

Abstract
The alpha-dystroglycanopathies are genetically heterogeneous muscular dystrophies that result from hypoglycosylation of alpha-dystroglycan (α-DG). Alpha-dystroglycan is an essential link between the extracellular matrix and the muscle fiber sarcolemma, and proper glycosylation is critical for its ability to bind to ligands in the extracellular matrix. We sought to identify the genetic basis of alpha-dystroglycanopathy in a family wherein the affected individuals presented with congenital muscular dystrophy, brain abnormalities and generalized epilepsy. We performed whole exome sequencing and identified compound heterozygous GMPPB mutations in the affected children. GMPPB is an enzyme in the glycosylation pathway, and GMPPB mutation were recently linked to eight cases of alpha-dystroglycanopathy with a range of symptoms. We identified a novel mutation in GMPPB (p.I219T) as well as a previously published mutation (p.R287Q). Thus, our work further confirms a role for GMPPB defects in alpha-dystroglycanopathy, and suggests that glycosylation may play a role in the neuronal membrane channels or networks involved in the physiology of generalized epilepsy syndromes.
Keywords: alpha-dystroglycanopathies, epilepsy, muscular dystrophy, GMPPB, glycosylation, exome sequencing
Introduction
The alpha-dystroglycanopathy congenital muscular dystrophies include a wide range of manifestations, ranging from the more severe Walker-Warburg syndrome (WWS) to muscle-eye-brain disease (MEB) and milder limb girdle muscular dystrophy (Bertini et al., 2011). Despite the clinical and genetic heterogeneity inherent in these disorders, all cases of alpha-dystroglycanopathy are defined by the hypoglycosylation of alpha-dystroglycan (α-DG), which is evident upon immunohistochemistry or immunoblot analysis of patient muscle samples.
The past several years have seen an explosion in the number of genes involved in alpha-dystroglycanopathy, rising from 6 genes in 2011 to 15 currently (Carss et al., 2013; Bertini et al., 2011). This is in large part due to the proliferation of next generation sequencing techniques, which have allowed for the relatively inexpensive and rapid sequencing of either whole exomes or genomes of affected families. This list of genes includes B3GALNT2, B3GNT1, DOLK, DPM2, DPM3, FKTN, FKRP, GTDC2, ISPD, LARGE, P0MGNT1, P0MT1, P0MT2, and TMEM5, with the most recent addition being GMPPB (guanosine diphosphate mannose pyrophosphorylase B) (Carss et al., 2013). Mutations in GMPPB manifest in a spectrum of clinical phenotypes, including alpha-dystroglycanopathy with structural brain and eye anomalies (type A, 14, OMIM 615350); alpha-dystroglycanopathy with developmental cognitive impairment (type B, 14, OMIM 615351); and alpha-dystroglycanopathy with a limb-girdle phenotype (type C, 14, limb-girdle phenotype, OMIM 615352).
Like other alpha-dystroglycanopathy genes, GMPPB encodes an enzyme in a pathway leading to α-DG glycosylation (Carss et al., 2013). α-DG is a cell surface membrane protein and part of the dystrophin complex that links intracellular signaling and the cytoskeleton to the extracellular matrix (Moore and Hewitt, 2009). Extracellular matrix association with α-DG depends on its glycosylation status, which affects binding to ligands including laminin (Moore and Hewitt, 2009). GMPPB catalyzes the formation of GDP-mannose, which is required for O-mannosylation of proteins including α-DG; mutations in two other enzymes in this pathway also cause alpha-dystroglycanopathy (Barone et al., 2012; Lefeber et al., 2009; Ning and Elbein, 2000). Previous work has demonstrated that diminished GMPPB activity, in human patients, cultured cells or animal models, reduces α-DG glycosylation (Carss et al., 2013). Mutations in the related protein GMPPA have recently been linked to a glycosylation disorder, characterized by achalasia, alacrima, and neurological defects (Koehler et al., 2013). Interestingly, none of the patients with GMPPA mutations presented with muscular dystrophy.
Here we present our analysis of an Ashkenazi Jewish family in which two of five children are affected by with congenital muscular dystrophy, generalized seizures, developmental delay, and elevated serum creatine kinase. Muscle pathology conclusively demonstrated alpha-dystroglycanopathy in the affected siblings, with the stereotypical reduction of glycosylated α-DG staining. Clinical screening of genes associated with alpha-dystroglycanopathies was non-diagnostic, so we performed whole exome sequencing on affected and unaffected family members in search of the causative mutation (Figure 2A). Both affected children, but no unaffected family members, carried GMPPB compound heterozygous mutations: p.R287Q, which was was previously identified in two Italian patients (Carss et al., 2013), and p.I219T, which is novel. This study confirms the role of GMPPB mutations in alpha-dystroglycanopathy associated with generalized epilepsy, and identifies a novel mutation in GMPPB.
