

Abstract
Activating mutations in TRPV4 are known to cause a spectrum of skeletal dysplasias ranging from autosomal dominant brachyolmia to lethal metatropic dysplasia. To develop an animal model of these disorders, we created transgenic mice expressing either wild-type or mutant TRPV4. Mice transgenic for wild-type Trpv4 showed no morphological changes at embryonic day 16.5, but did have a delay in bone mineralization. Overexpression of a mutant TRPV4 caused a lethal skeletal dysplasia that phenocopied many abnormalities associated with metatropic dysplasia in humans, including dumbbell-shaped long bones, a small ribcage, abnormalities in the autopod, and abnormal ossification in the vertebrae. The difference in phenotype between embryos transgenic for wild-type or mutant Trpv4 demonstrates that an increased amount of wild-type protein can be tolerated and that an activating mutation of this protein is required to produce a skeletal dysplasia phenotype.
Keywords: TRPV4, metatropic dysplasia, SMDK, skeletal dysplasia, calcium channel, transgenic mice
Introduction
TRPV4 is a tetrameric, calcium-selective, gated ion channel which is selectively (1), but not exclusively, expressed in chondrocytes where it plays a role in chondrocyte differentiation (2, 3). Mutations in TRPV4, which encodes the transient receptor potential vanilloid family member 4 calcium channel, result in a spectrum of dominantly-inherited skeletal dysplasia phenotypes (1, 4-7) as well as a group of peripheral neuropathies (8). At the mild end of the spectrum is familial digital arthropathy-brachydactyly (FDAB), a condition primarily characterized by osteoarthropathy in the hands (7). Autosomal dominant brachyolmia (BO) is a more generalized, but relatively mild condition characterized by short stature, a short trunk, and scoliosis (1). Spondylometaphyseal dysplasia, Kozlowski type (SMDK) is a more severe disorder with pronounced short stature, kyphoscoliosis, and short limbs. Radiographically, overfaced pedicles of the vertebral bodies, delayed epiphyseal ossification (particularly of the carpal bones) and irregular metaphyses characterize this phenotype (5). TRPV4 mutations can also produce non-lethal metatropic dysplasia, in which there is severe short stature, progressive kyphoscoliosis and marked shortening of the long bones, which frequently manifest a dumbbell shape (5, 6). Finally, the perinatal lethal form of metatropic dysplasia has extremely severe skeletal abnormalities including a small chest, and very short long bones and hands. Radiographically, there is a wafer-thin appearance to the vertebrae, a rounded pelvis and long bones with short, narrow diaphyses and flared ends (4). With the exception of FDAB, which appears to involve reduced channel activity (7), the majority of mutations result in single amino acid substitutions that activate the channel. Activation of the channels has been established by expression of mutations in cultured cells (1, 4, 5), demonstrating increased basal and agonist-stimulated channel activity as well as increased calcium concentrations within the cells (9). The same mutation in different individuals usually produces a similar phenotype (10); however, the substitutions found in each of the disorders are distributed across multiple domains of the TRPV4 molecule and the phenotype cannot be predicted from a previously unobserved genotype.
Because these conditions are mainly caused by a gain-of-function in TRPV4, a transgenic mouse approach was used to model the TRPV4 disorders. Wild-type or mutant Trpv4 was selectively expressed in cartilage using the Col2a1 promoter to test whether overexpression of wild-type Trpv4 was sufficient to generate a phenotype or if the expression of a mutant protein was necessary to produce the skeletal abnormalities which characterize the disorders in this spectrum.
