

Abstract
Purpose
To investigate the potential role for CD44+ and CD90+ hepatocellular carcinoma (HCC) cellular subpopulations in biological response to thermal ablation-induced heat stress.
Methods
This study was approved by the institutional animal care committee. The N1S1 rat HCC cell line was subjected to sublethal heat stress (45 °C) or control (37 °C) for 10 min, costained with fluorescent-labeled antibodies against CD44, CD90, and 7-AAD after a 48-h recovery and analyzed by flow cytometry to assess the percentage of live CD44+ and CD90+ HCC cells (n = 4). Experiments were repeated with pretreatment of N1S1 cells with a dose titration of the dual PI3K-mTOR inhibitor BEZ235 or vehicle control (n = 3). Rats bearing orthotopic N1S1 tumors were subjected to ultrasound-guided partial laser ablation (n = 5) or sham ablation (n = 3), euthanized 24 h after ablation, and liver/tumor analyzed for immunohisto-chemical staining of CD44 and CD90. Differences between groups were compared with an unpaired t test.
Results
Sublethal heat stress induced a significant increase in the relative proportion of live CD44+ and CD90+ HCC cells compared to the control group: CD44+ CD90− (5.3-fold; p = 0.0001), CD44−CD90+ (2.4-fold; p = 0.003), and CD44+ CD90+ (22.0-fold; p < 0.03). Inhibition of PI3K-mTOR prevented heat stress-induced enrichment of the population of live CD44+ HCC cells (p < 0.01), but not CD90+ cells (p > 0.10). Immunohistochemical analysis demonstrated preferential localization of clusters of CD44+ cells at both the tumor margin and ablation margin.
Conclusion
These studies provide experimental evidence supporting a role for HCC cells expressing the putative stem cell marker CD44 in HCC response to heat stress.
Keywords: Hepatocellular carcinoma, Cancer stem cell, Thermal ablation, CD44
Introduction
Stem cells are present in many tissues and play an important role in normal tissue physiology and homeostasis [1]. Although stem cells play a key role in normal tissue homeostasis, studies over the past 30 years have demonstrated that stem cell-like cells may play a key role in oncogenesis [2, 3]. Relatively rare subpopulations of malignant cells with stem cell-like features have been isolated from both hematopoietic and solid organ malignancies, resulting in development of the cancer stem cell (CSC) hypothesis [1, 3, 4]. The CSC hypothesis holds that mutations in normal stem or progenitor cells give rise to CSC with the capacity to initiate and sustain tumor growth; they have been implicated in tumor metastasis and resistance to anticancer therapies [1, 4–6]. For example, it is hypothesized that tumor recurrence after initial treatment response with cytotoxic chemotherapy results from preferential killing of differentiated cancer cells while residual CSC can repopulate the tumor [1]. As such, efforts are underway to better understand the cytoprotective mechanisms of CSC in order to identify therapeutic targets for enhancing the killing of CSC with anticancer treatments [7, 8].
In hepatocellular carcinoma (HCC), subpopulations of HCC cells that have stem cell-like properties have been isolated from both primary tumors and immortalized cell lines by their differential expression of cell surface cluster differentiation (CD) markers [9]. Currently recognized putative HCC CSC markers include CD13, CD44, CD90 (Thy-1), CD133, and CD326 (EpCAM) [9–19]. CSC from various malignancies including HCC have been shown to be regulated by diverse oncogenic pathways such as the PI3K-AKT-mTOR signaling, a critical cell survival mechanism [20–32]. In HCC, a previous study demonstrated that HCC CSC were more chemoresistant to doxorubicin as a result of up-regulation of AKT-mediated survival signaling [13].
Percutaneous thermal ablative therapies have greatly expanded treatment options for nontransplantable HCC patients while achieving short-term outcomes similar to surgical resection with less morbidity [33–36]. However, as tumor size increases, thermal ablation of HCC has increasing rates of local recurrence and tumor progression, particularly for tumors beyond 3 cm in size, and overall survival remains poor for these patients [34, 37–40]. As such, there remains a need to identify potential biological mechanisms mediating HCC response to thermal ablation. However, it is not known whether putative CSC may play a role in HCC response to heat stress from thermal ablative therapies. The aim of the present study was to investigate the potential role for CD44+ and CD90+ HCC cellular subpopulations in biological response to thermal ablation induced heat stress
Methods
Cell Lines
The N1S1 (ATCC, Manassas, VA) and AS30D (DSMZ, Braunschweig, Germany) rat HCC cell lines were cultured according to supplier recommendations.
