

Abstract
A method for structural elucidation of biomolecules dating to the 1980s utilized high-energy collisions (~10 keV, laboratory frame) that induced charge-remote fragmentations (CRF), a class of fragmentations particularly informative for lipids, steroids, surfactants, and peptides. Unfortunately, the capability for high-energy activation has largely disappeared with the demise of magnetic sector instruments. With the latest designs of tandem time-of-flight mass spectrometers (TOF/TOF), however, this capability is now being restored to coincide with the renewed interest in metabolites and lipids including steroid-sulfates and other steroid metabolites. For these metabolites, structure determinations are required at concentration levels below that appropriate for NMR. To meet this need, we explored CRF with TOF/TOF mass spectrometry for two groups of steroid sulfates, 3-sulfates and 21-sulfates. We demonstrated that the current generation of MALDI TOF/TOF instruments can generate charge-remote-fragmentations for these materials. The resulting collision-induced dissociation (CID) spectra are useful for positional isomer differentiation and very often allow the complete structure determination of the steroid. We also propose a new nomenclature that directly indicates the cleavage sites on the steroid ring with carbon numbers.
Introduction
Steroids are among the main regulators in mammalian physiology [1], and as a result, their metabolites are attractive candidates for conveying many aspects of an animal’s status. Steroid sulfates were first recognized as naturally occurring metabolites in 1938 when Schachter and Marrian [2] isolated estrone sulfate from the urine of pregnant mares. Since then, many more steroid sulfates have been isolated from different biological sources and identified. Until the 1960s, they were considered as biologically inactive end-products of metabolism appropriate for elimination [3]. Many efforts have been made since then to search for the physiological role for steroid sulfates, and these efforts revealed the significance of sulfated steroids for structural, signaling and regulatory functions [4–8]. That they are biologically active is made clear by the commonly used drug Premarin, which is a complex mixture of steroid sulfates. Recently, with the emergence of lipidomics and metabolomics, the interest in steroid conjugate analysis is again growing. An example is the work of one of the authors (T. Holy), who found steroid sulfates of the glucocorticoid family as “social odors” or “pheromones” in mouse urine [9, 10]. Given the low levels of these materials in mammalian fluids (at sub-micro molar concentration in mouse urine [9]), we need improved methods for structure determinations of steroid sulfates to deal with materials at concentrations below the levels at which NMR can be used.
In early studies of analysis of steroids and their conjugates, GC/MS with electron ionization (EI) was the dominant approach [11–13]. Although EI affords rich fragmentation, its utility is limited because most steroids require derivatization to form volatile, thermally stable analytes [14–17], sometimes compromising sensitivity. In the 1980s, the introduction of fast atom bombardment (FAB) [18] and tandem mass spectrometry (MS/MS) [19, 20], afforded a new approach for structural characterizations of steroid conjugates with mass spectrometry, but the approach required magnetic-sector instruments and high collision energies to generate charge-remote fragmentation. The concept of charge-remote (originally called remote-site fragmentation) was first described in 1983 as observed on a unique three-sector mass spectrometer [21, 22]. This instrumentation was later used to demonstrate the charge-remote fragmentations of a series of bile acids and bile salt conjugates [23] and other steroids [24, 25]. Tomer and Gross [26] then reported in 1988 that both charge-remote and charge-proximate fragmentations occur for bile salts, steroid sulfates, and glucuronides, showing that nearly complete structure determination of steroid rings could be achieved. Unfortunately, there has been a marked decline in the application of high-energy CID with the demise of sector instruments and the current dominance of ion traps, QTOFs, and hybrid ion-trap FT spectrometers that employ low-energy CID. Although the latter has been applied over years in structural characterization of organic compounds, including steroid sulfates [10, 27], the structural elucidation is complicated and incomplete because the major fragment ions usually result from losses of small molecules, and MS3 is often required to get a more complete set of fragment ions. Furthermore, low-energy CID spectra tend to show more rearrangement reactions and are not as reproducible as high-energy spectra. Because low-energy CID strongly depends on instrument characteristics and settings, the prospects of building an MS2 database for structure searches has become more complex [28].