Materials and Methods
Transgenic Mice
The transgenic construct was created by PCR amplification of mouse Trpv4 from a plasmid generously provided by the Bernd Nilius Laboratory, Leuven, Belgium. The primers used for amplification incorporated a consensus Kozak sequence containing in-frame ATG translation initiation and TAG termination codons, flanked by PvuII endonuclease sites to facilitate cloning into the pCol2a1-Tyr transgenic vector (11). Mutant Trpv4 sequence was generated by means of site-directed mutagenesis, utilizing overlapping PCR primers containing the desired c.1781G>A mutation (M. musculus Trpv4 mRNA NM_022017). Constructs were fully sequenced after production to confirm orientation and eliminate clones containing PCR errors. The linearized transgenic construct (Figure 1) was generated by PacI digestion and provided to the University of California, Irvine, transgenic facility for purification and microinjection into fertilized albino strain FVB oocytes. The microinjected zygotes were then implanted into pseudo-pregnant surrogate dams. Embryos were harvested 16 days after injection. Due to the presence of a tyrosinase reporter in the transgenic construct, successful incorporation and expression of the construct was initially determined by presence of black eye color (12) and then confirmed by PCR analysis. All experiments were approved by the UCLA and UCI animal care and use committees, as appropriate.
Figure 1. Linearized transgenic construct.
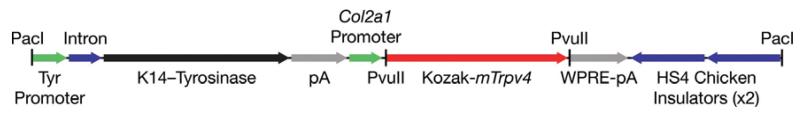
Wild-type and mutant murine Trpv4 cDNA containing an in-frame consensus Kozak translation initiation sequence (red) cloned downstream of a Col2a1 promoter sequence (green). A K14-Tyrosinase reporter minigene (black) is independently expressed via the tyrosinase promoter (green). A Woodchuck hepatitis virus Posttranscriptional Regulatory Element (WPRE, grey) is used to stabilize transcripts, leading to enhanced expression. Endonuclease sites (vertical black lines) used for Trpv4 cloning (PvuII) and construct linearization (PacI).
Cleared skeleton preparation
After harvest, skin and organs were removed from the embryos which were fixed in 95% ethanol overnight. Cartilage was stained overnight using 0.03% alcian blue dye (Sigma) in 80% ethanol and 20% acetic acid. After destaining in 95% ethanol, mineralized bone was stained in 0.005% alizarin red in 1% KOH for 4 hours. All embryos were then rinsed and cleared in 1% KOH at 4°C overnight before being transferred to 0.5% KOH in 50% glycerol for additional clearing and then 0.2% KOH in 80% glycerol for storage. Prior to microscopy, remaining soft tissue was excised and embryos were placed on gelatin-coated plates for imaging on a Leica dissecting microscope. Femur length measurements were made using a pinwheel reticle (Swift Microscope World).
Paraffin-embedded tissues
After removal of skin and organs, embryos were dissected and fixed in formalin overnight at 4°C. After decalcification in formic acid solution (ImmunoCal), samples were rinsed, dried, and dehydrated in graded ethanol solutions (50-100%). Samples were then cleared with xylene and embedded in paraffin in a vacuum oven. 0.5μm sections were obtained using a Leica microtome and placed on precoated slides (Fisher).
Immunostaining of paraffin-embedded sections
Slide-mounted sections were deparaffinized in xylene and rehydrated in graded ethanol solutions (100-50%). To improve antigen accessibility, sections were treated with 1mg/mL hyaluronidase (Sigma) at 37°C for 45 minutes. After washing, samples were blocked using a commercial blocking reagent (PerkinElmer) and treated overnight at 4°C with a primary antibody in blocking reagent. Sections were next treated with a secondary biotinylated anti-rabbit antibody (Invitrogen, diluted 1:250) for 30 minutes in blocking buffer. Fluorescent signal was generated by staining with Alexa 555-tagged streptavidin (Invitrogen). Samples were then counter-stained with DAPI and imaged on an Olympus fluorescent microscope. Anti-type II collagen antibody was purchased from Abcam (Cat. No. ab34712), and anti-type X collagen antibody was purchased from Calbiochem (Cat. No. 234196). Both antibodies were used at 1:500 dilutions and were generated in rabbit. Rabbit anti-SOX9 antibody was purchased from Novus Biologicals (Cat. No. 85551) and used at a dilution of 1:200. Rabbit anti-rat TRPV4 antibody was generously provided by the Nilius Laboratory in Leuven, Belgium (13). Scale bars were determined using a pinwheel reticle (Swift Microscope World).