Materials
Reagents
NVP-BEZ235 was purchased from Selleck Chemicals; 7-AAD (#559925) was purchased from BD Biosciences.
Antibodies
For flow cytometry, PE Mouse Anti-CD13 (#560998), FITC Mouse Anti-Rat CD44H (#550974), APC Mouse Anti-Rat CD90 (#561409), PE Anti-EpCAM, CD326 (#347198), FITC Mouse IgG2a, κ Isotype Control (#553456), APC Mouse IgG1, κ Isotype Control (#550854), and PE Mouse IgG1, κ Isotype Control (#555749) antibodies were purchased from BD Biosciences.
Immunohistochemistry
Rat anti-CD44 polyclonal antibody (#251174) and rat anti-CD90 (Thy-1) polyclonal antibody (#251285) were purchased from Abbiotec.
In Vitro Heat Stress Protocol
To determine in vitro sublethal heat stress conditions that induce a significant reduction in cell survival at 48 h after heat stress but still result in some viable HCC cells, N1S1 and AS30D cells were suspended in complete media in 1.5 ml microcentrifuge tubes and heat stressed at temperatures from 37 to 60 °C for 10 min in an isothermic water bath. Treatment temperature was monitored with an Omega HH41 digital thermometer (Omega Engineering, Stamford, CT) and maintained to within ±0.05 °C. Cells were then plated in 96-well tissue culture plates and incubated in a 37 °C, 5 % CO2 incubator. At 48 h after heat stress, a WST-1 viability assay (Roche, Manheim, Germany) was performed per the manufacturer’s instructions. Absorbance was measured on a DTX 880 microplate reader (Beckman Coulter). Absorbance data were normalized to the non-heat stressed (37 °C) control to determine relative cell viability and plotted versus temperature. Nonlinear regression curve fitting was used to calculate a heat stress IT50 by Prism 5.0 (GraphPad Software, Inc., La Jolla, CA). IT50 was defined as the temperature that induced a 50 % reduction in cell viability relative to 37 °C control for a 10-min exposure time. The experimental conditions based on the IT50 data were used to recapitulate an incomplete thermal ablation in vitro for subsequent experiments.
For heat stress only experiments, N1S1 cells were heat stressed (45 °C) or control (37 °C) for 10 min in an isothermic water bath (n = 4 independent N1S1 cell cultures). For combination drug-heat stress experiments, N1S1 cells were pretreated with a dose titration of NVP-BEZ235 (0.02, 0.1, or 0.5 μM) or vehicle control (0.1 % DMSO) for 1 h followed by sublethal heat stress (45 °C) or control (37 °C) for 10 min (n = 3 independent N1S1 cell cultures). Cells were recovered in complete media in a 37 °C, 5 % CO2 humidified incubator for 48 h followed by analysis by fluorescence activating cell sorting (FACS). N1S1 cells were gated on the live cell population (7-AAD negative), and the percentage of CD44+ CD90−, CD44−CD90+ , and CD44+ CD90+ cells from the live cell population was determined by dividing the number of positive cells by the total number of cells in the parent population.
Stem Cell Marker-Based Flow Cytometry
For initial screening of HCC stem cell markers, N1S1 and AS30D cells were rinsed in 1× PBS and stained with fluorescent-labeled antibodies against CD13, CD44, CD90, CD326, or corresponding isotype controls for 30 min on ice protected from light (n = 3 independent cell cultures). Cells were then rinsed with ice-cold 1× PBS and resuspended in phenol-red free complete media to a final concentration of 1 × 106 cells/ml. For heat stress experiments, N1S1 cells were rinsed in 1× PBS and costained with fluorescent-labeled antibodies against CD44 and CD90 or corresponding isotype controls for 30 min on ice protected from light. Cells were then rinsed with ice-cold 1× PBS, stained with the live–dead cell stain 7-AAD for 10 min, and resuspended in phenol-red free complete media to a final concentration of 1 × 106 cells/ml. All cells were analyzed with a FACSCanto digital flow cytometer (BD Biosciences). Gating parameters were adjusted according to negative and positive single-stain controls. Data were analyzed by BD CellQuest Pro software (BD Biosciences).
