

Abstract
Both commensal bacteria and infiltrating inflammatory cells play essential roles in the pathogenesis of inflammatory bowel disease. The molecular mechanisms whereby these pathogenic factors are regulated during the disease are not fully understood. We report here that a member of the TNFAIP8 (tumor necrosis factor-α-induced protein 8) family called TIPE2 (TNFAIP8-like 2, or TNFAIP8L2) plays a crucial role in regulating commensal bacteria dissemination and inflammatory cell function in experimental colitis induced by dextran sodium sulfate (DSS). Following DSS treatment, TIPE2-deficient mice, or chimeric mice that are deficient in TIPE2 only in their hematopoietic cells, lost less body weight and survived longer than wild type controls. Consistent with this clinical observation, TIPE2-deficient mice exhibited significantly less severe colitis and colonic damage. This was associated with a marked reduction in the colonic expression of inflammatory cytokines such as TNF-α, IL-6, and IL-12. Importantly, the ameliorated DSS-induced colitis in TIPE2−/− mice was also associated with reduced local dissemination of commensal bacteria and a weaker systemic inflammatory response. Combined with our previous report that TIPE2 is a negative regulator of anti-bacterial immunity, these results indicate that TIPE2 promotes colitis by inhibiting mucosal immunity to commensal bacteria.
Keywords: Colitis, Inflammation, Innate immunity, TNFAIP8L2 (TIPE2)
Introduction
Human inflammatory bowel diseases (IBD), represented mainly by ulcerative colitis and Crohn’s disease, cause considerable morbidity and increase significantly the risk of cancer in the colon and rectum (1). Ulcerative colitis exhibits a characteristic profile of chronic, relapsing and remitting inflammation involving the distal colon and rectum, and is generally considered as an immune-mediated disorder resulting from abnormal interactions between colonic microflora and mucosal immune cells (2). The relapsing and remitting course of the disease vary greatly among patients, with about 15% of them developing an ‘acute severe’ form some time in their lives (3). Although the precise etiology of ulcerative colitis is not well understood, it is now widely accepted that both genetic and environmental factors are involved (4). Several animal models of experimental colitis have been developed to help investigate the molecular and cellular mechanisms of the disease. Of these, the dextran sodium sulfate (DSS)-induced experimental colitis is one of the best studied. It is initiated by DSS-induced damage to the intestinal epithelial cells, but is dependent on the presence of both commensal microflora and myeloid (but not lymphoid) cells (5–7). Similar to human IBD, DSS-induced colitis is limited to the colonic mucosa and is characterized by diarrhoea, bloody faeces, weight loss, colonic ulceration, and a histopathological picture of inflammation, consisting of mainly infiltrating macrophages and granulocytes (5–6).
TIPE2, the tumor necrosis factor (TNF)-α-induced protein 8 (TNFAIP8) like-2 or TNFAIP8L2, is a member of the TNFAIP8 family (8), which is preferentially expressed in hematopoietic cells (8–10). TIPE2 serves as a negative regulator of immunity that maintains immune homeostasis (8, 11). Abnormal expression of TIPE2 has been found in patients with systemic lupus erythematosus, hepatitis B, diabetic nephropathy, and childhood asthma (12–15). TIPE2 is also implicated in the development of atherosclerosis and experimental stroke (16–18). TIPE2 may control innate immunity to bacteria and dsRNA viruses by targeting the Rac GTPases (19–20). However, the role of TIPE2 in inflammatory bowel diseases has never been reported. To address this issue, we studied DSS-induced colitis in TIPE2-deficient mice. We report here that TIPE2 plays an important role in the development of acute colitis by promoting dissemination of commensal bacteria in the colon.
Materials and Methods
Mice
Wild type (WT) C57BL/6 (B6) and CD45.1+ B6 mice were purchased from The Jackson Laboratory. B6 mice that carry a TIPE2 gene null mutation were generated by backcrossing TIPE2−/− 129 mice to B6 mice for 12 generations, as described previously (8, 19). All mice used were male and 8–12 weeks old, and were maintained under pathogen-free conditions in the University of Pennsylvania Animal Care Facilities. All animal procedures were preapproved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Induction and evaluation of DSS-induced colitis
Experimental colitis was induced by adding DSS (molecular weight 36–50KD, MP Biomedicals, Solon, OH) to the drinking water to a final concentration of 4% (w/v). Mice were then switched to regular drinking water until the end of the experiment. Mice were examined daily to determine their clinical Disease Activity Index (DAI), which was based on the degree of body weight loss, stool consistency, and fecal blooding (ranging from 0 to 12), as described previously (21). Briefly, DAI was scored as follows: weight loss (no change=0; <5%=1; 6–10%=2; 11–20%=3; >20%=4); stool (normal=0; soft, well-formed=1; soft without pellets=2; diarrhea=4); blood (no blood=0; visible blood in rectum=1; grossing bleeding in rectum=2; visible blood on fur =4). For histological analysis, the distal colonic specimens were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin (HE), and pathological scores, ranging from 0 to 6 (combining inflammatory cell infiltration score and tissue damage score), determined as follows (22). Inflammatory cell infiltration in the lamina propria: occasional inflammatory cells=0; increased inflammatory cells=1; confluence of inflammatory cells extending to the submucosa=2; transmural extension=3; tissue damage: no mucosal damage=0; lymphoepithelial lesions=1; surface mucosal erosion=2; extensive mucosal damage and extension into deeper structures of the bowel wall=3.