Hematoxylin and eosin staining
Sections were deparaffinized and rehydrated as described above. After washing with deionized water, samples were stained with hematoxylin QS (Vector Laboratories) for 3 minutes, rinsed in water and destained in acid ethanol before rinsing again. Samples were then stained with 0.1% eosin for 30 seconds before being destained in ethanol and imaged on an Olympus light microscope.
Statistical analysis
All results are shown as the mean ± S.E. Differences in length were analyzed with a Student’s t-test (Microsoft Excel).
Results
We used transgenic mice to determine whether overproduction of wild-type Trpv4 is sufficient to produce skeletal abnormalities or if expression of a mutant protein is required to generate a phenotype. Mice were generated that expressed either wild-type or mutant Trpv4 (R594H) under the control of the cartilage-selective Col2a1 promoter. The Col2a1 promoter was selected because it is highly expressed in cartilage and has been used in many studies to direct expression in chondrocytes (14), permitting analysis of the skeletal phenotype exclusive of other tissues. The R594H mutation was selected because it produces the mild to moderately severe, but non-lethal, SMDK phenotype in humans (5, 15). The transgenic construct also incorporated a tyrosinase reporter gene (Figure 1) to facilitate monitoring of gene expression in transgenic albino embryos (12).
No visible abnormalities were observed in the appearance of E16.5 embryos transgenic for wild-type Trpv4 (n = 7) compared to non-transgenic controls (n = 15). Morphologically, the whole embryos of both control and wild-type Trpv4 transgenic mice were of normal size (Figures 2A and B). Additionally, the whole skeletons (Figures 3A and B), vertebrae (Figures 3E and F), long bones (Figures 4A and B), and autopods (Figure 5A and B) of both control and wild-type transgenic embryos were of normal size and appearance. While the overall lengths of the femurs, like the patterning of the other skeletal elements, were nearly identical between these two groups of mice (Figure 6A), both the absolute mineralized length (Figure 6A) and the percent mineralized portion of the femurs (Figure 6B) were statistically significantly lower in the wild-type transgenic embryos. Additionally, while not reaching statistical significance, trends toward delayed metacarpal and vertebral ossification could also be seen (data not shown), further suggesting a delay in skeletal mineralization.
Figure 2. Gross mouse embryo phenotypes.

A. A non-transgenic embryo (littermate of C and D) at embryonic day 16.5. B. A Trpv4WT transgenic embryo. Presence and expression of the transgenic construct was confirmed by the dark pigmentation of the eyes due to expression of the tyrosinase reporter. C. A moderately affected Trpv4R594H transgenic embryo with shortened limbs, tail, and snout. D. A more extreme phenotype can be seen in the severely affected Trpv4R594H transgenic. (Tick marks at bottom of each image indicate 1mm).
Figure 3. Whole skeletal preparations and lumbar spines stained with alcian blue (cartilage) and alizarin red (mineralized bone).

A and B. Skeletons of non-transgenic and wild-type transgenic control mice, respectively, at E16.5. C. The skeleton of a moderately affected Trpv4R594H transgenic embryo showing shortened long-bones, an abnormal ribcage, and a shortened spine. D. A severely affected Trpv4R594H transgenic embryo with almost no mineralization of endochondral bones and normal mineralization of intramembranous bones such as the lateral clavicle and flat bones of the skull. E and F. Stained lumbar vertebrae of control and wild-type transgenic mice. G. Stained lumbar vertebrae of moderately affected Trpv4R594H transgenic embryos with flattened mineralized vertebral bodies and an abnormal pattern of mineralization. H. Lumbar vertebrae from a severely affected Trpv4R594H transgenic embryo showing extreme vertebral abnormalities and only a small amount of ectopic mineralization.
Figure 4. Alcian blue (cartilage)- and alizarin red (mineralized bone)-stained femur preparations.

A and B. Femurs from control embryos at E16.5. C. The femur of a moderately affected embryo with a shortened diaphysis, widened epiphyses, and only a small area of mineralization at the periphery of the diaphysis. D. The femur of a severely affected Trpv4R594H transgenic embryo with a short, narrow diaphysis, marked epiphyseal flaring, and no mineralization.
