

Abstract
Electron capture dissociation (ECD) has shown great potential in structural characterization of glycans. However, our current understanding of the glycan ECD process is inadequate for accurate interpretation of the complex glycan ECD spectra. Here, we present the first comprehensive theoretical investigation on the ECD fragmentation behavior of metal-adducted glycans, using the cellobiose-Mg2+ complex as the model system. Molecular dynamics simulation was carried out to determine the typical glycan-Mg2+ binding patterns and the lowest-energy conformer identified was used as the initial geometry for density functional theory-based theoretical modeling. It was found that the electron is preferentially captured by Mg2+ and the resultant Mg+• can abstract a hydroxyl group from the glycan moiety to form a carbon radical. Subsequent radical migration and α-cleavage(s) result in the formation of a variety of product ions. The proposed hydroxyl abstraction mechanism correlates well with the major features in the ECD spectrum of the Mg2+-adducted cellohexaose. The mechanism presented here also predicts the presence of secondary, radical-induced fragmentation pathways. These secondary fragment ions could be misinterpreted, leading to erroneous structural determination. The present study highlights an urgent need for continuing investigation of the glycan ECD mechanism, which is imperative for successful development of bioinformatics tools that can take advantage of the rich structural information provided by ECD of metal-adducted glycans.
Introduction
Tandem mass spectrometry (MS/MS) has become an indispensable tool for structural analysis of a variety of biomolecules. Characterization of glycan structures poses one of the greatest analytical challenges, not only because of the frequent limitation in sample quantities due to the lack of glycan amplification methods, but also because of the structural diversity and heterogeneity in most naturally-occurring glycans as a result of their non-template-driven biosynthesis [1–2]. The structural diversity of glycans arises from their varied branching patterns, and the existence of many possible linkage and stereochemical isomers. While slow-heating fragmentation methods, such as collisionally activated dissociation (CAD) [3–13] and infrared multiphoton dissociation (IRMPD) [14–15], can generate an abundance of glycosidic fragments for deduction of the glycan topology, they do not normally produce sufficient numbers of the cross-ring fragments that are crucial for determining the linkage configuration. Over the past few years, a number of unconventional fragmentation methods have been applied to tandem MS analysis of glycans, including ultraviolet photodissociation (UVPD) [16–19], free radical-activated glycan sequencing (FRAGS) [20], and various electron activated dissociation (ExD) methods, such as electron capture dissociation (ECD) [15, 21–24], electron transfer dissociation (ETD) [25], electronic excitation dissociation (EED) [22, 26], electron-induced dissociation (EID) [27–28], electron detachment dissociation (EDD) [29–30], and negative electron transfer dissociation (NETD) [31]. In particular, ECD appears to be a promising tool for glycomics research as it can provide richer structural information than CAD-based methods, and is fairly straightforward to implement in online liquid chromatography-MS/MS studies. However, the glycan ECD mechanisms are poorly understood, and this factor, in conjunction with the presence of a large number of glycan fragmentation channels, makes spectral interpretation a very challenging task.
To date, ECD mechanistic studies have been mainly focused on peptides. The classic ECD mechanism (the Cornell Mechanism or the hot hydrogen mechanism) [32–35] assumes that for protonated peptides, electron capture occurs at a protonated site, forming a hypervalent radical cation, which in turn transfers a hydrogen to a spatially accessible amide carbonyl, leading to cleavage of the adjacent N-Cα bond and formation of c and z• ions. The hot hydrogen mechanism was subsequently challenged by the Utah-Washington (UW) Mechanism [36–41], which claims that electron attachment to an amide π* orbital can occur either directly, with sufficient Coulomb stabilization, or, during a cascade of relaxation events that lead to lower-energy Rydberg levels, via electron transfer from an excited Rydberg orbital initially located at one of the positively charged sites. Once an amide bond acquires substantial electron density, it becomes a super-base which can abstract a proton from an accessible site, resulting in the formation of a labile aminoketyl radical that subsequently undergoes N-Cα bond cleavage. Because the electron transfer and proton migration are decoupled in time (partially) and in space, the UW mechanism can be applied to ECD of multiply charged ions with non-proton charge carriers (e.g. metal-adducted peptide ions). For ECD of metalated peptides, the fragmentation pattern has been found to be dependent on both the size and the electronic configuration of the metal charge carriers [42–47]. Whereas peptides cationized with alkaline earth metal ions (e.g. Ca2+), or first-row divalent transition metal ions with half-filled (e.g. Mn2+) or fully-filled d-orbitals (e.g. Zn2+), produced primarily c- and z•-type ions under ECD, those adducted with metal cations with partially filled d-orbitals (e.g. Co2+, Ni2+, and Cu2+) generated predominantly a-, b-, and y-type ions. Chan et al. postulated that metal cations with stable electronic configuration (e.g. Ca2+, Mn2+, and Zn2+) are bystanders during ECD, which proceeds via the radical-directed pathway, whereas in the latter case, metal ion reduction competes favorably, and the released energy is dissipated into the peptide moiety to generate slow-heating fragment ions [45]. Protonation in metal-adducted peptides likely results from formation of zwitterions with the metal cation participating in a salt bridge structure. On the other hand, larger cations can be effectively solvated by the peptide without forming zwitterions. Consequently, second- and third-row group IIB cations (Cd2+, Hg2+) are preferentially reduced during ECD to produce fragment ions via an ergodic process, despite their presumably stable electronic configuration [46]. Similar fragmentation behavior was also observed in ECD of trivalent metal ion-peptide complexes by Williams et al., who suggested that it is the electrochemical properties of the metal ion in the peptide environment rather than in isolation that determine the initial electron capture site and the fragmentation outcomes [47]. Thus, through input from many laboratories, the search for understanding of the ECD fragmentation of peptides and proteins has made substantial progress.