Real-time quantitative PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Two micrograms of total RNA was reverse transcribed using SuperScript II transcriptase (Invitrogen). Real time quantitative PCR was carried out in an Applied Biosystems 7500 System with Power SYBR Green PCR Master Mix (Applied Biosystems). The Quantitect Primers for mouse GAPDH, TIPE1, TIPE2, and TIPE3 were purchased from Qiagen. TIPE primer sequences used were as follows: forward, 5′-AGCAGTCCAACTCCGGGGAACAG-3′, reverse, 5′-GTCGATCAGCGTGGTGGCGATG-3′. Each sample was run in triplicate. The relative changes in gene expression were calculated using GAPDH as the loading control. For interrogating gut microbial diversity, quantitative real-time PCR amplification of 16S rRNA gene sequences was performed as previously reported (23). The total DNA in stool was extracted using the QIAamp DNA Stool Mini Kit (Qiagen) and real-time PCR was conducted using specific 16S rRNA primers for the following major groups: the Eubacterium rectale-Clostridium coccoides, Bacteroides sp. and Enterobacteriaceae. In addition, the total bacterial (eubacteria) numbers were also determined using standard curves constructed with reference bacteria specific for each group. Real-time PCR measures 16S gene copies per sample.
ELISA
Sera were collected and stored at −80°C before analysis. Colon was homogenized in CellLyticM buffer (Sigma-Aldrich, St. Louis, MO) supplemented with complete protease inhibitors mixture (Roche, Indianapolis, IN). Protein concentration was measured by BCA assay (Pierce). Cytokine content was presented as pg/mg of total colon protein. Antibodies were purchased from ebioscience and BD Biosciences Pharmingen, which include purified and biotinylated rat anti-mouse TNF-α, IL-6 and IL-12. Quantitative ELISA was performed according to the manufacturer’s instructions.
Flow cytometry
Colon was collected and washed with cold washing buffer (1×PBS) supplemented with 2% FBS, 100 units/ml penicillin/ streptomycin. To isolate lamina propria leukocytes, intestinal epithelial cells and intraepithelial lymphocytes were first stripped by shaking colonic sections in 2mM EDTA/1mM DTT for 30 minutes at 37°C. The remaining colon tissue was cut into small pieces and incubated in DMEM containing penicilin/streptomycin, 0.5 mg/ml collagenase (Sigma), 0.02 mg/ml Dnase (Roche), and 0.1 mg/ml Dispase (Invitrogen), for 20 minutes at 37°C with gentle shaking. Supernatant was filtered with 70 μm strainers (BD Biosciences) to obtain single cell suspensions. Next, cells were labeled with a combination of the following fluorescence-conjugated monoclonal antibodies: anti-CD11b (eBioscience) conjugated with fluorescein isothiocyanate (FITC) (dilution 1:100); anti-Ly6G (BD Biosciences) conjugated with phycoerythrin (PE) (dilution1:100); anti-CD11c (BD Biosciences) conjugated with PE-cyanine (Cy)-7 (dilution 1:200). Data were acquired by muticolor flow cytomery using a BD FACSCanto system (BD Biosciences), and analyzed with the FlowJo software (v9.2; TreeStar).