The introduction of TOF/TOF instruments, specifically designed for high-energy CID [29, 30], has triggered a “renaissance” of interest in high-energy fragmentation processes. MALDI coupled to an appropriate TOF/TOF-instrument can accommodate the need for sufficient mass resolving power, precursor-ion selection, and high collision energies for the structural analysis of biomolecules [31–34]. The first evidence comes from Trimpin et al. [31] who showed that metal-cationized fatty acids desorbed from a solvent-free preparation undergo CRF in a TOF/TOF. Additional evidence is from Allmeier and Pittenauer [33, 34] who induced CRF of trialkylglycerols (TAGs). Other applications of MALDI TOF/TOF were also reported for carbohydrates and peptides [35, 36]. Whereas fatty acids have been investigated by MALDI TOF/TOF and their CID spectra are well understood, steroids and their conjugates have been far less investigated [37]. Here, we demonstrate, for the first time to our knowledge, the application of MALDI TOF/TOF instrumentation to induce CRF and enable structural characterization of sulfated steroids. The results are comparable to those obtained on the older tandem magnetic sector instruments and strongly suggest that TOF/TOF instruments offer a new opportunity for lipid and steroid structure-determination studies and for lipidomics in general.
Experiment
Materials
All steroids and sulfated steroids, cholesteryl sulfate, 3β-hydroxy-5-pregnen-20-one-3-sulfate (termed SS395, for sulfated steroid 395, the number of which is the molecular mass of [M - H]−), etiocholan-3α-ol-11,17-dione sulfate (SS383), 4-pregnen-11β,21-diol-3,20-dione-21-sulfate (SS425), 4- pregnen-11β, 17,21-triol-3,20-dione-21-sulfate (SS441), 4-pregnen-17,21-diol-3,11,20-trione-21-sulfate (SS439), 5β-pregnan-11β,21-diol-3,20-dione, and 4-pregnen-11β, 20β,21-triol-3-one were purchased from Steraloids Inc (Newport, RI), and dissolved in methanol to give a 5 mM stock solution. SS427 and iSS427 were prepared by sulfating 5β-pregnan-11β, 21-diol-3,20-dione and 4-pregnen-11β,20β,21-triol-3-one as described previously [38]. The structures of all steroid sulfates used are listed in Figure 1. All other chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO).
Figure 1.

Structures of steroid sulfates investigated here.
Mass Spectrometry
A MALDI-TOF/TOF mass spectrometer (SpiralTOF™, JEOL Ltd., Tokyo, Japan) was used for all measurements. This MALDI-TOF/TOF mass spectrometer has unique multi-turn spiral ion optics with a 17 m long flight path to provide ultrahigh mass resolving power for MALDI-TOF measurements [39–41]. An ion gate positioned at the 15 m point in the spiral ion flight path provided mono-isotopic precursor-ion isolation for tandem mass spectrometry upon high-energy CID [42, 43]. Selected monoisotopic precursor ions undergo 20 kV collisions with the helium collision gas at a pressure of 1.7×10−4 Pa. Ions exiting the collision chamber underwent 9 kV post-acceleration and focusing to ensure efficient detection of low m/z fragments.
A 349 nm Nd-YLF laser operated at a laser repetition rate of 1 kHz was used to desorb and ionize the samples. The extraction delay was optimized to 50 nsec. α-Cyano-4-hydroxycinnamic acid (CHCA) matrix was dissolved in 1:1 water/acetonitrile containing 0.1% trifluoroacetic acid at a concentration of 10 mg/mL (Trifluoroacetic acid, normally removed for the negative-ion mode detection, caused the signal intensity to decrease by two fold compared to its absence. It was utilized here to show its compatibility with negative-ion mode detection and to keep open the possibility for adoption when analyzing a biological sample mixture in both the positive- and negative-ion modes.). The steroid sulfate and matrix solutions were mixed together 1:1 by volume. A 1.0 μL aliquot of this mixture was deposited onto the MALDI target plate for analysis with the MALDI-TOF/TOF mass spectrometer.
Results and Discussion
We used MALDI TOF/TOF to investigate the fragmentation of two groups of steroid sulfates, namely 3-sulfates and 21-sulfates. These two groups were chosen because hydroxyl sites on neutral steroids suitable for sulfonation by steroid sulfotransferase are commonly present at carbons 3 and 21 (or 17) for C-21 steroids (or C-19 steroids). Furthermore, they have the sulfate group fixed at either end of the steroid [44], allowing us to demonstrate the charge-remote nature of the fragmentation. The fixed charge at one end of the precursor ion after ionization should allow CRF to be induced remotely as the predominant reaction channel under high-energy CID for both 3-sulfates and 21-sulfates. With the fixed sulfate group, these steroid sulfates can be readily desorbed and ionized by MALDI as [M–H]− species (Figure 2a shows one example of the negative-ion mass spectrum of SS441). With the SpiralTOF, accurate mass measurements are achieved in MS1 scans, and monoisotopic precursor-ion selection is made possible for fragmentation (Fig 2b), allowing straightforward interpretation of the product-ion spectra (TOF/TOF). The major fragmentations for members of each subclass are similar and often involve through-ring cleavages, as is described below.