In contrast to the vast literature on ECD mechanisms of peptides and proteins, no chemically explicit mechanistic study has been reported for ECD of glycans. Metalation is essential for generating extensive glycan fragmentation and for minimizing proton-induced rearrangements. However, neither the hot hydrogen nor the amide super base mechanism can be directly applied to metal-adducted glycans, since both require the presence of protonation sites. Furthermore, many glycans lack the amide moiety that plays a crucial role in ECD of peptides. Our preliminary theoretical study revealed that competition between glycosidic cleavage and metal loss is influenced by not only the recombination energy of the metal cation, but also the detailed potential energy surfaces of various dissociation channels [22]. It remains unclear how cross-ring fragments are generated and which fragmentation pathways are accessible. This paper presents the first theoretical investigation into the ECD fragmentation behavior of metal-adducted oligosaccharides, using the cellobiose-Mg2+ complex as the model system.
Experimental
Mass Spectrometry Analysis
HPLC-grade water and methanol were purchased from Fisher Scientific (Pittsburgh, PA). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). The reducing end 18O-labeling was performed based on the method introduced by Viseux et al. [12] Briefly, native glycans were incubated in H218O (97%) in the presence of 2-aminopyridine as the catalyst at 55 °C overnight. The 18O-labeled glycans were dissolved to a concentration of 2 μM in 50:50:1 water: methanol: formic acid electrospray solution containing 20 μM magnesium acetate, and directly infused into the mass spectrometer with a nanoelectrospray ionization source.
The ECD experiment was performed on a 12-T solariX™ hybrid Qh-Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker Daltonics, Bremen, Germany). The Mg2+-adduct was isolated by the front end quadrupole with an isolation window of 4 m/z, which allowed transmission of both the 18O-labeled and unlabeled glycans whose presence was due to incomplete isotopic labeling and back exchange during storage. Ions of interest were accumulated in the collision cell for 1 s before being transferred to the ICR cell, where they were irradiated by low-energy (~1.5 eV) electrons for 100 ms to generate ECD fragments. The product ions were detected with a 0.577-s transient, and 24 transients were summed to improve the S/N ratio. Fragment ion peak lists were generated using the Bruker Daltonics Sophisticated Numerical Annotation Procedure (SNAP™) algorithm [48]. Internal calibration using several fragment ions assigned with high confidence typically provided a mass measurement accuracy within 0.5 ppm.
Theoretical Modeling
The Mg2+ binding characteristics and the steric energies of the corresponding conformations of the cellobiose-Mg2+ complex were explored by molecular dynamics (MD) simulation, using the CHARMm force field [49]. Various initial geometries of cellobiose with different Mg2+ binding patterns were utilized for MD simulation. Each geometry was subjected to a two-step initial optimization, using the steepest descent and the conjugate gradient methodologies, respectively. The preliminarily optimized structure was heated gradually from 50 K to 1000 K in 2000 steps, at 1 fs duration per step, followed by system equilibration at 1000 K for 1 ps, and then subjected to MD trajectory calculation. Each trajectory run lasted for 200 ps, or 200,000 steps with an integration step size of 1 fs. The un-equilibrated geometries were recorded at a time interval of 5 ps, resulting in 40 collected geometries for each trajectory. All geometries were then subjected to temperature-independent geometry optimization, using the conjugate gradient algorithm, with 2000 optimization steps and a convergence criterion of 0.0001 RMS. The relative stability of different conformers generated from the final geometric optimization was characterized by the potential energy. A cellobiose-Mg2+ binding pattern was deemed typical if it appeared repeatedly in different trajectories with different initial conditions. The conformation space was then clustered based on the cellobiose-Mg2+ binding patterns. All MD simulations were carried out using the standard dynamics cascade and minimization modules of the Discovery Studio 2.5.2 software (Accelrys, San Diego, CA).
Density functional theory (DFT)-based calculations were performed using the hybrid of Becke’s exchange and Lee-Yang-Parr’s correlation functionals (B3LYP) [50–52]. For each important binding cluster found by the MD simulation, the conformer with the lowest potential energy was selected for DFT calculations at the B3LYP/6-31+G(d,p)//B3LYP/6-31G(d) level of theory, in which the B3LYP/6-31+G(d,p) electronic energy was corrected by the B3LYP/6-31G(d) zero point vibrational energy with no scaling, and used to compare the relative stability of different conformers. For mechanistic studies, all potential energy surfaces were calculated at the B3LYP/6-31G(d) level, and the B3LYP/6-31G(d) electronic energy was corrected by the zero point vibrational energy with a scaling factor of 0.9806 [53], and used as the zero Kelvin enthalpy. All reactants, intermediates, and products were verified to be local minima by normal mode frequency analysis (no imaginary frequency). The transition states were verified to be the desired critical configurations by frequency analysis (one vibration mode with imaginary frequency which leads to the formation of product ions) and intrinsic reaction coordinate calculation. All quantum mechanical (QM) calculations were performed using the Gaussian 03 program suite [54] at the Scientific Computing Facilities at Boston University.
Results and Discussion
Electron Capture Dissociation of the Cellohexaose-Mg2+ Complex
The ECD spectrum of Mg2+-adducted cellohexaose is shown in Figure 1. The fragment ions were assigned using the Domon-Costello nomenclature [55]. As no attempts were made to isolate the 18O-labeled precursor ions from the unlabeled ones (Figure 1, inset a), all reducing-end fragment ions would be present as doublets with a 2-Da splitting. Interestingly, none of the fragment ions was observed as a doublet (e.g. Figure 1, inset b), suggesting that they are either non-reducing-end fragments or internal fragments, and their assignments have been confirmed by accurate mass measurements. The most abundant product ion in the ECD spectrum is the 2,4A6 ion. Similar preferential formation of the 2,4An ion, where n is the degree of polymerization, has also been reported previously in ECD of other metal-adducted 1→4-linked oligosaccharides [21]. Several series of A ions, including the 2,4A, 0,2A, and 0,3A (or the isomeric 1,4A) ions, are present, and their abundance generally decreases with size. Other types of cross-ring fragments, 3,5A - 2H and 1,5A - 2H ions, are present only at low abundance. Glycosidic cleavages generated primarily B-ions (B3-5), as well as some low-abundance C-ions (C3-5). All fragment ions incorporated magnesium.