Animal Model
All studies were approved by the institutional animal care and use committee (IACUC). The N1S1 orthotopic HCC model was developed as previously described (n = 8) [41]. Rats were randomized to ultrasound (US)-guided partial laser ablation (n = 5) or sham laser ablation (n = 3) using methods previously described [41]. Briefly, all ablation experiments were performed using an US Food and Drug Administration-approved 980-nm laser generator (Visualase, Houston, TX). Under ultrasound guidance with an L8-18i transducer (logiq E9 Ultrasound, GE Healthcare), a bare 400-μm core optical laser fiber with a 1.0-cm diffusing tip was percutaneously inserted through a 22-gauge introducer sheath at the tumor margin, and a 22-gauge needle with a 25-gauge wire thermocouple (Valleylab, Boulder, CO) was inserted 4–5 mm from the laser fiber tip within the tumor for intraprocedural temperature monitoring. For the ablation group, tumors were ablated at a power setting of 3 W under continuous US monitoring, and the ablation stopped when the thermocouple reached 45 °C in order to generate an intentional partial ablation. The laser was not turned on for sham-ablated animals. Rats were euthanized by CO2 inhalation 24 h after laser or sham ablation.
Immunohistochemistry
Liver/tumor tissue was removed, and 2-mm cross sections were cut encompassing tumor and background liver. All liver/ tumor specimens were placed in 10 % neutral buffered formalin, embedded in paraffin, and sectioned with a microtome for histopathologic and immunohistochemical analysis. Paraffin-embedded sections were stained with antibodies against CD44 (1:250) or CD90 (1:100) using methods previously described [42]. All sections were reviewed by an experienced pathologist (>20 years) in a blinded and random fashion to assess tumor/liver immunostaining [42]. Digital images were captured with a Leica DMLB microscope (Leica Microsystems) equipped with a MicroPublisher 3.3 RTV camera (Q-Imaging, Surrey, BC) and the MetaVue Imaging System (v.6.3r2; Universal Imaging Corp., Downington, PA).
Statistical Analysis
Statistical analyses were performed by Prism 5.0 (GraphPad Software Inc., La Jolla, CA). Differences between groups of live CD44+ CD90−, CD44−CD90+ , and CD44+ CD90+ N1S1 HCC cells were compared with an unpaired t test or one-way analysis of variance followed by post-hoc pairwise comparison by unpaired t test (or exact Mann–Whitney test). p <0.05 was considered statistically significant.
Results
Heat Stress Dose–Response
Analysis of heat stress dose–response curves 48 h after heat stress showed a 50 % reduction in HCC cell viability (IT50) at 45.6 °C for N1S1 (Fig. 1A) and 44.4 °C for AS30D (Fig. 1B) HCC cell lines for a 10-min heat stress exposure time. On the basis of the IT50 data, 45.0 °C for 10 min was chosen for subsequent experiments to recapitulate an incomplete thermal ablation in vitro.
Fig. 1.
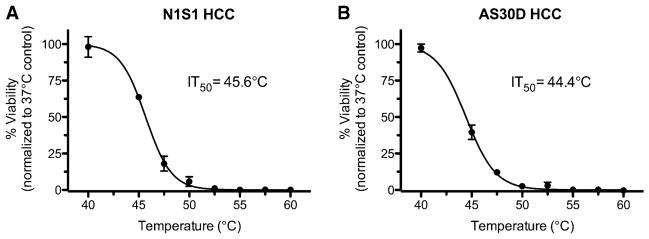
Effect of heat stress temperature on N1S1 and AS30D HCC cell viability. N1S1 and AS30D cells heat stressed at indicated temperatures from 37 to 60 °C for 10 min were assessed for viability with WST-1 assay at 48 h after heat stress. Data were normalized to 37 °C control and presented as mean ± SEM of 3 independent experiments. Nonlinear regression curve fitting was used to calculate heat stress IT50. IT50 was defined as temperature that induces 50 %reduction in cell viability relative to 37 °C control for 10-min exposure time
Subpopulations of N1S1 HCC Cells are CD44+ and CD90+ Positive
Flow cytometric analysis identified CD44+ and CD90+ cells in the N1S1 cell line but not the AS30D cell line. There were no cells staining positive for CD13 or CD326 in either cell line. The mean ± SEM for the percentage N1S1 cells positive for CD44 and CD90 out of the total cell population was 0.12 ± 0.03 % and 0.15 ± 0.02 %, respectively (n = 3). These data demonstrate that sub-populations of rare CD44+ and CD90+ cells exist within the N1S1 cell line at a frequency of ~7–18 per 10,000 cells (CD44) and 9–20 per 10,000 cells (CD90).