Figure 2.

Monoisotopic precursor ion selection of SS441 for TOF/TOF measurements in Spiral TOF.
Fragmentation nomenclature
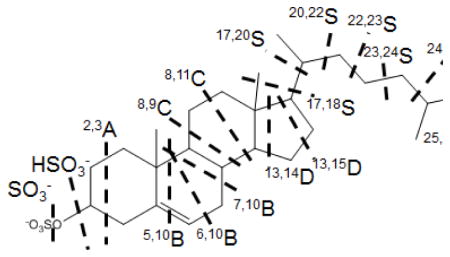
Given that this fragmentation by through-ring cleavages may be general, we provide a new nomenclature designating the steroid ring cleavages. Shown for cholesteryl sulfate (Table 1) is the nomenclature, where each cleavage is named for the ring in which it occurs and the positions of two carbon atoms constituting the two bonds that must be broken to cause a through-ring cleavage (the designated carbon atoms are to the left of the cleavage, and ring A is on the left, ring D on the right, as per convention). For example, a cleavage that breaks ring B at C9-C10 and C7-C8 is designated as 7,10B. In addition to major ring cleavages, high-mass ions are formed by cleavages of the side chain attached to ring D, remote from the charge site (Figure 3). Cleavages occurring at the side chain are named by using the two carbon positions at the C-C bond being cleaved. For example, the cleavage at bond C20-C22 is named as 20,22S (“S” refers to the side-chain).
Table 1.
Major fragment ions from high-energy CID of [M - H]− ions of 3-sulfates.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Steroid sulfate | SO3− HSO3− | 2,3A | 5,10B | 6,10B | 7,10B | 8,9C | 8,11C | 13,14D | 13,15D | 17,18S |
| Cholesteryl sulfate | 80,97 | 123 | N/A | N/A | 217 | 243 | 257 | 311 | 325 | 337 |
| SS395 | 80,97 | 123 | N/A | N/A | 217 | 243 | 257 | 311 | N/A | 337 |
| SS383 | 80,97 | 123 | 191 | 204 | N/A | 245 | N/A | 327 | 341 | N/A |
Figure 3.

High-energy CID fragmentation in the negative-ion mode of 3-sulfates: a. cholesteryl sulfate, b. SS395, c. SS383. The highly broadened [M–H]− signals indicate this precursor is off scale presumably because the product-ion yield is low and dispersed over many fragments.
3-Sulfates
We investigated three steroids substituted with sulfate at the 3-position (A ring) by MALDI TOF/TOF and obtained highly informative MS/MS data, with rich fragmentation occurring through the steroid ring (Table 1). As indicated in the product-ion spectra (Table 1 and Figure 3), 3-sulfates undergo predominantly charge-remote fragmentation upon high-energy collisions, providing information about the nature of the substituent and its location, especially if the substituent is an alkyl chain. Taking cholesteryl sulfate as an example, we find rich fragmentation of the ring structure and the side chain at C17 (Figure 3a) to give a detailed “footprint” of the ring junctions and the side-chain branching of this compound. For example, the mass difference for the B-ring cleavage, 7,10B, and the C ring cleavage, 8,9C, is 26 Da, which corresponds to C2H2 and shows that the ring junction is unsubstituted. Comparing the three 3-sulfates, we find that processes 2,3A, 8,9C, and 13,14D occur for the three compounds, and the mass differences between 2,3A and 8,9C are 120 Da (C9H12) for cholesteryl sulfate and SS395 whereas for SS383, the difference is 122 Da (C9H14), consistent with the absence of a double bond in the latter. Similarly, the differences between 8,9C and 13,14D are consistent with the substitution patterns (Table 1 and Figure 3). For cholesteryl sulfate and SS383, the differences between 13,14D and 13,15D are 14 Da, as expected whereas for SS395, the 13,15D fragment is missing, and replaced by an ion m/z 323 formed by loss of C4H8O by an unknown mechanism. On the contrary, the appearance of product ions from cleavages across the B ring are significantly different for SS383 and the other two, mainly because the C5-C6 bond is a single bond for SS383 and a double bond for the other two (Figure 3a and 3b). A fragment ion is formed by the nearby cleavage, 7,10B, of both cholesteryl sulfate and SS395, although this fragment is missing in the product-ion spectrum of SS383 (Figure 3c). Besides B-ring cleavages, cleavages of the C ring show the expected differences among the three compounds. For example, 8,11C is absent in SS383 fragmentation, consistent with the presence of the C11 keto group that prevents this cleavage (Table 1), and the differences between 8,9C and 8,11C for cholesteryl sulfate and SS395 is 14 Da, consistent with a CH2 group at position 11. This detailed analysis of ring cleavages was not presented in the early papers on CRF of steroids, and the prospects for a nearly complete structural analysis were not understood.