Figure 1.
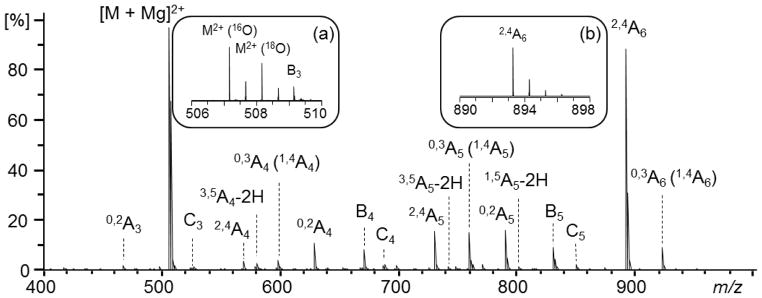
Low-energy (1.5 eV) ECD spectrum of the partially 18O-labeled cellohexaose-Mg2+ complex.
The Binding Characteristics and the Conformation Space Clustering of the Cellobiose-Mg2+ Complex
The first step in carrying out the ECD mechanistic study was determination of the population distribution of different conformers of the cellobiose-Mg2+ precursor ion, as its instantaneous geometric character is likely preserved during electron capture. The study of oligosaccharide conformations has been a major research field in carbohydrate chemistry for quite some time. The critical parameters for describing the conformation of disaccharides are the extent of rotation of each monosaccharide residue about its bond to the glycosidic oxygen atom, as indicated by the two dihedral angles: Φ (O5′-C1′-O4-C4 for cellobiose, Supplemental Scheme S1) and Ψ (C1′-O4-C4-C5). For native disaccharides, the conformation space can be explored systematically by grid search of the two dihedral angles, within the frame of either molecular mechanics or quantum mechanics [56–58]. Molecular mechanics and MD studies on oligosaccharides have also been carried out using the specifically developed force field [59]. However, for metal-adducted glycans, the presence of non-covalent interactions prevents systematic investigation of the conformation space, although ab initio or semi-empirical quantum mechanics studies can be performed on very simple monosaccharide and disaccharide systems [9, 60]. In this work, MD simulation was used to landscape the conformation space of the cellobiose-Mg2+ complex, and to determine the most probable Mg2+-binding characteristics and their corresponding lowest energy conformers, which were then used for the DFT-based molecular modeling to derive the ECD mechanism.
MD simulation produced a large number of conformers, of which only a small portion were likely populated. For instance, any conformer in which Mg2+ interacts with two adjacent hydroxyl groups of the same glucosyl ring likely has a high potential energy, and therefore negligible population. Excluding these irrelevant binding patterns, the conformation space can be clustered into three sub spaces, each characterized by a typical cellobiose-Mg2+ binding pattern. For each cluster, the conformer with the lowest potential energy was re-optimized using the density functional theory, and the optimized geometries are shown in Figure 2.
Figure 2.
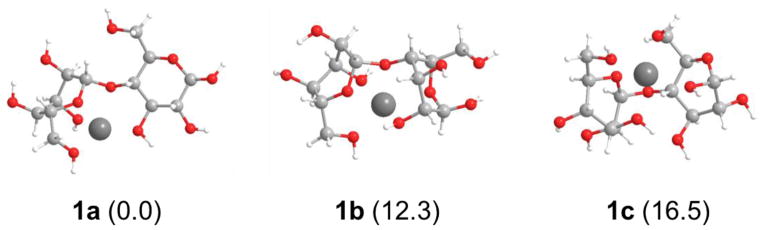
Typical low-energy conformers of the cellobiose-Mg2+ complex. The number in parentheses indicates the potential energy (in kcal/mol) of each structure relative to structure 1a.
The first binding pattern of the cellobiose-Mg2+ complex is represented by structure 1a, in which Mg2+ interacts with five oxygen atoms: O6′, O3′, O5′, O4, and O3. The corresponding O-Mg2+ distances are 2.05, 2.05, 2.04, 2.22, and 1.99 Å, respectively. This geometry is comparable to the lowest energy conformer of the native cellobiose previously determined using the MM3 force field [57]. The two characteristic dihedral angles Φ and Ψ for structure 1a are −85.8° and −94.9°, respectively, and may be compared with −87.9° and −142.0° for native cellobiose. In structure 1a, the non-reducing glucosyl ring is in a boat configuration rather than in a chair configuration as is the case for native cellobiose. Despite its higher steric strain, adopting the boat configuration in structure 1a allows stronger glycan-Mg2+ interaction, resulting in an overall decrease in potential energy. Structure 1b represents the second type of glycan-Mg2+ binding pattern, in which Mg2+ also interacts with five oxygen atoms: O6′, O3′, O5′, O4, and O2. The corresponding O-Mg2+ distances are 2.10, 2.16, 2.17, 2.05, and 2.09 Å, respectively, with Φ = −117.8° and Ψ = −168.2°. Similar to structure 1a, the non-reducing glucosyl ring in structure 1b adopts the boat configuration while the reducing glucosyl ring remains in the chair configuration. The third major binding pattern is represented by structure 1c, which involves Mg2+-binding to the O6′, O3′, O5′, O6, and O1 atoms to achieve its maximum coordination number of 5. The corresponding O-Mg2+ distances are 2.02, 2.14, 2.15, 2.08, and 2.18 Å, respectively, and the dihedral angles Φ = 49.3° and Ψ = −118.9°. Both glucosyl rings are in the boat configuration. In contrast to its role in structures 1a and 1b, the glycosidic oxygen in structure 1c no longer binds Mg2+; instead, the C6 primary hydroxyl and the C1 secondary hydroxyl from the glucosyl ring at the reducing end participate in the Mg2+-glycan binding.