Generation of bone marrow chimeric mice
Bone marrow cells were harvested by flushing the femurs and tibias from donor WT and TIPE2−/− mice that express either CD45.1 or CD45.2, as previously described (18, 24). Recipient mice were sublethally irradiated twice, at a dose of 4.5 Gy, 3 hours apart. Following the second irradiation, bone marrow cells were transferred into WT and TIPE2−/− mice, 10 million cells/mouse, intravenously via the tail vein. Four chimera groups were generated: WT→WT (WT cells expressing CD45.2 into WT mice expressing CD45.1); TIPE2−/−→WT (TIPE2−/− cells expressing CD45.2 into WT mice expressing CD45.1); WT→TIPE2−/− (WT cells expressing CD45.1 into TIPE2−/− mice expressing CD45.2); TIPE2−/−→TIPE2−/− (TIPE2−/− cells expressing CD45.2 into TIPE2−/− mice expressing CD45.2). For the first two weeks after bone marrow transfer, recipient mice received antibiotics in their drinking water, followed by a 5-week engraftment recovery period. Six weeks after the transplantation, the degree of bone marrow reconstitution was determined by staining peripheral blood leukocytes with PE-conjugated anti-CD45.1 and PercpCy5.5-conjugated anti-CD45.2 (BD Biosciences). As we reported, in the chimeric mice so generated, more than 90% of the hematopoietic cells were derived from donor bone marrow (18, 24).
Bacterial culture
After euthanizing the mice, the entire colon was removed under aseptic conditions. The terminal 3-cm segments of distal colon was washed, weighed, homogenized, and serially diluted. Different dilutions of the suspensions were plated in triplicate on Brain Heart Infusion (BHI) agar and Blood agar (BD Biosciences) plates, and incubated at 37°C for 24 hours, to quantify the bacterial colonies.
Assessment of intestinal permeability
Intestinal barrier permeability in vivo was measured with a FITC-labeled dextran method as previously described (25). In brief, WT and TIPE2−/− mice were deprived of water and food overnight, and were fed with permeability tracer FITC-dextran (molecular weight, 4kDa; Sigma-Aldrich) at 400mg/kg body weight by oral gavage. Blood was collected four hours later by retro-orbital bleeding. Fluorescence intensity of the serum was measured using a fluorescence spectrophotometer with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. FITC-dextran concentrations were calculated using standard curves generated by a serial dilution of FITC-dextran.
Blood cell counts
Before euthanizing the DSS-treated mice, blood was collected, and whole blood cell counts were determined by Drew Hemavet 950FS (Drew Scientific, Oxford, U.K.).
Statistical analysis
Quantitative data are presented as means±SEM of two or three experiments. The survival curves were plotted according to the Kaplan-Meier method and compared by the log-rank test. Two-tailed Student t test was used for all other cases and p<0.05 was considered statistically significant. All statistical analysis was performed with the Prism 5.0 for Windows (Graphpad Software, San Diego, CA).
Results
Expression of TIPE family members in the murine colon
Unlike TIPE2−/− 129 mice that develop systemic inflammation early in their lives, young TIPE2−/− mice of the B6 background are relatively healthy with a normal gastrointestinal tract (Fig S1). TIPE2 mRNA was readily detected in the colon tissue homogenates from WT but not TIPE2−/− mice (Fig. 1A). By contrast, similar levels of TNFAIP8, TIPE1, and TIPE3 were detected in both WT and TIPE2−/− colon, indicating that TIPE2 deficiency did not affect the expression of these related genes (Fig. 1B).
FIGURE 1. Expression of TNFAIP8 family of genes in murine colon.

(A) The mRNA levels of TNFAIP8 family members were determined by RT-PCR using total RNA isolated from the distal colon of wild type (WT) and TIPE2−/− mice. GAPDH was used as a loading control. (−), samples lacking reverse transcriptase were used as negative controls for the PCR. (B) Quantification of mRNA expression by quantitative realtime PCR. Data shown are relative gene expression levels, as mean ± SEM (n=3).
TIPE2−/− mice are resistant to DSS-induced colitis
Experimental colitis, induced by oral feeding with DSS, is a well-established model for IBD (26–28). To determine the potential role of TIPE2 in colonic inflammation, WT and TIPE2−/− mice were fed with 4% DSS for 5 days, to induce acute colitis. TIPE2 deficiency did not affect water consumption (Fig. S2), but markedly reduced the severity of the colitis (Fig. 2A). The mortality rate was reduced from 80% in the WT group to 0% in the TIPE2−/− group. When a higher DSS dose (5%) was used, TIPE2−/− mice died as well but with a significant delay (Fig. S3). Body weight loss is considered a surrogate marker of morbidity. Consistent with the mortality data, TIPE2−/− mice lost considerably less weight than the WT controls commencing on day 5. The body weight difference increased gradually until the end of the study (Fig. 2B). No weight loss was observed of mice drinking regular water (Fig. 2B). Consistent with these results, the clinical manifestation of the disease, reflected by DAI, was significantly less severe for TIPE2−/− mice compared with WT mice (Fig. 2C). Differences in colon weight and colon length were also apparent between the two groups. Without DSS treatment, TIPE2−/− mice had colon weights and colon lengths similar to those of WT mice. After DSS treatment, colon weight of TIPE2−/− mice was 23% more than that of WT mice (Fig. 2D), while colon length of TIPE2−/− mice was 27% longer than that of WT mice (Fig. 2E; Fig. 2F, left panel).