Importantly, branching sites on the side chain are easily determined by examining the pattern for CnH2n+2 eliminations, because cleavages happening at either side of a branch point are more facile than those occurring via double cleavages at a branch point.
In addition to the common fragment ions due to ring or side-chain cleavage, some unexpected fragments are also formed; these are consistent with our early findings on CRF of steroid sulfates [26]. For example, besides the losses of neutral molecules in which the C17 substituent is cleaved with H-transfer, cleavages also occur to eliminate directly a CH3 radical, presumably forming a distonic ion, rather than H transfer and elimination of neutral molecules (this cleavage is termed 17,18S, as shown in Table 1). The mechanism for this cleavage was previously proposed to occur through a six-member transition state (Scheme 1 in ref 26). Although obtaining detailed mechanistic data is not the purpose of this paper and must await future work, we show the fragmentation for clarity. Another example is the fragmentation of the three 3-sulfates to undergo a CH4 loss upon CID. This process most likely arises from the loss of an angular methyl group (C18 or C19) with H rearrangement and indicates the presence of methyl substituents.
21-Sulfates
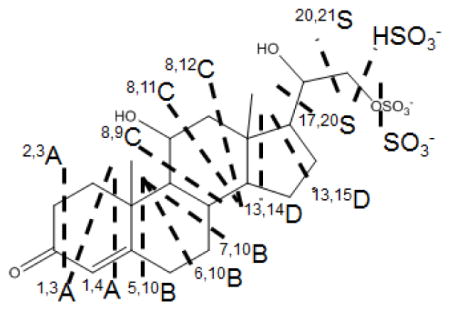
We investigated five different compounds with different substitutions on the ring or the side chain, as listed in Table 2. Similar to 3-sulfates, 21-sulfates give rich charge-remote fragmentation as cross-ring cleavages and of the side chain upon high-energy CID (Figure 4). Ions of m/z 80 and 97 are also abundant in product-ion spectra of 21-sulfates, representing SO3− and HSO4− from releases of small, charged sulfur-containing groups. Although we adopted the same nomenclature for all the cleavages of 21-sulfates as for the 3-sulfates, the designations for 21-sulfates are of portions of the steroid that are lost as neutrals whereas for the 3-sulfates, they are part of the fragment ion. The scheme in Table 2 shows an example of the cleavages of SS427.
Table 2.
Major fragment ions from high-energy CID of [M - H]− ions of 21-sulfates.
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Steroid sulfate | 2,3A | 1,3A | 1,4A | 5,10B | 6,10B | 7,10B | 8,9C | 8,11C | 8,12C | 13,14D | 13,15D | 17,20S | 20,21S |
| iSS427 | 383 | 371 | N/A | 317 | 303 | 291 | 263 | 235 | 221 | 180 | 167 | 139 | 110 |
| SS427 | 383 | 371 | 357 | 315 | 301 | 288 | 261 | 233 | 219 | 178 | 165 | N/A | 110 |
| SS425 | 381 | 369 | N/A | 315 | 301 | 289 | 261 | 233 | 219 | 178 | 165 | N/A | 110 |
| SS441 | 381 | 367 | N/A | 331 | 317 | 305 | N/A | N/A | 235 | N/A | N/A | 139 | 110 |
| SS439 | 395 | 383 | N/A | 329 | N/A | 303 | N/A | N/A | 235 | N/A | 181 | 139 | 110 |
Note:
All 21-sulfates produce fragment ions at m/z 80 and 97, representing SO3− and HSO4− respectively.
2,3A and 1,3A product ions from SS441 is combined with a consecutive lost of H2O, which is revealed in m/z.
Figure 4.

High-energy CID fragmentation in the negative-ion mode of 21-sulfates: a. iSS427, b. SS427 c, SS425 d, SS441, and e. SS439. The highly broadened [M–H]− signals indicate this precursor is off scale presumably because the product-ion yield is low and dispersed over many fragments.