The relative energies of structures 1a, 1b and 1c are listed in Figure 2. Since geometry optimization at the B3LYP/6-31+G(d,p) level was beyond our computational capability, the B3LYP/6-31+G(d,p) single point energies were calculated on the B3LYP/6-31G(d) geometries to investigate the basis set augmentation effect. The result indicated that the basis set augmentation did not change the order of the relative stability of these three typical cellobiose-Mg2+ conformers. Therefore, further theoretical modeling was carried out based upon structure 1a, which should be the predominant conformer at room temperature.
The Hydroxyl Abstraction Mechanism for ECD of the Cellobiose-Mg2+ Complex
Unlike the peptide cations, the cellobiose-Mg2+ complex contains neither protonation sites nor unsaturated bonds. Thus, the initial electron capture likely occurs at the Mg2+-binding site due to large second ionization potential of Mg (15.04 eV). The actual electron-Mg2+ recombination energy in the cellobiose-Mg2+ system (7.08 eV, or 163.2 kcal/mol, based on structure 1a) is smaller because Mg2+ is solvated by the glycan. The frontier singly occupied molecular orbital (SOMO) of the charge reduced species 2 is illustrated in Supplemental Figure S1. The unpaired electron is localized at the Mg nucleus, as confirmed by the population analysis which shows that the Mulliken spin density at the Mg nucleus is 0.9035.
As electron transfer occurs on a much faster time-scale than nuclear motion, the initial geometry of the cellobiose-Mg2+ complex is likely preserved in the charge-reduced cellobiose-Mg+• species. Under such circumstance, as depicted in Supplemental Figure S1, three hydroxyl groups, located at the C3, C3′, and C6′ positions, respectively, are spatially accessible to Mg+•. In contrast, direct hydrogen abstraction by Mg+• is not favored, as it would require disruption of the electrostatic interaction between Mg+• and one or more hydroxyl groups to accommodate hydrogen transfer. Thus, it seems most likely that hydroxyl abstraction by Mg+• forms a MgOH+ closed-shell cation, which is still electrostatically bound to the cellobiose moiety, and serves as a charge tag in MS measurements. Meanwhile, a carbon radical is formed, which can initiate further α-cleavages or β-eliminations, producing different fragment ions. Note that in structure 1a, four out of the five oxygen atoms that bind Mg2+ are contributed by the nonreducing glucosyl unit, since O4 can also be regarded as O1′. Thus, following bond dissociation, nonreducing end fragments would be more likely to retain the charge and be detected, and, indeed, this preference is observed experimentally. The large recombination energy release ensures that hydroxyl abstraction from any of the three positions is energetically and kinetically accessible. Here, the discussion will be focused on the reaction pathways following the C3-hydroxyl migration.
Scheme 1a illustrates a possible pathway for the 2,4A2 ion formation. The number in parenthesis next to each intermediate or transition state indicates its potential energy (in kcal/mol) relative to the precursor ion. The initial C3-hydroxyl abstraction is associated with an energy barrier of 16.3 kcal/mol, and the resultant C3 radical (intermediate 3) is 7.7 kcal/mol more stable than the charge reduced specie 2. The C3 radical can induce α-cleavage of the C4-C5 bond, with an energy barrier of 23 kcal/mol, forming intermediate 4 with a reaction endothermicity of 11.3 kcal/mol. Intermediate 4 then undergoes consecutive α-cleavages and loses two small neutral species, 2-hydroxyacetaldehyde and 1,2-trans-dihydroxyethylene, respectively, to form the 2,4A2 + H ion, 6. Loss of 2-hydroxyacetaldehyde is associated with an energy barrier of 20.7 kcal/mol, close to the reaction endothermicity, whereas no canonical transition state can be found for loss of dihydroxyethylene at the B3LYP/6-31G(d) level of theory. Such a phenomenon is common for direct bond dissociation, which often involves a loose transition state that is dependent on either temperature or overall internal energy. Since a loose transition state is usually a late transition state that resembles the product rather than the reactant, its number of states can be approximated by the number of states of the product, from the point of view of the RRKM theorem. Therefore, in cases where the transition state cannot be identified, the energy barrier is estimated to be approximately the same as the reaction endothermicity (e.g. 5→6). The 2,4A2 + H ion can further lose a hydrogen atom to form the final product, the closed-shell 2,4A2 ion, 7.
Scheme 1.

Proposed mechanisms for formation of the 2,4A2 ion.
Although all processes in Scheme 1a are energetically accessible, the dihydroxyethylene loss is associated with a substantial energy barrier and reaction endothermicity (41.9 kcal/mol), as it requires the formation of a vinyl radical. Moreover, hydrogen loss in the final step is also associated with a large energy barrier (53.6 kcal/mol) and endothermicity (48 kcal/mol), since the hydrogen is eliminated from an sp2 carbon. An alternative pathway for 2,4A2 ion formation is shown in Scheme 1b. In intermediate 5, internal rotation about the C2-C3 bond can lead to a planar conformation, 5a, which facilitates C4→C1 hydrogen migration. Without ring constraint, this rotation about a single bond only has a 2 kcal/mol energy barrier. The resultant intermediate 8 then loses a 1,2-dihydroxylethyl radical to form the 2,4A2 ion. The mechanism presented in Scheme 1b is much more appealing than the one in Scheme 1a, as the key 1,4-hydrogen migration step only poses a moderate reaction energy barrier of 27.8 kcal/mol, and the final product is 39.9 kcal/mol lower in energy.
Formation of the other prominent A ion series, the 0,3A (1,4A) ions, however, cannot be explained by the mechanism shown in Scheme 1, as consecutive α-cleavages can only result in breakage of bonds that are separated by two atoms, whereas the 0,3A or 1,4A ion formation requires cleavage of bonds that are three atoms apart. In CID of hexoses, neither 0,3- nor 1,4-cross ring cleavage was observed for 1→4 linked residues, although cleavage of a 1→3 linked hexose can produce an isomer of the 0,3An ion, where n is the degree of polymerization, possibly via a retro-aldol or retro-ene rearrangement of an open-ring structure [9, 61]. Here, however, observation of the 0,3A ions (or their isomers) is not limited to the reducing end, and they were likely generated by a different, radical induced, mechanism, such as the one illustrated in Scheme 2. The C3-radical in intermediate 3 can undergo 1,2-hydrogen migration which generates a C4-radical species, 10. Although 1,2-hydrogen migration is associated with a substantial energy barrier (42.7 kcal/mol), this restriction is still easily surmountable owing to the large energy release during electron capture. The C4 radical can induce consecutive α-cleavages, releasing a dihydroxyethylene molecule to form a radical species, 12, which then either loses a hydrogen atom to produce the 0,2A2 ion, product 13, or loses a hydroxymethyl radical to form an ion that is isomeric to the 0,3A2 ion, product 14. The latter pathway is both energetically and kinetically favored, and is consistent with the observation of the (apparent) 0,3A6 ion but not the 0,2A6 ion.