Sublethal Heat Stress Enriches the Population of Live CD44+ and CD90+ N1S1 HCC Cells In Vitro
In order to determine whether sublethal heat stress enriches the proportion of live HCC subpopulations expressing putative HCC stem cell markers in vitro, sublethal heat stress of N1S1 cells followed by flow cytometric analysis for CD44 and CD90 expression at 48 h after heat stress was performed. Flow cytometric analysis showed that sublethal heat stress induced a small but significant increase in the relative proportion (% ± SEM) of live CD44+ and CD90+ cells compared to the 37 °C control group, as evidenced by the enrichment of CD44+ CD90− (5.3-fold, 0.16 ± 0.004 % vs. 0.03 ± 0.01 %, respectively, p = 0.0001), CD44−CD90+ (2.4-fold, 0.12 ± 0.007 % vs. 0.05 ± 0.01 %, respectively, p = 0.003), and CD44+ CD90+ (22.0-fold, 13.2 × 10−6 % ± 4.3 × 10−6 % vs. 0.6 × 10−6 % ± 0.6×10−6 %, respectively, p <0.03) subpopulations (Fig. 2). These data demonstrate that the populations of CD44+ and CD90+ N1S1 HCC cells, although rare, are enriched by sublethal heat stress.
Fig. 2.
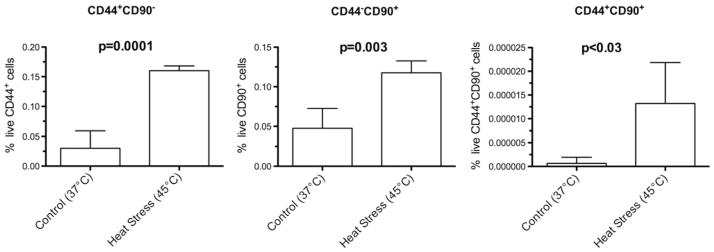
Effect of sublethal heat stress on proportion of live CD44+ , CD90+ , and CD44+ CD90+ N1S1 HCC cells. Graph shows that sublethal heat stress (45 °C, 10 min) resulted in significant increase in proportion of live CD44+ CD90− (5.3-fold), CD44−CD90+ (2.4-fold), and CD44+ CD90+ (22.0-fold) N1S1 HCC cell subpopulations relative to control group (37 °C, 10 min) after 48-h recovery. Treatment groups compared by unpaired t test. Data are presented as mean ± SEM from 4 independent cell cultures
Inhibition of PI3K-mTOR Prevents the Enrichment of live CD44+ N1S1 HCC Cells by Sublethal Heat Stress In Vitro
In order to determine whether inhibition of the PI3K-AKT-mTOR pathway prevents the enrichment of live HCC subpopulations expressing putative HCC stem cell markers after sublethal heat stress in vitro, pretreatment of N1S1 cells with a dose titration of the dual PI3K-mTOR inhibitor NVP-BEZ235 followed by sublethal heat stress and flow cytometric analysis for CD44 and CD90 expression at 48 h after heat stress was performed. Treatment with PI3K-mTOR inhibitor at 0.1 and 0.5 μM prevented heat stress-induced enrichment of the population of live CD44+ N1S1 cells (p < 0.01) but did not significantly prevent the enrichment of the population of live CD90+ N1S1 cells (p > 0.10) (Fig. 3).
Fig. 3.