The CID spectra of [M - H]− ions can be used to delineate clearly the locations and type of different substitutions. For example, the positional isomers, iSS427 and SS427 differ in the position of a double bond; iSS427 has the double bond between C4 and C5, whereas SS427 has the unsaturation located at C20 as a keto group. As a result, their product-ion spectra (Figure 4a and 4b) show that ions formed by 1,4A cleavage are among the most abundant fragments formed from SS427 but undetectable for iSS427. The same types of fragmentations involving rings B, C, and D occur for both compounds, but they show a consistent mass shift of 2 Da. In addition, the C20 keto groups in SS427 and in SS425 (Figure 4b and 4c) prevent the 17,20S cleavage, but replacement of the keto group with C20-OH in iSS427 promotes this fragmentation and gives the ion of m/z 139 (Figure 4a). An m/z 139 ion is observed, however, although less abundant, in the product-ion spectra of SS441 and SS439 (Figure 4d and 4e), indicating that the effect of a C20 keto group on 17,20S is eased when substituted by a hydroxyl group at C17 (the only difference between SS441 and SS425). Although SS441 differs from SS425 only by the C17 hydroxyl group, it undergoes fewer cleavages of the C and D rings, suggesting that a C17 hydroxyl group suppresses cleavages of the C and D ring. Likewise, SS439 also undergoes nearly no cleavages across the C and D ring under CID.
Expulsions of neutral molecules, CH4 or CO, are dominant reactions for most 21-sulfates being studied. The loss of CH4 is similar to that in 3-sulfates, which likely involves an angular methyl group. Loss of CO is also a charge-remote cleavage on the A ring of 21-sulfates, which occurs similarly at the D ring of SS383, a 3-sulfate steroid. The other two 3-sulfates undergo no such fragmentation owing to the lack of keto groups on the ring system. Besides losses of CH4 and CO, SS441 also gives abundant ions by the loss of H2O, which are followed by other consecutive fragmentations such as 2,3A and 1,3A, as indicated in Table 2.
We wish to note that the cleavages are not always as simple as indicated by the dashed lines. Sometimes the cleavage occurs as designated (e.g., 1,3A, 1,4A, 7,10B, 8,11C, 8,12C, and 13,15D) whereas other cleavages occur as designated but are accompanied by additional H transfer(s). Consider, for example, 2,3A. A simple through-ring cleavage would give loss of C2H2O, but the actual fragmentation is to lose C2H4O. The mechanistic reasons for these differences are interesting and remain to be elucidated. Nevertheless, structural assignments can still be made in the absence of a complete understanding of the ion chemistry.
Approximate Detection Limits
In addition to studying the effectiveness of MS/MS fragmentation for structure elucidation, we evaluated the detection limit of the 3- and 21-sulfates on this MALDI TOF/TOF instrument, requiring sufficient fragmentation information for structure characterization. We took one example from each group of steroids, cholesteryl sulfate and SS425, and evaluated the signal-to-noise ratio (S/N) of the base peaks in their product-ion spectra when different amounts of sample were applied to the MALDI plate. As shown in Table 3, S/N of the base peak at m/z 311 in the product-ion mass spectrum of cholesteryl sulfate decreased from 200 to 80 when the applied sample amount is reduced from 25 pmol to 4 pmol. Further reduction of the sample applied to 1 pmol caused the S/N for detecting the ion of m/z 331 to decrease to 20. Although the intensity for the base peak detection is still acceptable with 1 pmol of sample application, some of lower-abundance product ions disappeared below the S/N at this condition (e.g., the ion of m/z 217), making a structure assignment incomplete. Similarly, for SS425, S/N of the base peak at m/z 219 decreased from 500 to 60 in the product-ion spectrum when the sample applied was decreased from 25 to 1 pmol; further, the lower abundance fragment ions began to disappear with 1 pmol of sample application. Although the S/N tracks only approximately the applied sample quantity, we can conclude that the approximate detection limit sufficient for structure assignment of steroid sulfates is low pmol levels, with some variations for different samples. This low sample-size requirement demonstrates the applicability of MALDI TOF/TOF for analyzing biological samples where only trace amounts can be isolated (e.g., SS425 was shown to exist in mouse urine at micro-molar concentrations in a previous study [9]).
Table 3.
Signal-to-noise ratios of base peaks in the product-ion spectra of cholesteryl sulfate and SS425 as a function of the amount of sample applied
| Steroid sulfate | Cholestery sulfate | SS425 | ||||||
|---|---|---|---|---|---|---|---|---|
| Base peak (m/z) | 311 | 219 | ||||||
| Applied amount (pmol) | 25 | 10 | 4 | 1 | 25 | 10 | 4 | 1 |
| S/N | 200 | 100 | 80 | 20 | 500 | 400 | 200 | 60 |
Conclusion
The effectiveness of charge-remote-fragmentation for primary structure identification of lipids and steroids has been known for some time. The approach can determine locations of double bonds, identify various functional groups, locate branching and ring junctions directly from product-ion spectra. Instrumentation and ionization methods traditionally used for CRF, however, have become rare, diminishing the importance of CRF in structural studies. It continues to be recognized and invoked, however, as one class of fragmentation, a “thermal-like” process that occurs with little or no involvement of a charge or radical site. Low-energy MS/MS fragmentation of steroid sulfates [38] produce fewer cross-ring cleavages and is less informative than the spectra from high-energy activation, which induces extensive fragmentation across the steroid ring at the ring junctures.