Figure 2. Mutations in GMPPB Cause Alpha-Dystroglycanopathy.
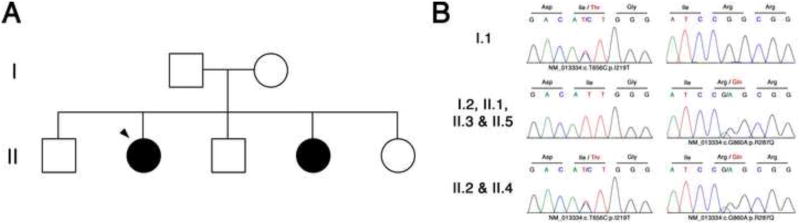
(A) An Ashkenazi Jewish family of Eastern European descent; affected individuals are indicated by a black circle, and the proband is indicated with an arrowhead. Whole exome sequencing was performed on all individuals except II.5. (B) Compound heterozygous mutations identified in GMPPB. Affected individuals (II.2 and II.4) carry both mutations in GMPPB, p.I219T (c.T>C656) and p.R287Q (c.G>A860), while I.1 (father) carries one copy of the p.I219T variant and the remaining family members carry one copy of the p.R287Q variant.
Patients and Methods
The Stanford University Institutional Review Board approved all aspects of this study, with written informed consent received for all participating family members.
Clinical Presentation of Subjects
The patients are sisters who presented to the pediatric neuromuscular clinic for evaluation at ages 4 and 8 years. Both were born at full-term following normal pregnancies. They had microcephaly that was noted early in life and had experienced early feeding difficulties resulting in failure to thrive. The older child had severe reflux requiring short-term placement of a percutaneous gastrostomy tube for nutrition. She also developed plagiocephaly (attributed to gross motor delay and positioning required to manage her reflux) and strabismus, neither of which was present in her sister. Both children displayed mild global delay and walked at 18 months. The older child also displayed truncal and limb ataxia and had a tendency to fall. Both children met criteria for childhood apraxia of speech. On neurological exam, they displayed hypotonia, proximal weakness, unsustained clonus at the ankles, and equivocal Babinski signs.
The younger child developed refractory generalized epilepsy at 30 months, with attacks that at different times were clinically characterized as atypical absence seizures, myoclonic seizures, and drop attacks. The older child has not had clinical seizures, but EEGs of both children revealed generalized 3-Hz spike-and-wave discharges.
Family History
The children’s parents are both of Ashkenazi Jewish, Ukrainian, and Polish ancestry. Their father is also of Irish descent. Both parents are in good health, as are the patients’ two brothers and one sister. There is no history of myopathy, seizures, or any central nervous system (CNS) disease in other family members.
Evaluation
Serum creatine kinase (CK) was repeatedly above 4000 U/L in both affected children. A brain MRI of the older girl at age 2 years revealed hypoplasia of the cerebellar vermis, pons, and medulla, as well as gyral abnormalities of the bilateral cerebellar hemispheres consistent with mild polymicrogyria (Figure 1A). Brain MRI of the younger girl at age 3 years did not reveal definite abnormalities.
Figure 1. Clinical Presentation of Proband.