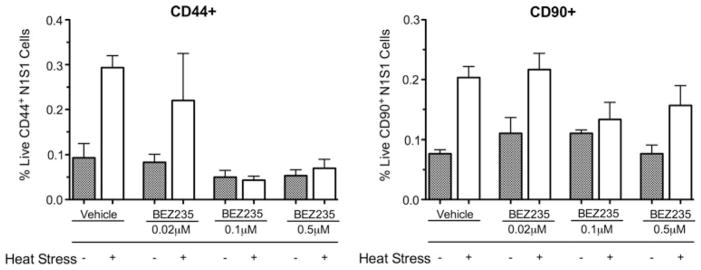
Effect of PI3K-mTOR inhibition on proportion of live CD44+ and CD90+ N1S1 HCC cells after sublethal heat stress. Graph shows that pretreatment with dual PI3K-mTOR inhibitor BEZ235 at 0.1 and 0.5 μM prevented heat stress-induced enrichment of population of live CD44+ N1S1 cells after 48-h recovery (p < 0.01) but not CD90+ N1S1 cells (p > 0.10). Treatment groups were compared by 1-way analysis of variance followed by post-hoc pairwise comparison by unpaired t test. Data are presented as mean ± SEM from 3 independent cell cultures
CD44+ N1S1 HCC Cells Preferentially Localize to the Tumor Margin In Vivo
Finally, in order to determine the localization of HCC subpopulations expressing putative stem cell markers in HCC tumors in vivo, immunohistochemical staining of paraffin-embedded sections from N1S1 tumors orthotopically implanted in the livers of immunocompetent Sprague Dawley rats was performed. Immunohistochemical analysis demonstrated preferential localization of CD44+ cells at the tumor margin (Fig. 4). There was no evidence of cells staining positive for CD44+ in the background liver. Immunohistochemical staining for CD90 did not detect any positively staining cells in the tumor or liver (data not shown). Finally, CD44 immunohistochemical staining of N1S1 tumors that had undergone intentional partial laser thermal ablation demonstrated clusters of CD44+ cells near the tumor ablation margin (Fig. 5).
Fig. 4.
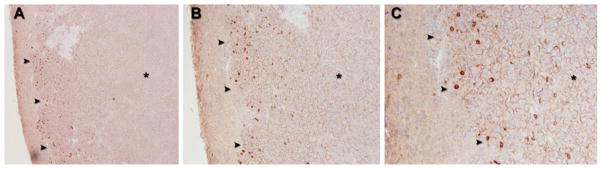
Representative CD44 immunostaining in orthotopic N1S1 HCC tumor model. A Low-power (50×), B medium-power (100×), and C high power (200×) photomicrographs of N1S1 tumors (black asterisk) demonstrate rare cells staining positive for CD44 (brown) preferentially localizing at tumor margin (black arrowheads) 24 h after sham ablation
Fig. 5.

Representative CD44 immunostaining at tumor ablation margin in orthotopic N1S1 HCC tumor model from 2 different rats (A, B and C, D, respectively). A, C Low-power (40×) and B, D high-power (100×) photomicrographs of ablation zone (ablated tissue denoted by black asterisk) from both rats demonstrate clusters of rare cells staining positive for CD44 (brown) at tumor ablation margin (black arrowheads) 24 h after partial laser ablation
Discussion
CSC have been implicated in resistance to various anti-cancer therapies, including cytotoxic and molecular-targeted agents and ionizing radiation, but have yet to be examined in the context of thermal ablation [1, 13]. These proof-of-concept studies provide experimental evidence supporting a role for HCC cells expressing putative CSC makers CD44 and CD90 in HCC biological response to heat stress.
The initial screen identified CD44- and CD90-positive cells in the N1S1 cell line but not the AS30D cell line. These findings are consistent with previous studies that have demonstrated that HCC cell lines, primary HCC tumors, and blood samples from HCC patients harbor rare subpopulations of cells expressing putative CSC markers in a patient-, tumor-, cell line-, and marker-dependent manner [9–24]. The findings that the AS30D cells do not express any of the putative CSC markers tested is consistent with the absence of these markers in other cell lines. Nonetheless, this does not rule out the possibility of other CSC markers, such as CD133 [14]. Specifically, the percentage of CD44 and CD90 cells out of the total N1S1 cell population was ~0.12 and 0.15 %, respectively, findings consistent with other studies reporting a range of 0.07–3.29 % for CD44+ HCC cells and 0.02–0.19 % for CD90+ HCC cells [12, 18]. A pioneering study by Yang et al. [12] demonstrated that CD90+ but not CD90− HCC cells were strongly tumorigenic in immunodeficient mice and that CD44+ cells developed tumors more quickly than CD44− cells. Moreover, they found that CD44+ CD90+ HCC cells demonstrated a more aggressive, metastatic phenotype compared to the CD44−CD90+ cells and that inhibition of CD44 using a neutralizing antibody prevented local and metastatic tumor formation by CD90+ cells [12]. In short, these findings support a role for CD44+ and CD90+ cells in HCC tumor progression and biological heterogeneity and suggest that CD44 may represent a potential therapeutic target.