The ability to induce CRF coupled with the advantage of monoisotopic precursor-ion selection afforded by new TOF/TOF instruments, permits relatively straight-forward interpretation of product-ion spectra because all product ions are monoisotopic. These characteristic ions formed under high-energy CID are nearly identical to those observed many years ago on tandem sector instruments when the sample was introduced by FAB ionization. Cleavages through the steroid rings occur whether the steroid is a 3- or a 21-sulfate, consistent with the classification that the processes are “charge-remote” as would be expected because the intervening rigid steroid ring makes it impossible for the charge site to interact with the reaction site. Although the earlier measurements on steroids proved that the fragmentation is indeed charge-remote and showed the nature of the remote side chain, this work also shows that there is more information in these spectra (e.g., on ring junctions and substituent locations).
In spite of the commonality of ring cleavages, 3-sulfates can be easily differentiated from 21-sulfates by the low m/z product ions. For example, 3-sulfates give a product ion of m/z 123 by 2,3A cleavage, whereas 21-sulfates give product ions of m/z 110 arising from the side-chain cleavage, 20,21S. 3-Sulfates containing alkyl side chains on the D ring can also be differentiated from 21-sulfates, because the former is rich in fragments in the high mass range, owing to cleavages of the side chain, to give product ions that reflect its structure and locate the branch points. Locating functional groups is also straightforward by considering the mass differences between nearby cleavages. An important caveat is that certain substitutions on the ring (e.g., a C11 keto group) can prevent nearby fragmentations. In this case, the absence of cleavages is also informative and should be taken into account when elucidating unknown steroid structures.
We project that an important application of this approach will be in imaging steroids, lipids, and related materials.
Acknowledgments
This research was supported by the National Institute of General Medical Sciences (8 P41 GM103422-35 to MLG) and the National Institute on Deafness and Other Communication Disorders (R01 DC005964 to TEH), both of the National Institutes of Health.
References
- 1.Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones--a focus on rapid, nongenomic effects. Pharmacological reviews. 2000;52:513–556. [PubMed] [Google Scholar]
- 2.Schachter B, Marrian GF. The isolation of estrone sulfate from the urine of pregnant mares. J Biol Chem. 1938;126:663–669. [Google Scholar]
- 3.Wang DY, Bulbrook RD. Steroid sulphates. Advances in Reproductive Physiology. 1968;3:113–146. [Google Scholar]
- 4.Strott CA. Sulfonation and molecular action. Endocrine reviews. 2002;23:703–732. doi: 10.1210/er.2001-0040. [DOI] [PubMed] [Google Scholar]
- 5.Noordam C, Dhir V, McNelis JC, Schlereth F, Hanley NA, Krone N, Smeitink JA, Smeets R, Sweep FC, Claahsen-van der Grinten HL, Arlt W. Inactivating papss2 mutations in a patient with premature pubarche. The New England journal of medicine. 2009;360:2310–2318. doi: 10.1056/NEJMoa0810489. [DOI] [PubMed] [Google Scholar]
- 6.Gibbs TT, Russek SJ, Farb DH. Sulfated steroids as endogenous neuromodulators. Pharmacology, biochemistry, and behavior. 2006;84:555–567. doi: 10.1016/j.pbb.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 7.Strott CA, Higashi Y. Cholesterol sulfate in human physiology: What’s it all about? Journal of lipid research. 2003;44:1268–1278. doi: 10.1194/jlr.R300005-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Hill M, Parizek A, Kancheva R, Jirasek JE. Reduced progesterone metabolites in human late pregnancy. Physiological research / Academia Scientiarum Bohemoslovaca. 2011;60:225–241. doi: 10.33549/physiolres.932077. [DOI] [PubMed] [Google Scholar]
- 9.Nodari F, Hsu FF, Fu XY, Holekamp TF, Kao LF, Turk J, Holy TE. Sulfated steroids as natural ligands of mouse pheromone-sensing neurons. J Neurosci. 2008;28:6407–6418. doi: 10.1523/JNEUROSCI.1425-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu FF, Nodari F, Kao LF, Fu XY, Holekamp TF, Turk J, Holy TE. Structural characterization of sulfated steroids that activate mouse pheromone-sensing neurons. Biochemistry-Us. 2008;47:14009–14019. doi: 10.1021/bi801392j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryhage R, Stenhagen E. Mass spectrometry in lipid research. Journal of lipid research. 1960;1:361–390. [PubMed] [Google Scholar]