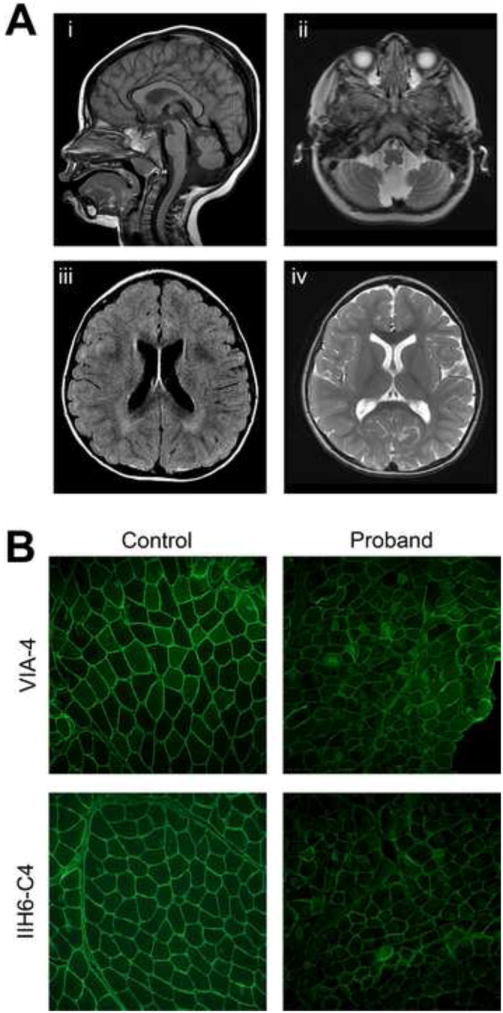
A) Brain MRI of the older child at age 2 years showing hypoplasia of the cerebellar vermis and lower brainstem and cerebellar polymicrogyria on sagittal T1 (i) and axial T2 (ii) views. Axial FLAIR (iii) and axial T2 (iv) images show periventricular white matter T2 hyperintensities suggestive of mild cerebral parenchymal underdevelopment. (B) Immunofluorescence for two epitopes of glycosylated alpha-dystroglycan both show variably diminished staining in the muscle biopsy as compared with control muscle.
Initial extensive genetic studies did not yield a diagnosis. This included karyotyping, array comparative genomic hybridization (aCGH), and evaluations for fragile X syndrome, metachromatic leukodystrophy, Pompe disease, carbohydrate deficient transferrin, and various causes of congenital muscular dystrophy and myopathy (ISPD, COL6A1, COL6A2, COL6A3, FKRP, FKTN, ITGA7, LAMA2, LARGE, P0MGNT1, P0MT1, P0MT2, SEPN1, ACADL, ACADM, ACADVL, ACAD9, AGL, C10orf2, CPT1B, CPT2, GAA, GYS1, HADHA, HADHB, LPIN1, OPA1, 0PA3, PFKM, PGAM2, PGM1, PHKA1, POLG, P0LG2, PYGM, RRM2B, SUCLA2, TK2, and TYMP).
Exome Sequencing
Genomic DNA samples were isolated from all family members using Oragene•DISCOVER (OGR-500) saliva collection kits. Exomes were generated for all family members (except for II.5) using the SureSelect Human All Exon 50Mb kit (Agilent, Santa Clara, CA). Sequencing was performed with 250bp paired-end reads on an Illumina MiSeq platform (Illumina Inc., San Diego, CA). Reads were aligned to the human reference genome (UCSC hgl9, GRCh37, February 2009 release) using bowtie2 (Langmead and Salzberg, 2012; Langmead and Salzberg, 2012) and SAMtools (Li et al., 2009; Li et al., 2009). We applied GATK base quality score recalibration, indel realignment, duplicate removal, and performed coverage calculations, SNP and INDEL discovery and genotyping across each sample using standard hard filtering parameters or variant quality score recalibration (DePristo et al., 2011). Variants were filtered against dbSNPvl37,1000 genomes and ESP 6500 databases and were then annotated using ANNOVAR (Wang et al., 2010). We assessed segregation of candidate mutations by Sanger sequencing using standard methods in all family members.
Muscle Pathology
Skeletal muscle biopsy cryosections were prepared using standard techniques and were stained with the IIH6-C4 (Millipore #05-593), VIA4-1 (Millipore #05-298), 4H8-2 (abeam #ab11576) and 5H2-4 monoclonal antibodies (Millipore #mab1922) directed respectively against glycosylated α-DG, the alpha 2 chain of laminin and the M-chain of human merosin. A non-dystrophic muscle biopsy was used as a control.
Results
Pathology
The proband (II.2) presented in clinic with a pattern of weakness and CNS involvement, consistent with alpha-dystroglycanopathy. After initial genetic test for congenital muscular dystrophy was negative, a muscle biopsy was taken to perform pathology to confirm the diagnosis. Immunohistochemistry revealed a reduction in glycosylated α-DG signal compared to a non-dystrophic control sample (Figure 1B). Additionally, the muscle fibers of patient II.2 show mild variation in fiber size, a mild focal increase in endomysial fibrosis and rare regenerative fibers, consistent with congenital muscular dystrophy. Additional staining against the alpha 2 chain of laminin and the M-chain of human merosin did not show any defects (Figure S1).