Figure 5. Alcian blue (cartilage)- and alizarin red (mineralized bone)-stained autopod preparations.

A and B. The paws of non-transgenic and wild-type Trpv4 transgenic embryos at E16.5, respectively. C. The paw of a moderately affected embryo with poorly-defined carpal borders as well as abnormal metacarpals and phalanges. D. The paw of a severely affected Trpv4R594H transgenic embryo, showing markedly abnormal metacarpals and phalanges as well as fused carpals.
Figure 6. Comparisons of femur length and growth plate histology.

A. A comparison of non-transgenic (n = 10) and Trpv4WT (n = 8) total femur length and mineralized length measurements. B. A percent comparison of femur mineralization to total femur length in the same embryos shown in Panel A. A significant reduction in ossification was seen. C and D. Well-organized hematoxilyn and eosin-stained growth plates in non-transgenic (C) and wild-type transgenic (D) controls. E. Narrowing of the hypertrophic zone with widening of the reserve zone as well as a lack of well-defined boundaries between the zones in a moderately affected Trpv4R594H transgenic embryo. F. A profoundly disorganized growth plate from a severely affected Trpv4R594H transgenic embryo showing marked widening of the reserve zone, a lack of columnar structure in the proliferating zone, and a narrow hypertrophic zone with poor margins between each zone. (*p < 0.05, **p < 0.01)
In contrast to mice overexpressing wild-type Trpv4, mice transgenic for the SMDK-causing (5) Trpv4R594H allele exhibited perinatal lethal skeletal abnormalities. Because the dam often consumed mutant pups (data not shown), subsequent analyses were carried out on E16.5 embryos. The embryos expressing mutant Trpv4 (n = 8), exhibited a range of skeletal abnormalities, with the most severely affected embryos (n = 3) appearing hydropic at the time of collection (Figures 2C and D). The phenotype included several features commonly seen in the human TRPV4 skeletal dysplasias, such as small size with pronounced midface hypoplasia and short limbs. Mutant animals also had shortened tails consistent with axial abnormalities. While there was a spectrum of severity for Trpv4R594H transgenic mice, overall the phenotype was much more severe than would be expected for a mutation that causes SMDK in humans. The cleared and stained skeletons of Trpv4R594H transgenic mice were much smaller (Figures 3C and D) than those of the control animals, with abnormalities in the craniofacial, axial and appendicular skeletal elements. The most severely affected mice showed a nearly complete lack of mineralization of all endochondral bones, although membranous bones appeared to ossify normally (Figure 3D).
Compared with controls, the spines of most mice expressing Trpv4R594H had an abnormal pattern of mineralization with merging of the ossified regions of the central core and lateral processes (Figure 3G). In the most severely affected Trpv4R594H transgenic animals (Figure 3H), there was a complete lack of appropriate ossification of the vertebral bodies, but a small amount of ectopic ossification in a portion of the lumbar spine.
The long bones of Trpv4R594H transgenic embryos were uniformly shorter than controls with narrow diaphyses and proportionally wider epiphyses (Figures 4C and D). The more severely affected embryos also showed complete absence of mineralization of the long bones (Figure 4D). There were marked irregularities in the shapes of the metacarpals and phalanges of both moderately (Figure 5C) and severely (Figure 5D) affected Trpv4R594H transgenic mice compared to controls. Additionally, the boundaries of the carpals in moderately affected Trpv4R594H transgenic mice appeared poorly-defined, with the severely affected embryos exhibiting carpal fusions. Although the human TRPV4 disorders are characterized by delayed carpal ossification, mineralization does not occur in the mouse autopod until after E16.5 (see the control in Figure 5A), so this aspect of phenotype could not be assessed in the mice.