FIGURE 2. Reduced experimental colitis in TIPE2−/− mice.

(A) WT (n=5) and TIPE2−/− (n=5) mice were fed with 4% DSS in the drinking water for 5 days and then switched to regular drinking water. Survival was monitored until day 14 after the start of the DSS feeding. This experiment was repeated three times with similar results. The differences between the two groups are statistically significant as determined by Kaplan-Meier analysis (P<0.01). (B–G) WT (n=5) and TIPE2−/− (n=5) mice were fed with 4% DSS or water ad lib for 5 days followed by recovery on regular drinking water for 2 days prior to euthanasia. (B) Body weight loss was measured daily. (C) Disease activity index (DAI) was scored daily. (D–E) Mice were sacrificed on day 7, and both colon length and colon weight were examined. (F) Representative images of colon (left panel) and its HE-stained sections (right panel, original magnification ×100) from WT and TIPE2−/− mice on day 7. (G) Colonic histopathological scores of mice shown in the right panel of F. Data shown are means ± SEM of one representative experiment of three. *P<0.05; **P<0.01; ***P<0.001.
Histological examination was also performed to validate the clinical data. DSS treatment induced significant histopathological changes in the colons of WT mice characterized by massive inflammatory infiltrates and disruption of mucosal structures (Fig. 2F, right panel), consistent with previous reports (29). However, TIPE2−/− mice displayed less severe injury compared with WT mice (Fig. 2F, right panel). The histopathological score of TIPE2−/− mice was significantly lower than that of WT mice (4.6±0.6 in WT versus 2.6±0.4 in TIPE2−/− mice)(Fig. 2G). Taken together, these results demonstrate that TIPE2−/− mice may be significantly less susceptible to DSS-induced colitis.
Reduced inflammatory cytokine expression and inflammatory cell infiltration in TIPE2−/− colon
DSS-induced colitis is an inflammatory disease mediated by many proinflammatory cytokines (30). To determine the effect of TIPE2 deficiency on the production of proinflammatory cytokines at the site of DSS-induced inflammation, we examined the expression levels of several cytokines by ELISA. We found that proinflammatory cytokines such as TNF-α, IL-6, and IL-12 were markedly increased in WT mice after DSS treatment. However, TIPE2−/− mice produced significantly less of these cytokines relative to WT mice (Fig. 3, A–C). On the other hand, TIPE2−/− mice produced comparable amounts of IL-1β, IL-4, and IFN-γ as compared with WT mice (data not shown).
FIGURE 3. Reduced inflammatory cytokine production and inflammatory cell infiltration in TIPE2−/− colon.

WT (n=5) and TIPE2−/− (n=5) mice were fed with 4% DSS or water ad lib for 5 days followed by recovery on regular drinking water for 2 days. (A–C) Colonic lysates from control and DSS-fed WT and TIPE2−/− mice were analyzed by ELISA for the amounts of TNF-α, IL-6, and IL-12. Data shown are means ± SEM of pg of indicated cytokines per mg of total colon proteins. (D) Flow cytometric profiles of neutrophils (Ly6G+CD11b+), macrophages (Ly6G−CD11b+), and dendritic cells (CD11c+) isolated from the colonic lamina propria of WT (top panels) and TIPE2−/− mice (bottom panels). Numbers indicate the frequencies of cells of each subset. (E–G) The total numbers of neutrophils (Ly6G+CD11b+), macrophages (Ly6G−CD11b+), and dendritic cells (CD11c+) per mg of colon were determined by flow cytometry. Data shown are means ± SEM of one representative experiment. The experiments were repeated three times with similar results. *P<0.05; **P<0.01; ***P<0.001.
To characterize the inflammatory cells involved, leukocytes isolated from lamina propria of the colon were analyzed by flow cytometry. Various inflammatory cell subsets were found in WT and TIPE2−/− colons after DSS feeding. In comparison with WT colon, the TIPE2−/− colon had significantly less neutrophils (8.89±0.55 v.s. 2.92±0.32, p<0.001), macrophages (12.63±0.63 v.s. 6.92±0.69, p<0.001), and dendritic cells (24.04±3.65 v.s. 11.97±0.27, p<0.05) (Fig. 3, D–F). Importantly, a significant proportion of the leukocytic infiltrate in both WT and TIPE2−/− mice consisted of Ly6G− CD11b+ macrophages (Fig. 3, D–F).