- 12.Budzikiewicz H, Djerassi C, Williams DH. Structure elucidation of natural products by mass spectrometry. Vol. Ii. steroids, Terpenoids, Sugars and Miscellaneous classes. 1964. [Google Scholar]
- 13.Sjovall J. Fifty years with bile acids and steroids in health and disease. Lipids. 2004;39:703–722. doi: 10.1007/s11745-004-1288-1. [DOI] [PubMed] [Google Scholar]
- 14.Wudy SA, Hartmann M, Homoki J. Determination of 11-deoxycortisol (reichstein’s compound s) in human plasma by clinical isotope dilution mass spectrometry using benchtop gas chromatography-mass selective detection. Steroids. 2002;67:851–857. doi: 10.1016/s0039-128x(02)00052-1. [DOI] [PubMed] [Google Scholar]
- 15.Reddy S, Iden CR, Brownawell BJ. Analysis of steroid conjugates in sewage influent and effluent by liquid chromatography-tandem mass spectrometry. Analytical chemistry. 2005;77:7032–7038. doi: 10.1021/ac050699x. [DOI] [PubMed] [Google Scholar]
- 16.Bean KA, Henion JD. Direct determination of anabolic steroid conjugates in human urine by combined high-performance liquid chromatography and tandem mass spectrometry. Journal of chromatography. B, Biomedical sciences and applications. 1997;690:65–75. doi: 10.1016/s0378-4347(96)00403-3. [DOI] [PubMed] [Google Scholar]
- 17.Bowers LD, Sanaullah Direct measurement of steroid sulfate and glucuronide conjugates with high-performance liquid chromatography-mass spectrometry. Journal of chromatography B, Biomedical applications. 1996;687:61–68. doi: 10.1016/s0378-4347(96)00232-0. [DOI] [PubMed] [Google Scholar]
- 18.Barber M, Bordoli RS, Sedgwick RD, Tyler AN. Fast atom bombardment of solids (fab) - a new ion-source for mass-spectrometry. J Chem Soc Chem Comm. 1981:325–327. [Google Scholar]
- 19.Cooks RG, Glish GL. Mass-spectrometry mass-spectrometry. Chem Eng News. 1981;59:40–52. [Google Scholar]
- 20.Levsen K, Schwarz H. Gas-phase chemistry of collisionally activated ions. Mass Spectrom Rev. 1983;2:77–148. [Google Scholar]
- 21.Gross ML, Chess EK, Lyon PA, Crow FW, Evans S, Tudge H. Triple analyzer mass-spectrometry for high-resolution ms/ms studies. Int J Mass Spectrom. 1982;42:243–254. [Google Scholar]
- 22.Tomer KB, Crow FW, Gross ML. Location of double-bond position in unsaturated fatty-acids by negative-ion ms/ms. J Am Chem Soc. 1983;105:5487–5488. [Google Scholar]
- 23.Tomer KB, Jensen NJ, Gross ML, Whitney J. Fast atom bombardment combined with tandem mass spectrometry for determination of bile salts and their conjugates. Biomedical & environmental mass spectrometry. 1986;13:265–272. doi: 10.1002/bms.1200130602. [DOI] [PubMed] [Google Scholar]
- 24.Cheng MT, Barbalas MP, Pegues RF, Mclafferty FW. Tandem mass-spectrometry - structural and stereochemical information from steroids. J Am Chem Soc. 1983;105:1510–1513. [Google Scholar]
- 25.Kingston EE, Beynon JH, Newton RP, Liehr JG. The differentiation of isomeric biological compounds using collision-induced dissociation of ions generated by fast atom bombardment. Biomedical mass spectrometry. 1985;12:525–534. doi: 10.1002/bms.1200120915. [DOI] [PubMed] [Google Scholar]
- 26.Tomer KB, Gross ML. Fast atom bombardment and tandem mass spectrometry for structure determination: Remote site fragmentation of steroid conjugates and bile salts. Biomedical & environmental mass spectrometry. 1988;15:89–98. doi: 10.1002/bms.1200150206. [DOI] [PubMed] [Google Scholar]
- 27.Strahm E, Saudan C, Sottas PE, Mangin P, Saugy M. Direct detection and quantification of 19-norandrosterone sulfate in human urine by liquid chromatography-linear ion trap mass spectrometry. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2007;852:491–496. doi: 10.1016/j.jchromb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Werner E, Heilier JF, Ducruix C, Ezan E, Junot C, Tabet JC. Mass spectrometry for the identification of the discriminating signals from metabolomics: Current status and future trends. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2008;871:143–163. doi: 10.1016/j.jchromb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Cotter RJ. High energy collisions on tandem time-of-flight mass spectrometers. Journal of the American Society for Mass Spectrometry. 2013;24:657–674. doi: 10.1007/s13361-012-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornish TJ, Cotter RJ. A curved-field reflectron for improved energy focusing of product ions in time-of-flight mass spectrometry. Rapid communications in mass spectrometry: RCM. 1993;7:1037–1040. doi: 10.1002/rcm.1290071114. [DOI] [PubMed] [Google Scholar]