To examine the organization of the growth plates, we performed histochemistry on samples of developing bone stained with hematoxilyn and eosin. Consistent with the properly-formed skeleton, both the non-transgenic (Figure 6C) and wild-type transgenic (Figure 6D) embryos had well-organized growth plates with clearly defined reserve, proliferative, and hypertrophic zones and clear boundaries between each zone. The moderately affected Trpv4R594H transgenic embryos (Figure 6E) had a much narrower proliferative zone with a widened reserve zone and less well-defined boundaries between the different zones. In the severely affected Trpv4R594H transgenic embryos (Figure 6F), the growth plate was highly disorganized with much more pronounced widening of the reserve zone, lack of an organized proliferative zone, and a narrow hypertrophic zone, with very poorly defined margins between each zone. These findings are similar to the growth plates studied from human fetuses with lethal metatropic dysplasia, which show fewer hypertrophic cells, overgrowth of the poorly organized reserve zone cartilage, and significantly decreased mineralization (4).
To define the pattern of expression of Trpv4 in the transgenic mice, growth plates from the distal femur were stained with anti-TRPV4 antibody. While expression of native TRPV4 was too low to detect in non-transgenic mice (Figure 7A), TRPV4 expression was detected in the reserve, proliferating and hypertrophic zones of the growth plates of mice transgenic for wild-type Trpv4, and a low level of expression was apparent in perichondrium (Figure 7B). A similar pattern of expression of mutant TRPV4 was observed in moderately affected (Figure 7C) and severely affected (Figure 7D) embryos, although in the severely affected mutant the zones were difficult to distinguish.
Figure 7. TRPV4, type II, and type X collagen immunostaining of decalcified and paraffin-embedded distal femur growth plates.

A. Non-transgenic embryos did not have detectable levels of TRPV4 expression in the growth plate. B. TRPV4 protein could be seen throughout the growth plate of a wild-type transgenic embryo. C and D. TRPV4 protein could be seen throughout the growth plates of Trpv4R594H transgenic embryos. E-H. Type II collagen could be seen in the extracellular matrix of the reserve and proliferating zones of the growth plates of all mice examined, including the severely affected Trpv4R594H transgenic embryos (H). I and J. Type X collagen could be seen in the hypertrophic zones of control embryos. K. Type X collagen could be seen in the hypertrophic zone of moderately affected Trpv4R594H transgenic embryos L. Diffuse type X collagen was observed at low levels in the growth plates of severely affected Trpv4R594H transgenic embryos.
All embryos tested had normal patterns of type II collagen expression within the growth plate (Figure 7E-H). The overall pattern of type X collagen expression was also normal (Figure 7I-L), but severely affected Trpv4R594H transgenic embryos (Figure 7L) had less type X collagen, consistent with the reduced number of hypertrophic chondrocytes in these mice (16, 17). The expression pattern for SOX9 showed the highest level of expression across all genotypes in late proliferating chondrocytes (Figure S1). Due to the highly disrupted growth plate architecture there was less expression in the most severely affected mutant embryos, consistent with a reduction in proliferating chondrocytes.
Based on normal TUNEL staining, there did not appear to be increased apoptosis (data not shown). Staining with PCNA and Phosphohistone H3 antibodies also gave a normal pattern (data not shown), suggesting that abnormal proliferation of growth plate chondrocytes did not explain the reduced number of cells.
Discussion
To better understand the role of TRPV4 in skeletal development, we created transgenic mice overexpressing either wild-type or mutant Trpv4 in chondrocytes. Embryos overexpressing wild-type Trpv4 had normally patterned bones with well-organized growth plates, but a mild delay in mineralization. These data are consistent with previous studies showing that overexpressing wild-type Trpv4 in cells increased intracellular calcium levels (4, 5) and indicate that there is a phenotypic consequence to overexpression of wild-type Trpv4. Thus in contrast with the normal expression levels of TRPV4 with an activating mutation that cause a range of skeletal dysplasias (1, 4-6, 10), overexpression of normal TRPV4 in vivo produces only subtle changes in skeletal mineralization, at least at the level of TRPV4 overexpression in the animals studied. There is evidence in zebrafish that more severe skeletal phenotypes can result from generalized overexpression of wild-type TRPV4 (18), presumed to correlate with the degree of overexpression. Currently, we do not know how this mild defect would affect skeletal development beyond E16.5 or at higher levels of overexpression.