TIPE2 deficiency in hematopoietic cells ameliorates colitis
Because both colonic epithelial and immune cells play important roles in the development of colitis, we next determined the cell populations that are critical for TIPE2-mediated effect. We detected no significant differences in colonic epithelial cell apoptosis, proliferation, and permeability between WT and TIPE2−/− mice fed 4% DSS for 3 days (Our unpublished data), suggesting that TIPE2 may not have a primary effect on colonic epithelium in DSS-induced colitis.
To determine the potential roles of hematopoietic cells, we generated four groups of bone marrow chimeras. Six weeks after bone marrow transplantation, we found that more than 90% of the blood leukocytes were of donor origin, confirming successful engraftment (Fig. S4). Following DSS treatment, KO → KO chimeric mice developed the least severe colitis (Fig. 4, A and B). Transplant of KO bone marrow to WT recipient mice (KO → WT) markedly reduced body weight loss (Fig. 4A), improved DAI (Fig. 4B), and preserved the colon length (Fig. 4C), as well as reduced histopathologcial score (Fig. 4, D and E). These results indicate that TIPE2-deficient hematopoietic cells are responsible for the reduced colonic inflammation in the TIPE2 knockout mice.
FIGURE 4. TIPE2 deficiency in hematopoietic cells reduces colonic inflammation.

Bone marrow chimeric mice (n=5) were fed with 4% DSS for 5 days followed by recovery on regular drinking water for 2 days. (A) Body weight loss was measured daily. (B) DAI was scored daily. (C) Colon lengths at the end of the experiment (Day 7). (D) Colonic histopathologic scores of mice treated with DSS. (E) Representative images of HE-stained colonic sections of chimeric mice on Day 7. Original magnification, ×100. Data are shown as means ± SEM of one representative experiment of three. *P<0.05; **P<0.01.
Reduced local dissemination of commensal microflora and systemic inflammation in TIPE2−/− mice
It is well established that commensal microflora in the lumen of the colon play an essential role in the development of human IBD and murine experimental colitis (31–33). Our previous study showed that TIPE2 knockout macrophages and neutrophils had enhanced phagocytic and bactericidal activities and TIPE2 knockout mice were resistant to Listeria monocytogenes and staphylococcus aureus infections (19). Because we observed reduced inflammatory response in the DSS-induced colitis of TIPE2−/− mice, we asked whether TIPE2 deficiency affected commensal microflora dissemination in mice. As shown in Fig. 5, A and B, significantly less bacteria were detected in the colon of TIPE2−/− mice relative to DSS-treated WT mice. We also found a significant decrease in plasma FITC fluorescence in TIPE2−/− mice after DSS-induced injury compared to WT mice (Fig. 5C), indicating less colon tissue disruption and less permeability (Fig. 2F). It is known that systemic dissemination of bacteria and bacterial components not only leads to increased peripheral blood leukocyte counts, but also triggers an exuberant cytokine inflammatory response. In agreement with the reduced bacterial dissemination in TIPE2−/− mice, the numbers of total peripheral blood leukocytes and monocytes were markedly reduced in TIPE2−/− mice relative to DSS-treated WT mice (Fig. 5E). Although there appeared to be a trend toward decreased neutrophils and lymphocytes in the knockout mice, however, the differences were not statistically significant (Fig. 5D). Furthermore, serum concentrations of the proinflammatory cytokine IL-6 was also lower in TIPE2−/− mice when compared to WT mice (Fig. 5D). Taken together, these results indicate that the reduced DSS-induced morbidity and mortality in the absence of TIPE2 may be caused by enhanced immunity to commensal bacteria.
FIGURE 5. Reduced local dissemination of commensal microflora and weakened systemic inflammatory responses in TIPE2−/− mice during colitis.

WT (n=5) and TIPE2−/− (n=4) mice were fed with 4% DSS or water ad lib for 5 days followed by recovery on regular drinking water for 2 days before sacrifice. (A and B) Bacterial counts in colonic tissues of control and DSS-fed WT and TIPE2−/− mice were determined by colony-forming assay using BHI agar (A) and blood agar (B). (C) Control and DSS-fed mice were fed by oral gavage with FITC-dextran (0.4 mg/g), and seral FITC-dextran concentrations were determined 4 hours later. (D) Seral concentrations of IL-6 in WT and TIPE2−/− mice were determined by ELISA at the end of the experiment. (E) White blood cell counts of WT and TIPE2−/− mice were determined using Drew Hemavet 950FS on Day 7. Data are shown as means ± SEM of one representative experiment of three. *P<0.05; **P<0.01; ***P<0.001.