- 31.Trimpin S, Clemmer DE, McEwen CN. Charge-remote fragmentation of lithiated fatty acids on a tof-tof instrument using matrix-ionization. Journal of the American Society for Mass Spectrometry. 2007;18:1967–1972. doi: 10.1016/j.jasms.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW. Shotgun metabolomics approach for the analysis of negatively charged water-soluble cellular metabolites from mouse heart tissue. Analytical chemistry. 2007;79:6629–6640. doi: 10.1021/ac070843+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pittenauer E, Allmaier G. The renaissance of high-energy cid for structural elucidation of complex lipids: Maldi-tof/rtof-ms of alkali cationized triacylglycerols. Journal of the American Society for Mass Spectrometry. 2009;20:1037–1047. doi: 10.1016/j.jasms.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 34.Pittenauer E, Allmaier G. High-energy collision induced dissociation of biomolecules: Maldi-tof/rtof mass spectrometry in comparison to tandem sector mass spectrometry. Combinatorial chemistry & high throughput screening. 2009;12:137–155. doi: 10.2174/138620709787315436. [DOI] [PubMed] [Google Scholar]
- 35.Narouz MR, Soliman SE, Fridgen TD, Nashed MA, Banoub JH. High-energy collision-induced dissociation tandem mass spectrometry of regioisomeric lactose palmitic acid monoesters using matrix-assisted laser desorption/ionization. Rapid communications in mass spectrometry: RCM. 2014;28:169–177. doi: 10.1002/rcm.6770. [DOI] [PubMed] [Google Scholar]
- 36.Baum F, Fedorova M, Ebner J, Hoffmann R, Pischetsrieder M. Analysis of the endogenous peptide profile of milk: Identification of 248 mainly casein-derived peptides. Journal of proteome research. 2013;12:5447–5462. doi: 10.1021/pr4003273. [DOI] [PubMed] [Google Scholar]
- 37.Hailat I, Helleur RJ. Direct analysis of sterols by derivatization matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and tandem mass spectrometry. Rapid communications in mass spectrometry: RCM. 2014;28:149–158. doi: 10.1002/rcm.6766. [DOI] [PubMed] [Google Scholar]
- 38.Yan Y, Rempel DL, Holy TE, Gross ML. Mass spectrometry combinations for structural characterization of sulfated-steroid metabolites. Journal of the American Society for Mass Spectrometry. 2014 doi: 10.1007/s13361-014-0836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh T, Tsuno H, Iwanaga M, Kammei Y. The design and characteristic features of a new time-of-flight mass spectrometer with a spiral ion trajectory. Journal of The American Society for Mass Spectrometry. 2005;16:1969–1975. doi: 10.1016/j.jasms.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Satoh T, Tsuno H, Iwanaga M, Kammei Y. A new spiral time-of-flight mass spectrometer for high mass analysis. J Mass Spectrom Soc Jpn. 2006;541:11–17. [Google Scholar]
- 41.Satoh T, Sato T, Tamura J. Development of a high-performance maldi-tof mass spectrometer utilizing a spiral ion trajectory. Journal of The American Society for Mass Spectrometry. 2007;18:1318–1323. doi: 10.1016/j.jasms.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Satoh T, Sato T, Kubo A, Tamura J. Tandem time-of-flight mass spectrometer with high precursor ion selectivity employing spiral ion trajectory and improved offset parabolic reflectron. Journal of The American Society for Mass Spectrometry. 2011;22:797–803. doi: 10.1007/s13361-011-0090-3. [DOI] [PubMed] [Google Scholar]
- 43.Kubo A, Satoh T, Itoh Y, Hashimoto M, Tamura J, Cody R. Structural analysis of triacylglycerols by using a maldi-ToF/ToF system with monoisotopic precursor selection. Journal of The American Society for Mass Spectrometry. 2013;24:684–689. doi: 10.1007/s13361-012-0513-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strott CA. Steroid sulfotransferases. Endocrine reviews. 1996;17:670–697. doi: 10.1210/edrv-17-6-670. [DOI] [PubMed] [Google Scholar]