Embryos transgenic for Trpv4R594H exhibited severe skeletal abnormalities that recapitulate what is seen in humans with metatropic dysplasia and much more severe than individuals with SMDK caused by the R594H mutation. These embryos exhibited multiple skeletal abnormalities including dumbbell-shaped long bones, midface hypoplasia, platyspondyly and a small ribcage, reminiscent of what is seen in humans with metatropic dysplasia. The findings demonstrate that the overexpression of Trpv4R594H is sufficient to cause a skeletal dysplasia. In humans, the SMDK phenotype produced by the R594H mutation is moderately severe, but many of the transgenic mouse embryos presented a phenotype more similar to lethal metatropic dysplasia.
The more severe phenotype is likely the result of several factors. Use of the Col2a1 promoter resulted in strong, chondrocyte-specific expression of Trpv4, resulting in a level of TRPV4 in excess of normal endogenous expression. Additionally, TRPV4 is a tetramer (2), and the overexpression of mutant TRPV4 would be predicted to result in the overwhelming majority of functional tetramers containing only mutant subunits, another possible explanation for the unexpected phenotypic severity. We also cannot rule out the possibility that mice may be more susceptible to the effects of either this particular mutation or to Trpv4 mutations in general.
Histological analysis showed impaired differentiation of hypertrophic chondrocytes in the growth plates of moderately affected Trpv4R594H transgenic embryos with a near absence of hypertrophic chondrocytes in the growth plates of severely affected Trpv4R594H transgenic embryos. In extreme cases, no distinct hypertrophic zone could be detected by immunostaining for type X collagen. This defect in the hypertrophic zone accounts for the decreased or complete lack of mineralization in severely affected embryos transgenic for mutant TRPV4.
This study has demonstrated two important principles. First, that increased levels of wild-type TRPV4 can have phenotypic consequences, but these appear to be mild. Second, that expression of mutant Trpv4 is sufficient to cause a skeletal dysplasia reminiscent of Trpv4-based skeletal dysplasias in humans. The ability to model Trpv4-based skeletal dysplasias in mice will now allow a deeper exploration of the in vivo molecular mechanisms that produce these disorders, which in turn should open the door to new levels of understanding of the TRPV4 diseases.
Supplementary Material
Acknowledgments
MMW, SWT and YC carried out the studies, which were overseen by BL and DHC. This study was supported in part by grant HD22657 from NICHD/NIH and funds from the Orthopaedic Hospital Research Center at UCLA. MMW was supported by an NIH training grant (5 T32 AR 59033-2). We thank Bernd Nilius and colleagues for providing a construct containing the mouse Trpv4 cDNA and for TRPV4 antibodies and Faith Hall-Glenn for her assistance with the skeletal preparations. We also thank Karen Lyons and Deborah Krakow for their help in reviewing this manuscript. The study was also supported by the BCM Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, and the Rolanette and Berdon Lawrence Bone Disease Program of Texas.
Nonstandard abbreviations
- FDAB
familial digital arthropathy-brachydactyly
- BO
brachyolmia
- SMDK
Spondylometaphyseal dysplasia, Kozlowski type
- TRPV4
transient receptor potential vanilloid family member 4
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jbmr.2220]
Additional Supporting Information may be found in the online version of this article.
Disclosures:
The authors state that they have no conflicts of interest.