To interrogate gut microbial diversity, we performed quantitative real-time PCR amplification of 16S rRNA gene sequences. The total DNA in stool was extracted and real-time PCR was conducted using specific 16S rRNA primers for the following major groups: the Eubacterium rectale-Clostridium coccoides, Bacteroides sp. and Enterobacteriaceae. In addition, the total bacterial (eubacteria) numbers were also determined using standard curves constructed with reference bacteria specific for each group. We found no significant differences in the numbers of total bacteria or the three representative bacterial groups, between naive WT and TIPE2−/− mice of 6–8-weeks of age (Fig. 6), indicating that bacterial composition was not affected by TIPE2 deficiency.
FIGURE 6. Analysis of microbiota composition in naive WT and TIPE2−/− mice.
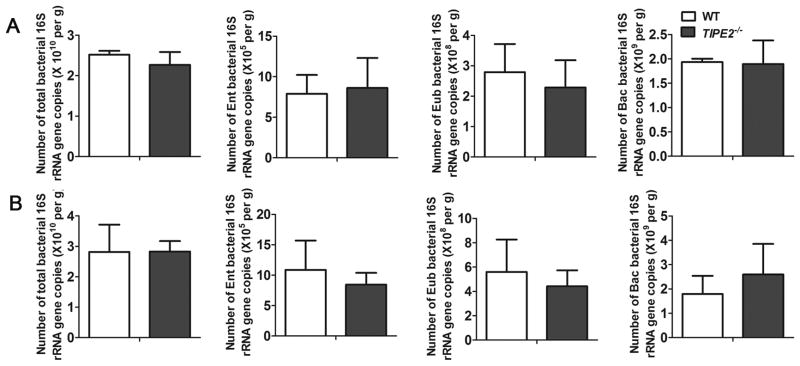
Stool genomic DNAs of 6-week-old (A) and 8-week-old (B) WT (n=3) and TIPE2−/− (n=3) mice were extracted using the QIAamp DNA Stool Mini Kit. Quantitative real-time PCR was performed to quantify the numbers of total bacteria, the Enterobacteriaceae (Ent) group, the Eubacterium rectale-Clostridium coccoides (Eub) group, and the Bacteroides (Bac) group per gram of stool. Data are shown as means ± SEM of one representative experiment of three.
Discussion
Results reported here indicate that TIPE2 is an important regulator of DSS-induced colitis. TIPE2−/− mice are resistant to DSS-induced colitis, and enhanced bacterial clearance after DSS treatment may be responsible. TIPE2 function has been studied in several diseases, including autoimmune diseases, bacterial infection, chronic inflammatory diseases, and cancer (12, 18–19, 34). TIPE2 is constitutively expressed at high levels in immune cells, especially in macrophages and neutrophils (8, 19–20), which play crucial roles in acute ulcerative colitis. To our knowledge, little is known of the function of TIPE2 in intestinal disorders. Acute ulcerative colitis induced by DSS is characterized by massive infiltration of inflammatory cells such as macrophages, neutrophils, and CD4+ T cells within the colonic walls, which destroy epithelium and shorten the colon length (35). These infiltrated inflammatory cells are major producers of inflammatory mediators such as TNF-α, IL-6, IL-12, IFN-γ, and IL-1β that contribute to pathogenesis (6). We found that TIPE2−/− mice produced markedly less TNF-α, IL-6, and IL-12, but not IFN-γ or IL-4 relative to WT mice (36).
Our previous studies showed that TIPE2 deficient macrophages produced significantly more IL-6 and IL-12 upon stimulation with lipopolysaccharide (LPS), and TIPE2 deficient mice were more susceptible to LPS-induced septic shock (8). We also found TIPE2 deficient mice were hypersensitive to Poly (I:C) lethality (20). In addition, TIPE2 deficiency exacerbates cerebral ischemia/reperfusion injury (16) and atherosclerosis in Ldlr−/− mice (18). Based on these findings, we expected TIPE2−/− mice to be more susceptible to colitis development than WT mice. On the contrary, TIPE2 deficiency rendered mice resistant to DSS-induced colitis. Adoptive transfer of TIPE2 deficient bone marrow cells was capable of rescuing colonic injury phenotype in WT mice, suggesting that TIPE2 expression in hematopoietic cells may play an important role in the development of colitis. Our previous studies also showed that TIPE2 serves as a negative regulator of phagocytosis and oxidative burst during infection. TIPE2 deficient mice exhibited resistance to bacterias challenge and TIPE2 deficient macrophages and neutrophils exhibited enhanced bacterial clearance (19). Indeed, significantly less bacteria were found in the colon of TIPE2−/− mice relative to DSS-treated WT mice. The number of leukocytes and serum concentrations of proinflammatory cytokine IL-6 were markedly reduced in TIPE2−/− mice, consistent with a reduced bacterial dissemination in these mice. Thus, in colitis, enhanced bacterial clearance in TIPE2−/− mice likely ameliorated the disease.