Scheme 2.

Proposed mechanism for formation of the “0,3A2” (0,2A2-CHOH) ion.
Alternatively, the 0,2A ion can be formed via consecutive α-cleavages induced by a C5-radical, which is generated following two 1,2 hydrogen migrations, as shown in Scheme 3. However, as was the case in Scheme 2, the final step involves hydrogen atom loss, which is thermodynamically and kinetically unfavored. Scheme 4a shows a third possible pathway, in which intermediate 5 loses its O1 hydrogen to form the 0,4A2 ion, which is isomeric to the 0,2A2 ion. At first glance, this mechanism offers little advantage over those presented in Schemes 2 and 3, as it also involves a hydrogen loss which is associated with a 33.2 kcal/mol reaction barrier. However, for internal residues in the larger cellohexaose-Mg2+ complex, the equivalent process would lead to glycosidic bond cleavage and formation of a 0,4Ai/Ci ion (i < n), instead of hydrogen loss. Using the model system shown in Scheme 4b, the corresponding reaction barrier for isopropyl loss is only 20.1 kcal/mol. This mechanism seems to agree with the observation that 0,2Ai ions were only observed for i < 6.
Scheme 3.

Proposed mechanism for formation of the 0,2A2 ion.
Scheme 4.
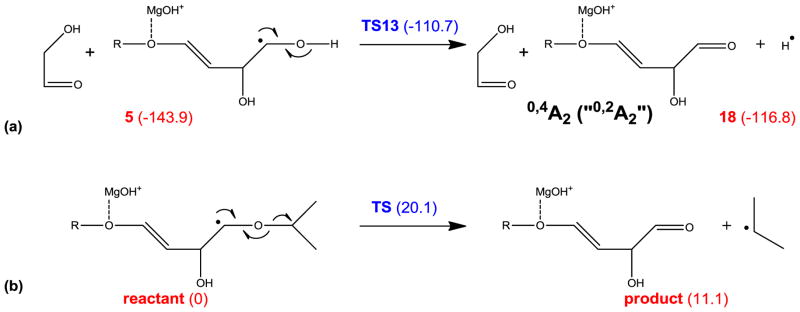
Proposed mechanism for formation of the 0,4A ions at the reducing end (a), and in internal residues (b).
Since QM calculation on the cellohexaose-Mg2+ system is beyond our current computational capability, MD simulation was performed to explore its conformational space. Because the β-1→4 linkage in cellohexaose affords less flexibility than other types of linkage (e.g. α-1→6 linkage), the access to the population of low-energy conformers involving Mg2+ binding to adjacent residues is entropically favored. Supplemental Figure S2 shows a typical Mg2+-binding pattern for the cellohexaose-Mg2+ complex, in which Mg2+ is solvated by residues 4 and 5, and interacts with the same five atoms (O3′, O5′ and O6′ on the non-reducing ring, and O3 and O4 on the reducing ring) as in the cellobiose system. Upon electron capture, conformers involving Mg2+-binding to internal residues will likely give rise to smaller Ai cross-ring fragments with i < 6. Thus, it appears to be reasonable to extrapolate the cellobiose model to the larger cellohexaose-Mg2+ system.
Implications of the Hydroxyl Abstraction Mechanism
The hydroxyl migration mechanism suggests that the fragmentation pattern of metal-adducted glycans is likely influenced by the choice of the initial position(s) of the abstracted hydroxyl group, which is determined by the detailed metal-glycan binding pattern. Thus, ECD may be used to differentiate stereo glycan isomers that differ in either their epimeric or anomeric configuration. However, hydrogen migration after hydroxyl abstraction can potentially move the radical to any of several positions, resulting in a “blurring” of the fragmentation pattern that would otherwise be unique to hydroxyl abstraction from a certain position.
A second, perhaps more important implication of the results presented here is that some fragments observed experimentally may not have the expected structure. Secondary radical-induced fragmentation, before or after radical migration, is a well-known phenomenon in the ECD of peptides [62–70], and evidence now becoming available suggests that this can also occur in ECD of metalated glycans. These secondary fragment ions may be misinterpreted as the products of simple cross-ring fragmentations. For example, the ECD spectrum of the laminarihexaose-Mg2+ complex (Supplemental Figure S3) contains a series of ions that have the same m/z values as the 0,3A ion series in the ECD spectrum of the cellobiose-Mg2+ complex. However, laminarihexaose contains exclusively 1→3 linked glucosyl rings, wherein 0,3 cross-ring cleavage should lead to loss of C3H6O3 in all residues. In light of the mechanism proposed here, these ions could potentially be formed in a reaction pathway similar to the one outlined in Scheme 2 (reaction 12→14), which generates 0,2A – CHOH ions. If one has no prior knowledge of the analyte’s identity, 0,2A CHOH ions could easily be mistaken as 0,3A ions characteristic of 1→4 and 1→6 linkages, leading to erroneous structural determination. Therefore, there is a practical need for continuing investigation into glycan ECD processes. Further experimental work, possibly involving selective isotope labeling at different ring positions, is needed to validate the mechanism proposed here.