References
- 1.Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40(8):999–1003. doi: 10.1038/ng.166. PubMed PMID: 18587396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, et al. Analysis of the Native Quaternary Structure of Vanilloid Receptor 1. Journal of Biological Chemistry. 2001;276(30):28613–9. doi: 10.1074/jbc.M103272200. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, et al. Functional Gene Screening System Identified TRPV4 as a Regulator of Chondrogenic Differentiation. Journal of Biological Chemistry. 2007;282(44):32158–67. doi: 10.1074/jbc.M706158200. doi: 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- 4.Camacho N, Krakow D, Johnykutty S, Katzman PJ, Pepkowitz S, Vriens J, et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A. 2010;152A(5):1169–77. doi: 10.1002/ajmg.a.33392. PubMed PMID: 20425821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krakow D, Vriens J, Camacho N, Luong P, Deixler H, Funari TL, et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet. 2009;84(3):307–15. doi: 10.1016/j.ajhg.2009.01.021. PubMed PMID: 19232556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura G, Dai J, Lausch E, Unger S, Megarbane A, Kitoh H, et al. Spondylo-epiphyseal dysplasia, Maroteaux type (pseudo-Morquio syndrome type 2), and parastremmatic dysplasia are caused by TRPV4 mutations. Am J Med Genet A. 2010;152A(6):1443–9. doi: 10.1002/ajmg.a.33414. doi: 10.1002/ajmg.a.33414. PubMed PMID: 20503319. [DOI] [PubMed] [Google Scholar]
- 7.Lamande SR, Yuan Y, Gresshoff IL, Rowley L, Belluoccio D, Kaluarachchi K, et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet. 2011;43(11):1142–6. doi: 10.1038/ng.945. doi: 10.1038/ng.945. PubMed PMID: 21964574. [DOI] [PubMed] [Google Scholar]
- 8.Landoure G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, Inada H, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet. 2010;42(2):170–4. doi: 10.1038/ng.512. doi: 10.1038/ng.512. PubMed PMID: 20037586; PubMed Central PMCID: PMC2812627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Progress in biophysics and molecular biology. 2010;103(1):2–17. doi: 10.1016/j.pbiomolbio.2009.10.002. doi: 10.1016/j.pbiomolbio.2009.10.002. PubMed PMID: 19835908. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura G, Lausch E, Savarirayan R, Shiba M, Spranger J, Zabel B, et al. TRPV4-associated skeletal dysplasias. Am J Med Genet C Semin Med Genet. 2012;160C(3):190–204. doi: 10.1002/ajmg.c.31335. PubMed PMID: 22791502. [DOI] [PubMed] [Google Scholar]
- 11.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, et al. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(50):19004–9. doi: 10.1073/pnas.0605170103. doi: 10.1073/pnas.0605170103. PubMed PMID: 17142326; PubMed Central PMCID: PMC1748167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overbeek PA, Aguilar-Cordova E, Hanten G, Schaffner DL, Patel P, Lebovitz RM, et al. Coinjection strategy for visual identification of transgenic mice. Transgenic research. 1991;1(1):31–7. doi: 10.1007/BF02512994. PubMed PMID: 1844573. [DOI] [PubMed] [Google Scholar]
- 13.Gevaert T, Vriens J, Segal A, Everaerts W, Roskams T, Talavera K, et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Invest. 2007;117(11):3453–62. doi: 10.1172/JCI31766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13689–94. doi: 10.1073/pnas.94.25.13689. PubMed PMID: 9391087; PubMed Central PMCID: PMC28367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai J, Kim OH, Cho TJ, Schmidt-Rimpler M, Tonoki H, Takikawa K, et al. Novel and recurrent TRPV4 mutations and their association with distinct phenotypes within the TRPV4 dysplasia family. Journal of medical genetics. 2010;47(10):704–9. doi: 10.1136/jmg.2009.075358. doi: 10.1136/jmg.2009.075358. PubMed PMID: 20577006. [DOI] [PubMed] [Google Scholar]
- 16.Kielty CM, Kwan AP, Holmes DF, Schor SL, Grant ME. Type X collagen, a product of hypertrophic chondrocytes. The Biochemical journal. 1985;227(2):545–54. doi: 10.1042/bj2270545. PubMed PMID: 4004779; PubMed Central PMCID: PMC1144874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid TM, Linsenmayer TF. Developmental acquisition of type X collagen in the embryonic chick tibiotarsus. Developmental biology. 1985;107(2):373–81. doi: 10.1016/0012-1606(85)90319-7. PubMed PMID: 3882482. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Fu X, Gaiser S, Kottgen M, Kramer-Zucker A, Walz G, et al. OS-9 regulates the transit and polyubiquitination of TRPV4 in the endoplasmic reticulum. The Journal of biological chemistry. 2007;282(50):36561–70. doi: 10.1074/jbc.M703903200. doi: 10.1074/jbc.M703903200. PubMed PMID: 17932042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.