Exome Sequencing
We performed whole exome sequencing on 6 of 7 family members to identify the cause of alpha-dystroglycanopathy in the two affected siblings (II.2 and II.4). Following exome sequencing we identified 8 candidate genes (CD24, DNAH1, GMPPB, LAMB1, MUC12, MUC17, PDZD2, and USP6). We used Sanger sequencing to analyze these genes in an additional family member, II.5, that had not been subjected to exome sequencing, and this allowed us to exclude all other genes except GMPPB. Subsequent Sanger sequencing in all family members confirmed that the two affected siblings carried compound heterozygous mutations in GMPPB, while the remaining family members only carried one mutation each (Figure 2B). A novel mutation, p.I219T, was present in the father (I.1) while the mother and all unaffected children (I.2, II.1, II.3, II.5) carried the p.R287Q mutation. Affected children (II.2 and II.4) carried both p.I219T and p.R287Q mutations (Figure 2B).
While our exome sequencing study was underway, Carss and colleagues reported mutations in GMPPB as a cause of alpha-dystroglycanopathy in 8 patients (Carss et al., 2013). One of the mutations (p.R287Q) that we identified in this study (Figure 2B) was also identified by Carss and colleagues (Carss et al., 2013), and is located within the C-terminal domain of the protein. The new mutation (p.I219T) that we identified is located in the nucleotidyl transferase domain. While the previously identified p.R287Q mutation is very rare in the general population (rs202160208), the p.I219T variant that we identified is novel and not present in any public databases. This residue is highly conserved in GMPPB orthologs from D. melanogaster to human. Our results provide an independent confirmation that mutations in GMPPB cause alpha-dystroglycanopathy and identify an additional mutation (p.I219T) that can also cause the disease.
Discussion
In this paper we provide further evidence that mutations in GMPPB lead to alpha-dystroglycanopathy with a range of phenotypes. We have identified the compound heterozygous mutations p.I219T and p.R287Q in the GMPPB gene of two siblings of Ashkenazi Jewish descent with alpha-dystroglycanopathy, with a phenotype including features of proximal weakness, generalized epilepsy, apraxia of speech, ataxia, strabismus, and cerebellar hypoplasia. When combined with other recently described mutations in GMPPB, a total of nine mutations have now been identified in ten individuals with alpha-dystroglycanopathy (Carss et al., 2013).
The p.I219T variant identified in our patients is novel and therefore has not been previously characterized. The p.R287Q variant was previously identified in two unrelated Italian girls with alpha-dystroglycanopathy who, like our patients, presented with proximal weakness, ataxia, speech impairment, generalized epilepsy, and cerebellar hypoplasia, albeit to a more severe degree (Messina et al., 2009; Di Rosa et al., 2011; Carss et al., 2013). Those children, also compound heterozygotes for mutations in GMPPB, had a second mutation at C.95C>T (p.Pro32Leu).
Epilepsy in alpha-dystroglycanopathies and other congenital muscular dystrophies has in some cases been attributed to defects in cortical neuronal migration that result in cobblestone lissencephaly and other focal structural malformations (Reed, 2009). Our patients have generalized 3-Hz spike-and-wave discharges without focal cerebral cortical abnormalities, indicative of a generalized seizure disorder. The presence of generalized epilepsy in both girls indicates that GMPPB, and possibly other glycosylation enzymes, can affect neuronal membrane channels or networks implicated in generalized epilepsies, which may clarify the molecular pathophysiology of both genetically determined forms of epilepsy and of the CNS dysfunction in alpha-dystroglycanopathies. Thoroughly evaluating and optimally managing seizures in alpha-dystroglycanopathies may improve behavioral and cognitive function in this otherwise untreatable disorder.
Next generation sequencing techniques such as whole exome sequencing have been enormously successful in identifying the causal mutations behind Mendelian diseases in affected families (Bamshad et al., 2011). This is clearly demonstrated by the marked increase in the number of genes associated with alpha-dystroglycanopathy in the past 3 years (Carss et al., 2013; Bertini et al., 2011). Here we also put this methodology to use to identify compound heterozygous mutations in GMPPB as a cause of alpha-dystroglycanopathy, highlighting the utility of whole exome sequencing in determining the causal mutations behind familial diseases.
Supplementary Material
Table 1. Clinical Summary of Patients.
| Younger child (II.4) | Older child (II.2) | |
|---|---|---|
| Microcephaly | + | + |
| Plagiocephaly | + | |
| Feeding difficulties | + | + |
| Proximal weakness | + | + |
| Epilepsy (absence, myoclonic, drop attacks) | + | |
| Strabismus | + | |
| MRI brain | Normal | Abnormal |
| CK | 4233 U/L | 4505 U/L |
Clinical summary of two affected siblings (II.2 and II.4), showing similarities in clinical features like microcephaly and elevated CK levels, yet difference in brain MRI and epilepsies. Abbreviations, CK: creatine kinase.