Conclusions
This theoretical study on the cellobiose-Mg2+ model system investigated the ECD fragmentation process of metal-adducted native glycans. It was found that the electron was initially attached to Mg2+, converting it to a Mg+• radical cation which could abstract a hydroxyl from an accessible site to form a carbon radical. Subsequent α-cleavages and hydrogen elimination induced by the carbon radical, either at the original hydroxyl abstraction site or at a different site following 1,2 hydrogen migration(s), could lead to formation of various glycosidic and cross-ring fragment ions. The hydroxyl abstraction mechanism can successfully explain the characteristic ECD fragmentation behavior of the cellohexaose-Mg2+ complex observed experimentally. Meanwhile, exploration of the proposed mechanism also revealed some unexpected fragmentation pathways which will likely have significant implications for the development of bioinformatics tools for interpretation of glycan ECD spectra. There is still much to be learned about the glycan ECD process. Further theoretical studies are needed to investigate the effects of different metal charge carriers on the ECD fragmentation behaviors of metal-adducted glycans, and change in fragmentation patterns due to structural variations, including alternative linkage and stereochemical configurations, as well as the consequences of chemical modifications such as permethylation and reduction and/or labeling of the reducing end.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the financial support from the National Institutes of Health via research grants P41 RR10888/GM104603, and S10 RR025082. The authors would also like to acknowledge the technical support and the computing resources provided by the Scientific Computing and Visualization Group at Boston University.
References
- 1.Helenius A, Aebi M. Intracellular Functions of N-Linked Glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 2.Varki A, Cummings RD, Esko JD. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2009. [PubMed] [Google Scholar]
- 3.Ashline DJ, Lapadula AJ, Liu YH, Lin M, Grace M, Pramanik B, Reinhold VN. Carbohydrate Structural Isomers Analyzed by Sequential Mass Spectrometry. Anal Chem. 2007;79:3830–3842. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancilla MT, Penn SG, Carroll JA, Lebrilla CB. Coordination of alkali metals to oligosaccharides dictates fragmentation behavior in matrix assisted laser desorption ionization Fourier transform mass spectrometry. J Am Chem Soc. 1996;118:6736–6745. [Google Scholar]
- 5.Cancilla MT, Wang AW, Voss LR, Lebrilla CB. Fragmentation reactions in the mass spectrometry analysis of neutral oligosaccharides. Anal Chem. 1999;71:3206–3218. doi: 10.1021/ac9813484. [DOI] [PubMed] [Google Scholar]
- 6.Harvey DJ. Collision-induced fragmentation of underivatized N-linked carbohydrates ionized by electrospray. J Mass Spectrom. 2000;35:1178–1190. doi: 10.1002/1096-9888(200010)35:10<1178::AID-JMS46>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Harvey DJ. Structural determination of N-linked glycans by matrix-assisted laser desorption/ionization and electrospray ionization mass spectrometry. Proteomics. 2005;5:1774–1786. doi: 10.1002/pmic.200401248. [DOI] [PubMed] [Google Scholar]
- 8.Harvey DJ, Bateman RH, Green MR. High-energy collision-induced fragmentation of complex oligosaccharides ionized by matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom. 1997;32:167–187. doi: 10.1002/(SICI)1096-9888(199702)32:2<167::AID-JMS472>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Hofmeister GE, Zhou Z, Leary JA. Linkage Position Determination in Lithium-Cationized Disaccharides - Tandem Mass-Spectrometry and Semiempirical Calculations. J Am Chem Soc. 1991;113:5964–5970. [Google Scholar]
- 10.Lemoine J, Fournet B, Despeyroux D, Jennings KR, Rosenberg R, Dehoffmann E. Collision-Induced Dissociation of Alkali-Metal Cationized and Permethylated Oligosaccharides - Influence of the Collision Energy and of the Collision Gas for the Assignment of Linkage Position. J Am Soc Mass Spectrom. 1993;4:197–203. doi: 10.1016/1044-0305(93)85081-8. [DOI] [PubMed] [Google Scholar]
- 11.Sheeley DM, Reinhold VN. Structural Characterization of Carbohydrate Sequence, Linkage, and Branching in a Quadrupole Ion Trap Mass Spectrometer: Neutral Oligosaccharides and N-Linked Glycans. Anal Chem. 1998;70:3053–3059. doi: 10.1021/ac9713058. [DOI] [PubMed] [Google Scholar]
- 12.Viseux N, deHoffmann E, Domon B. Structural analysis of permethylated oligosaccharides by electrospray tandem mass spectrometry. Anal Chem. 1997;69:3193–3198. doi: 10.1021/ac961285u. [DOI] [PubMed] [Google Scholar]
- 13.Kailemia MJ, Li L, Ly M, Linhardt RJ, Amster IJ. Complete Mass Spectral Characterization of a Synthetic Ultralow-Molecular-Weight Heparin Using Collision-Induced Dissociation. Anal Chem. 2012;84:5475–5478. doi: 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y, Lebrilla CB. Infrared Multiphoton Dissociation of Alkali Metal-Coordinated Oligosaccharides. Anal Chem. 2003;75:1590–1598. doi: 10.1021/ac026009w. [DOI] [PubMed] [Google Scholar]
- 15.Adamson JT, Hakansson K. Infrared multiphoton dissociation and electron capture dissociation of high-mannose type glycopeptides. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 16.Devakumar A, Mechref Y, Kang P, Novotny MV, Reilly JP. Laser-induced photofragmentation of neutral and acidic glycans inside an ion-trap mass spectrometer. Rapid Commun Mass Spectrom. 2007;21:1452–1460. doi: 10.1002/rcm.2981. [DOI] [PubMed] [Google Scholar]
- 17.Wilson JJ, Brodbelt JS. Ultraviolet Photodissociation at 355 nm of Fluorescently Labeled Oligosaccharides. Anal Chem. 2008;80:5186–5196. doi: 10.1021/ac800315k. [DOI] [PubMed] [Google Scholar]