In summary, we have discovered that TIPE2 deficiency reduces inflammatory responses in a murine acute colitis model through enhancement of immune responses to commensal bacteria. Our data suggest a potential role for TIPE2 in the pathogenesis of IBD, especially in patients with impaired protection against commensal bacteria. This finding may not only advance our understanding of the mechanisms of IBD, but also lead to the development of TIPE2-based strategies for treating the disease.
Supplementary Material
Acknowledgments
The authors thank Drs. Terry Cathopoulis, Ji Qi, Derek Johnson, Qingguo Ruan, Xionghong Liang, Svetlana Fayngerts, George Luo, and George Buchlis for reagents and/or valuable advices, the CHOP Pathology Core for technical assistance.
Funding This work was funded by National Institutes of Health, USA (AI-077533, AI-050059, and GM-085112 to YHC). Y.L. is supported by a fellowship from the China Scholarship Council (No. 201206220065).
Abbreviations used in this article
- B6
C57BL/6
- BHI
Brain Heart Infusion
- DAI
disease activity index
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel diseases
- LPS
lipopolysaccharide
- TIPE
TNFAIP8
- TIPE2
the tumor necrosis factor (TNF)-alpha-induced protein 8-like 2 (TNFAIP8L2)
- WT
wild type
Footnotes
Conflict of interest disclosure: The authors declare no competing financial interests.
References
- 1.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goyette P, Labbe C, Trinh TT, Xavier RJ, Rioux JD. Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med. 2007;39:177–199. doi: 10.1080/07853890701197615. [DOI] [PubMed] [Google Scholar]
- 3.Solberg IC, Lygren I, Jahnsen J, Aadland E, Hoie O, Cvancarova M, Bernklev T, Henriksen M, Sauar J, Vatn MH, Moum B. Clinical course during the first 10 years of ulcerative colitis: results from a population-based inception cohort (IBSEN Study) Scand J Gastroenterol. 2009;44:431–440. doi: 10.1080/00365520802600961. [DOI] [PubMed] [Google Scholar]
- 4.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 6.Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 7.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 8.Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, Kong L, Xu L, Hilliard B, Hu S, Shen H, Yang X, Chen YH. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–426. doi: 10.1016/j.cell.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang G, Hao C, Lou Y, Xi W, Wang X, Wang Y, Qu Z, Guo C, Chen Y, Zhang Y, Liu S. Tissue-specific expression of TIPE2 provides insights into its function. Mol Immunol. 2010;47:2435–2442. doi: 10.1016/j.molimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Shi Y, Wang Y, Zhu F, Wang Q, Ma C, Chen YH. The unique expression profile of human TIPE2 suggests new functions beyond its role in immune regulation. Mol Immunol. 2011;48:1209–15. doi: 10.1016/j.molimm.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Lou Y, Liu S. The TIPE (TNFAIP8) family in inflammation, immunity, and cancer. Mol Immunol. 2011;49:4–7. doi: 10.1016/j.molimm.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Li D, Song L, Fan Y, Li X, Li Y, Chen J, Zhu F, Guo C, Shi Y, Zhang L. Down-regulation of TIPE2 mRNA expression in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Immunol. 2009;133:422–427. doi: 10.1016/j.clim.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Zhang Y, Wei X, Zhen J, Wang Z, Li M, Miao W, Ding H, Du P, Zhang W, He M, Yi F. Expression and regulation of a novel identified TNFAIP8 family is associated with diabetic nephropathy. Biochim Biophys Acta. 2010;1802:1078–1086. doi: 10.1016/j.bbadis.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Xi W, Hu Y, Liu Y, Zhang J, Wang L, Lou Y, Qu Z, Cui J, Zhang G, Liang X, Ma C, Gao C, Chen Y, Liu S. Roles of TIPE2 in hepatitis B virus-induced hepatic inflammation in humans and mice. Mol Immunol. 2011;48:1203–1208. doi: 10.1016/j.molimm.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Liu X, Wei Z, Wang X, Wang Z, Zhong W, Li Y, Zhu F, Guo C, Zhang L. The Expression and Significance of TIPE2 in Peripheral Blood Mononuclear Cells from Asthmatic Children. Scand J Immunol. 2013;78:523–528. doi: 10.1111/sji.12110. