. doi: 10.2353/ajpath.2007.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, Esposito B, Teissier E, Daemen MJ, Leseche G, Boulanger C, Tedgui A, Mallat Z. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–2177. doi: 10.1161/CIRCULATIONAHA.106.662080. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda A, Jacob A, Wu R, Zhou M, Nicastro JM, Coppa GF, Wang P. Milk fat globule-EGF factor VIII in sepsis and ischemia-reperfusion injury. Mol Med. 2011;17:126–133. doi: 10.2119/molmed.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23:582–590. doi: 10.1016/j.tem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11:234–250. doi: 10.1038/nrd3669. [DOI] [PubMed] [Google Scholar]
- 14.Neutzner M, Lopez T, Feng X, Bergmann-Leitner ES, Leitner WW, Udey MC. MFG-E8/lactadherin promotes tumor growth in an angiogenesis-dependent transgenic mouse model of multistage carcinogenesis. Cancer Res. 2007;67:6777–6785. doi: 10.1158/0008-5472.CAN-07-0165. [DOI] [PubMed] [Google Scholar]
- 15.Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abe T, Hajishengallis G. Optimization of the ligature-induced periodontitis model in mice. J Immunol Meth. 2013;394:49–54. doi: 10.1016/j.jim.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, Ciero PA, Krauss JL, Li F, Rauner M, Hofbauer LC, Choi EY, Chung KJ, Hashim A, Curtis MA, Chavakis T, Hajishengallis G. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–473. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, Hajishengallis G. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol. 2012;189:5442–5448. doi: 10.4049/jimmunol.1202339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker PJ, Dixon M, Roopenian DC. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:5864–5868. doi: 10.1128/iai.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi N, Udagawa N, Tanaka S, Suda T. Generating murine osteoclasts from bone marrow. Meth Mol Med. 2003;80:129–144. doi: 10.1385/1-59259-366-6:129. [DOI] [PubMed] [Google Scholar]
- 21.Collin-Osdoby P, Osdoby P. RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Meth Mol Biol. 2012;816:187–202. doi: 10.1007/978-1-61779-415-5_13. [DOI] [PubMed] [Google Scholar]
- 22.Park-Min KH, Lee EY, Moskowitz NK, Lim E, Lee SK, Lorenzo JA, Huang C, Melnick AM, Purdue PE, Goldring SR, Ivashkiv LB. Negative regulation of osteoclast precursor differentiation by CD11b and beta2 integrin-B-cell lymphoma 6 signaling. J Bone Miner Res. 2013;28:135–149. doi: 10.1002/jbmr.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajishengallis G. Aging and its impact on innate immunity and inflammation: Implications for periodontitis. J Oral Biosci. 2014;56:30–37. doi: 10.1016/j.job.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajishengallis G, Lamont RJ. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol. 2014;44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther. 2011;13:234. doi: 10.1186/ar3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mantovani A, Garlanda C, Locati M, Rodriguez TV, Feo SG, Savino B, Vecchi A. Regulatory pathways in inflammation. Autoimmun Rev. 2007;7:8–11. doi: 10.1016/j.autrev.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Wang WY, Lim JH, Li JD. Synergistic and feedback signaling mechanisms in the regulation of inflammation in respiratory infections. Cell Mol Immunol. 2012;9:131–135. doi: 10.1038/cmi.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013;13:257–269. doi: 10.1038/nri3403. [DOI] [PubMed] [Google Scholar]
- 30.Garlet GP. Destructive and protective roles of cytokines in periodontitis: A reappraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 31.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 32.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 33.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, Uzel G, Chen W, Chavakis T, Holland SM, Hajishengallis G. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra240. doi: 10.1126/scitranslmed.3007696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Haese JG, I, Demir E, Kehl T, Winckler J, Giese NA, Bergmann F, Giese T, Buchler MW, Friess H, Hartel M, Ceyhan GO. The impact of MFG-E8 in chronic pancreatitis: potential for future immunotherapy? BMC Gastroenterol. 2013;13:14. doi: 10.1186/1471-230X-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khalifeh-Soltani A, McKleroy W, Sakuma S, Cheung YY, Tharp K, Qiu Y, Turner SM, Chawla A, Stahl A, Atabai K. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nat Med. 2014;20:175–183. doi: 10.1038/nm.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.