- 18.Ko BJ, Brodbelt JS. 193 nm Ultraviolet Photodissociation of Deprotonated Sialylated Oligosaccharides. Anal Chem. 2011;83:8192–8200. doi: 10.1021/ac201751u. [DOI] [PubMed] [Google Scholar]
- 19.Racaud A, Antoine R, Joly L, Mesplet N, Dugourd P, Lemoine J. Wavelength-Tunable Ultraviolet Photodissociation (UVPD) of Heparin-Derived Disaccharides in a Linear Ion Trap. J Am Soc Mass Spectrom. 2009;20:1645–1651. doi: 10.1016/j.jasms.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Thomas DA, Sohn CH, Beauchamp JL. Biomimetic Reagents for the Selective Free Radical and Acid Base Chemistry of Glycans: Application to Glycan Structure Determination by Mass Spectrometry. J Am Chem Soc. 2013;135:10684–10692. doi: 10.1021/ja402810t. [DOI] [PubMed] [Google Scholar]
- 21.Adamson JT, Hakansson K. Electron capture dissociation of oligosaccharides ionized with alkali, alkaline earth, and transition metals. Anal Chem. 2007;79:2901–2910. doi: 10.1021/ac0621423. [DOI] [PubMed] [Google Scholar]
- 22.Yu X, Huang Y, Lin C, Costello CE. Energy-Dependent Electron Activated Dissociation of Metal-Adducted Permethylated Oligosaccharides. Anal Chem. 2012;84:7487–7494. doi: 10.1021/ac301589z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C, Xie B, Chan SY, Costello CE, O’Connor PB. Collisionally activated dissociation and electron capture dissociation provide complementary structural information for branched permethylated oligosaccharides. J Am Soc Mass Spectrom. 2008;19:138–150. doi: 10.1016/j.jasms.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 24.Zhou W, Håkansson K. Electron Capture Dissociation of Divalent Metal-adducted Sulfated N-Glycans Released from Bovine Thyroid Stimulating Hormone. J Am Soc Mass Spectrom. 2013;24:1798–1806. doi: 10.1007/s13361-013-0700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han L, Costello CE. Electron Transfer Dissociation of Milk Oligosaccharides. J Am Soc Mass Spectrom. 2011;22:997–1013. doi: 10.1007/s13361-011-0117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu X, Jiang Y, Chen Y, Huang Y, Costello CE, Lin C. Detailed Glycan Structural Characterization by Electronic Excitation Dissociation. Anal Chem. 2013;85:10017–10021. doi: 10.1021/ac402886q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budnik BA, Haselmann KF, Elkin YN, Gorbach VI, Zubarev RA. Applications of electron-ion dissociation reactions for analysis of polycationic chitooligosaccharides in Fourier transform mass spectrometry. Anal Chem. 2003;75:5994–6001. doi: 10.1021/ac034477f. [DOI] [PubMed] [Google Scholar]
- 28.Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ. Electron-Induced Dissociation of Glycosaminoglycan Tetrasaccharides. J Am Soc Mass Spectrom. 2008;19:1449–1458. doi: 10.1016/j.jasms.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolff JJ, Amster IJ, Chi L, Linhardt RJ. Electron Detachment Dissociation of Glycosaminoglycan Tetrasaccharides. J Am Soc Mass Spectrom. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adamson JT, Håkansson K. Electron Detachment Dissociation of Neutral and Sialylated Oligosaccharides. J Am Soc Mass Spectrom. 2007;18:2162–2172. doi: 10.1016/j.jasms.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Wolff JJ, Leach FE, Laremore TN, Kaplan DA, Easterling ML, Linhardt RJ, Amster IJ. Negative Electron Transfer Dissociation of Glycosaminoglycans. Anal Chem. 2010;82:3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 33.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron Capture Dissociation of Gaseous Multiply-Charged Proteins Is Favored at Disulfide Bonds and Other Sites of High Hydrogen Atom Affinity. J Am Chem Soc. 1999;121:2857–2862. [Google Scholar]
- 34.Zubarev RA, Haselmann KF, Budnik B, Kjeldsen F, Jensen F. Towards an understanding of the mechanism of electron-capture dissociation: a historical perspective and modern ideas. Eur J Mass Spectrom. 2002;8:337–349. [Google Scholar]
- 35.Breuker K, Oh H, Lin C, Carpenter BK, McLafferty FW. Nonergodic and conformational control of the electron capture dissociation of protein cations. Proc Natl Acad Sci U S A. 2004;101:14011–14016. doi: 10.1073/pnas.0406095101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Syrstad EA, Turecek F. Toward a general mechanism of electron capture dissociation. J Am Soc Mass Spectrom. 2005;16:208–224. doi: 10.1016/j.jasms.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Sobczyk M, Anusiewicz I, Berdys-Kochanska J, Sawicka A, Skurski P, Simons J. Coulomb-Assisted Dissociative Electron Attachment: Application to a Model Peptide. J Phys Chem A. 2004;109:250–258. doi: 10.1021/jp0463114. [DOI] [PubMed] [Google Scholar]
- 38.Simons J. Mechanisms for S S and N Cα bond cleavage in peptide ECD and ETD mass spectrometry. Chem Phys Lett. 2010;484:81–95. [Google Scholar]
- 39.NCα F. Bond Dissociation Energies and Kinetics in Amide and Peptide Radicals. Is the Dissociation a Non-ergodic Process? J Am Chem Soc. 2003;125:5954–5963. doi: 10.1021/ja021323t. [DOI] [PubMed] [Google Scholar]
- 40.Chamot-Rooke J, Malosse C, Frison G, Tureček F. Electron Capture in Charge-Tagged Peptides. Evidence for the Role of Excited Electronic States. J Am Soc Mass Spectrom. 2007;18:2146–2161. doi: 10.1016/j.jasms.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Tureček F, Julian RR. Peptide Radicals and Cation Radicals in the Gas Phase. Chem Rev. 2013;113:6691–6733. doi: 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Håkansson K. Divalent Metal Ion-Peptide Interactions Probed by Electron Capture Dissociation of Trications. J Am Soc Mass Spectrom. 2006;17:1731–1741. doi: 10.1016/j.jasms.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 43.Kleinnijenhuis AJ, Mihalca R, Heeren RMA, Heck AJR. Atypical behavior in the electron capture induced dissociation of biologically relevant transition metal ion complexes of the peptide hormone oxytocin. Int J Mass spectrom. 2006;253:217–224. [Google Scholar]