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Wei X, Liu L, Liu S, Wang Z, Zhang B, Fan B, Yang F, Huang S, Jiang F, Chen YH, Yi F. TIPE2, a novel regulator of immunity, protects against experimental stroke. J Biol Chem. 2012;287:32546–32555. doi: 10.1074/jbc.M112.348755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, Zhang W, Lou Y, Xi W, Cui J, Geng M, Zhu F, Chen YH, Liu S. TIPE2 deficiency accelerates neointima formation by downregulating smooth muscle cell differentiation. Cell Cycle. 2013;12:501–510. doi: 10.4161/cc.23325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lou Y, Liu S, Zhang C, Zhang G, Li J, Ni M, An G, Dong M, Liu X, Zhu F, Zhang W, Gao F, Chen YH, Zhang Y. Enhanced atherosclerosis in TIPE2-deficient mice is associated with increased macrophage responses to oxidized low-density lipoprotein. J Immunol. 2013;191:4849–4857. doi: 10.4049/jimmunol.1300053. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Fayngerts S, Wang P, Sun H, Johnson DS, Ruan Q, Guo W, Chen YH. TIPE2 protein serves as a negative regulator of phagocytosis and oxidative burst during infection. Proc Natl Acad Sci U S A. 2012;109:15413–15418. doi: 10.1073/pnas.1204525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H, Zhuang G, Chai L, Wang Z, Johnson D, Ma Y, Chen YH. TIPE2 controls innate immunity to RNA by targeting the phosphatidylinositol 3-kinase-Rac pathway. J Immunol. 2012;189:2768–2773. doi: 10.4049/jimmunol.1103477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, Rojas M, Wang L, Oprea G, Garg P, Gewirtz AT, Roman J, Merlin D, Sitaraman SV. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 22.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croswell A, Amir E, Teggatz P, Barman M, Salzman NH. Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect Immun. 2009;77:2741–2753. doi: 10.1128/IAI.00006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, Hunter C, Chen YH. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. 2002;110:843–850. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, Braun J, Xia L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med. 2007;204:1417–1429. doi: 10.1084/jem.20061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tlaskalova-Hogenova H, Tuckova L, Stepankova R, Hudcovic T, Palova-Jelinkova L, Kozakova H, Rossmann P, Sanchez D, Cinova J, Hrncir T, Kverka M, Frolova L, Uhlig H, Powrie F, Bland P. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Ann N Y Acad Sci. 2005;1051:787–798. doi: 10.1196/annals.1361.122. [DOI] [PubMed] [Google Scholar]
- 27.Berndt BE, Zhang M, Chen GH, Huffnagle GB, Kao JY. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol. 2007;179:6255–6262. doi: 10.4049/jimmunol.179.9.6255. [DOI] [PubMed] [Google Scholar]
- 28.Ghia JE, Galeazzi F, Ford DC, Hogaboam CM, Vallance BA, Collins S. Role of M-CSF-dependent macrophages in colitis is driven by the nature of the inflammatory stimulus. Am J Physiol Gastrointest Liver Physiol. 2008;294:G770–777. doi: 10.1152/ajpgi.00453.2007. [DOI] [PubMed] [Google Scholar]
- 29.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 31.Turunen UM, Farkkila MA, Hakala K, Seppala K, Sivonen A, Ogren M, Vuoristo M, Valtonen VV, Miettinen TA. Long-term treatment of ulcerative colitis with ciprofloxacin: a prospective, double-blind, placebo-controlled study. Gastroenterology. 1998;115:1072–1078. doi: 10.1016/s0016-5085(98)70076-9. [DOI] [PubMed] [Google Scholar]
- 32.Rembacken BJ, Snelling AM, Hawkey PM, Chalmers DM, Axon AT. Non-pathogenic Escherichia coli versus mesalazine for the treatment of ulcerative colitis: a randomised trial. Lancet. 1999;354:635–639. doi: 10.1016/s0140-6736(98)06343-0. [DOI] [PubMed] [Google Scholar]
- 33.Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, Scholmerich J, Sartor RB. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun. 2001;69:2277–2285. doi: 10.1128/IAI.69.4.2277-2285.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gus-Brautbar Y, Johnson D, Zhang L, Sun H, Wang P, Zhang S, Chen YH. The anti-inflammatory TIPE2 is an inhibitor of the oncogenic Ras. Mol Cell. 2012;45:610–618. doi: 10.1016/j.molcel.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Won HY, Jang EJ, Lee K, Oh S, Kim HK, Woo HA, Kang SW, Yu DY, Rhee SG, Hwang ES. Ablation of peroxiredoxin II attenuates experimental colitis by increasing FoxO1-induced Foxp3+ regulatory T cells. J Immunol. 2013;191:4029–4037. doi: 10.4049/jimmunol.1203247. [DOI] [PubMed] [Google Scholar]
- 36.Strober W, I, Fuss J, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.