Acknowledgments
We thank the patients and their family for participating in this study. This work is supported by NIH grants NS065317 (A.D.G.) and NS073660 (A.D.G.). A.R.R. is supported by an Alzheimer’s Disease Research grant from the BrightFocus Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Barone R, Aiello C, Race V, Morava E, Foulquier F, Riemersma M, Passarelli C, Concolino D, Carella M, Santorelli F, Vleugels W, Mercuri E, Garozzo D, Sturiale L, Messina S, Jaeken J, Fiumara A, Wevers RA, Bertini E, Matthijs G, Lefeber DJ. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. 2012;72:550–558. doi: 10.1002/ana.23632. [DOI] [PubMed] [Google Scholar]
- Bertini E, D’Amico A, Gualandi F, Petrini S. Congenital muscular dystrophies: a brief review. Semin Pediatr Neurol. 2011;18:277–288. doi: 10.1016/j.spen.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carss KJ, Stevens E, Foley AR, Cirak S, Riemersma M, Torelli S, Hoischen A, Wilier T, van Scherpenzeel M, Moore SA, Messina S, Bertini E, Bönnemann CG, Abdenur JE, Grosmann CM, Kesari A, Punetha J, Quinlivan R, Waddell LB, Young HK, Wraige E, Yau S, Brodd L, Feng L, Sewry C, MacArthur DG, North KN, Hoffman E, Stemple DL, Hurles ME, van Bokhoven H, Campbell KP, Lefeber DJ, Lin YY, Muntoni F. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies. 2013 doi: 10.1016/j.ajhg.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa G, Messina S, D’Amico A, Bertini E, Pustorino G, Spanò M, Tortorella G. A new form of alpha-dystroglycanopathy associated with severe drug-resistant epilepsy and unusual EEG features. Epileptic Disord. 2011;13:259–262. doi: 10.1684/epd.2011.0461. [DOI] [PubMed] [Google Scholar]
- Koehler K, Malik M, Mahmood S, Gießelmann S, Beetz C, Hennings JC, Huebner AK, Grahn A, Reunert J, Nürnberg G, Thiele H, Altmüller J, Nürnberg P, Mumtaz R, Babovic-Vuksanovic D, Basel-Vanagaite L, Borck G, Brämswig J, Mühlenberg R, Sarda P, Sikiric A, Anyane-Yeboa K, Zeharia A, Ahmad A, Coubes C, Wada Y, Marquardt T, Vanderschaeghe D, Van Schaftingen E, Kurth I, Huebner A, Hübner CA. Mutations in GMPPA cause a glycosylation disorder characterized by intellectual disability and autonomic dysfunction. Am J Hum Genet. 2013;93:727–734. doi: 10.1016/j.ajhg.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeber DJ, Schönberger J, Morava E, Guillard M, Huyben KM, Verrijp K, Grafakou O, Evangeliou A, Preijers FW, Manta P, Yildiz J, Grünewald S, Spilioti M, van den Elzen C, Klein D, Hess D, Ashida H, Hofsteenge J, Maeda Y, van den Heuvel L, Lammens M, Lehle L, Wevers RA. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85:76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina S, Tortorella G, Concolino D, Spanö M, D’Amico A, Bruno C, Santorelli FM, Mercuri E, Bertini E. Congenital muscular dystrophy with defective alpha-dystroglycan, cerebellar hypoplasiab, and epilepsy. Neurology. 2009;73:1599–1601. doi: 10.1212/WNL.0b013e3181c0d47a. [DOI] [PubMed] [Google Scholar]
- Moore CJ, Hewitt JE. Dystroglycan glycosylation and muscular dystrophy. Glycoconj J. 2009;26:349–357. doi: 10.1007/s10719-008-9182-0. [DOI] [PubMed] [Google Scholar]
- Ning B, Elbein AD. Cloning, expression and characterization of the pig liver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glc pyrophosphorylases are different proteins. Eur J Biochem. 2000;267:6866–6874. doi: 10.1046/j.1432-1033.2000.01781.x. [DOI] [PubMed] [Google Scholar]
- Reed UC. Congenital muscular dystrophy. Part I: a review of phenotypical and diagnostic aspects. Arq Neuropsiquiatr. 2009;67:144–168. doi: 10.1590/s0004-282x2009000100038. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:el64. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.