- 44.Fung YME, Liu H, Chan TWD. Electron Capture Dissociation of Peptides Metalated with Alkaline-Earth Metal Ions. J Am Soc Mass Spectrom. 2006;17:757–771. doi: 10.1016/j.jasms.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 45.Chen X, Fung Y, Chan W, Wong P, Yeung H, Chan TWD. Transition Metal Ions: Charge Carriers that Mediate the Electron Capture Dissociation Pathways of Peptides. J Am Soc Mass Spectrom. 2011;22:2232–2245. doi: 10.1007/s13361-011-0246-1. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Chan W, Wong P, Yeung H, Chan T. Formation of Peptide Radical Cations (M+·) in Electron Capture Dissociation of Peptides Adducted with Group IIB Metal Ions. J Am Soc Mass Spectrom. 2011;22:233–244. doi: 10.1007/s13361-010-0035-2. [DOI] [PubMed] [Google Scholar]
- 47.Flick T, Donald W, Williams E. Electron Capture Dissociation of Trivalent Metal Ion-Peptide Complexes. J Am Soc Mass Spectrom. 2013;24:193–201. doi: 10.1007/s13361-012-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koster C, Holle A. A new intelligent annotation procedure: SNAP. ASMS annual conference; Dallas, TX. 1999. [Google Scholar]
- 49.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 51.Becke AD. Density-functional thermochemistry. III The role of exact exchange. The Journal of Chemical Physics. 1993;98:5648–5652. [Google Scholar]
- 52.Stephens P, Devlin F, Chabalowski C, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 53.Scott AP, Radom L. Harmonic Vibrational Frequencies: An Evaluation of Hartree–Fock, Moller–Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J Phys Chem. 1996;100:16502–16513. [Google Scholar]
- 54.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JAJ, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 55.Domon B, Costello CE. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms Ms Spectra of Glycoconjugates. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 56.Weimar T, Kreis UC, Andrews JS, Pinto BM. Conformational analysis of maltoside heteroanalogues using high-quality NOE data and molecular mechanics calculations. Flexibility as a function of the interglycosidic chalcogen atom. Carbohydr Res. 1999;315:222–233. [Google Scholar]
- 57.Mendonca S, Johnson GP, French AD, Laine RA. Conformational Analyses of Native and Permethylated Disaccharides. J Phys Chem A. 2002;106:4115–4124. [Google Scholar]
- 58.da Silva CO, Nascimento MAC. Ab initio conformational maps for disaccharides in gas phase and aqueous solution. Carbohydr Res. 2004;339:113–122. doi: 10.1016/j.carres.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 59.Woods RJ, Dwek RA, Edge CJ, Fraser-Reid B. Molecular Mechanical and Molecular Dynamic Simulations of Glycoproteins and Oligosaccharides. 1 GLYCAM_93 Parameter Development. J Phys Chem. 1995;99:3832–3846. [Google Scholar]
- 60.Zheng YJ, Ornstein RL, Leary JA. A density functional theory investigation of metal ion binding sites in monosaccharides. J Mol Struc Theochem. 1997;389:233–240. [Google Scholar]
- 61.Spengler B, Dolce JW, Cotter RJ. Infrared laser desorption mass spectrometry of oligosaccharides: fragmentation mechanisms and isomer analysis. Anal Chem. 1990;62:1731–1737. [Google Scholar]
- 62.Cooper HJ, Hudgins RR, Hakansson K, Marshall AG. Secondary fragmentation of linear peptides in electron capture dissociation. Int J Mass spectrom. 2003;228:723–728. [Google Scholar]
- 63.Leymarie N, Costello CE, O’Connor PB. Electron capture dissociation initiates a free radical reaction cascade. J Am Chem Soc. 2003;125:8949–8958. doi: 10.1021/ja028831n. [DOI] [PubMed] [Google Scholar]
- 64.O’Connor PB, Lin C, Cournoyer JJ, Pittman JL, Belyayev M, Budnik BA. Long-lived electron capture dissociation product ions experience radical migration via hydrogen abstraction. J Am Soc Mass Spectrom. 2006;17:576–585. doi: 10.1016/j.jasms.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 65.Savitski MM, Kjeldsen F, Nielsen ML, Zubarev RA. Hydrogen rearrangement to and from radical z fragments in electron capture dissociation of peptides. J Am Soc Mass Spectrom. 2007;18:113–120. doi: 10.1016/j.jasms.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 66.Kjeldsen F, Haselmann KF, Budnik BA, Jensen F, Zubarev RA. Dissociative capture of hot (3–13 eV) electrons by polypeptide polycations: an efficient process accompanied by secondary fragmentation. Chem Phys Lett. 2002;356:201–206. [Google Scholar]
- 67.Tsybin YO, He H, Emmett MR, Hendrickson CL, Marshall AG. Ion Activation in Electron Capture Dissociation To Distinguish between N-Terminal and C-Terminal Product Ions. Anal Chem. 2007;79:7596–7602. doi: 10.1021/ac071165u. [DOI] [PubMed] [Google Scholar]
- 68.Li X, Lin C, Han L, Costello CE, O’Connor PB. Charge Remote Fragmentation in Electron Capture and Electron Transfer Dissociation. J Am Soc Mass Spectrom. 2010;21:646–656. doi: 10.1016/j.jasms.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsybin YO, Ramström M, Witt M, Baykut G, Håkansson P. Peptide and protein characterization by high-rate electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry. J Mass Spectrom. 2004;39:719–729. doi: 10.1002/jms.658. [DOI] [PubMed] [Google Scholar]
- 70.Chalkley RJ, Brinkworth CS, Burlingame AL. Side-Chain Fragmentation of Alkylated Cysteine Residues in Electron Capture Dissociation Mass Spectrometry. J Am Soc Mass Spectrom. 2006;17:1271–1274. doi: 10.1016/j.jasms.2006.05.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.