Previous studies have suggested a model whereby CSC are enriched after treatment with cytotoxic therapies, thereby promoting tumor repopulation and progression [1]. In these experiments, sublethal heat stress enriched the population of live CD44+ 90−, CD44−CD90+ , and CD44+ CD90+ cells, although the double positive cells were extremely rare. These data support at least two potential hypotheses. First, sublethal heat stress selects for CD44+ and CD90+ cells, effectively decreasing the total HCC cell population while the absolute number of CD44+ and/or CD90+ cells does not change. This hypothesis would suggest that CD44+ and/or CD90+ cells are more resistant to heat stress than CD44− and CD90− cells. Second, sublethal heat stress may induce an increase in the absolute number of CD44+ and CD90+ cells and a decrease in the total HCC cell population. This hypothesis would suggest that sublethal heat stress induces the proliferation of CD44+ and/or CD90+ cells. Regardless of the mechanism, these data demonstrate that the populations of CD44+ and CD90+ N1S1 HCC cells, although rare, are enriched by sublethal heat stress. Given the enhanced capacity for tumor initiation, self-renewal, pluripotency, and chemoresistance in CSC, these findings may suggest that residual CD44+ and/or CD90+ cells promote tumor repopulation and progression after sublethal heat stress.
Diverse signaling pathways and regulatory mechanisms have been implicated in promoting the survival of CSC to anticancer therapies in HCC and other malignances [17, 43–45]. Previous studies have demonstrated that PI3K-AKT-mTOR signaling may play an important role in the enhanced survival of CSC to chemotherapy and ionizing radiation [13, 17, 25–29]. Interestingly, PI3K-mTOR inhibition prevented heat stress-induced enrichment of the population of live CD44+ N1S1 cells but not CD90+ N1S1 cells. These data demonstrate that PI3K-AKT-mTOR signaling may play different roles in CD44+ and CD90+ cells, suggesting further biological heterogeneity within the N1S1 cell line. Moreover, these data suggest that PI3K-mTOR may be a potential therapeutic target in these cell subpopulations, findings consistent with previous studies in HCC and other malignancies [13, 25].
Finally, the presence of CD44+ and CD90+ cells was investigated in orthotopic N1S1 tumors in vivo [33]. Interestingly, cells staining positive for CD44 were rare and preferentially localized within the N1S1 tumor at the tumor margin. Additionally, rare CD44+ cells were found at the tumor ablation margin. On the other hand, cells staining positive for CD90 were not identified in the N1S1 tumor or background liver. However, the presence of CD90+ cells in the N1S1 cell line in vitro but not when orthotopically implanted in vivo suggests that there may be a technical issue with the anti-CD90 antibody, and therefore the presence of CD90+ cells in vivo cannot be ruled out. Such variation in immunostaining for HCC CSC is consistent with previous reports [46]. Nonetheless, given the localization of CD44+ cells at the leading edge of the tumor, these findings raise the question, are CD44+ HCC cells tumor propagating cells, and if so, do they promote local recurrence after an incomplete thermal ablation?
There are limitations to these studies. First, it is recognized that there are limitations in currently available techniques for identifying and studying CSC, including stem cell marker-based flow cytometry and immunohistochemistry [5, 46–48]. Second, the experiments were limited to one cell line. These findings warrant further validation not only in other HCC cell lines known to harbor CD44+ and CD90+ cells but also to other CSC markers in order to better understand the generalizability of these findings. Additionally, the mechanism or mechanisms whereby heat stress enriches the population of CD44+ and CD90+ cells warrants further investigation. Third, although CD44+ and CD90+ cells were identified in the N1S1 cell line and enriched by sublethal heat stress, these experiments do not confirm that these cells were in fact bona fide CSC. As such, CD44+ and CD90+ N1S1 HCC cells require further characterization using limiting dilution and serial transplantation assays in order to validate their role as CSC and determine whether they play a role in enhanced HCC tumorigenesis. Fourth, although these experiments suggest that PI3K-AKT-mTOR signaling may play a differential role in CD44+ and CD90+ HCC cellular response to heat stress, the cellular and molecular heterogeneity between CD44+ and CD90+ cells warrants further characterization in order to better understand the role of PI3K-AKT-mTOR in mediating cell proliferation and survival in these cellular subpopulations.
Residual HCC CSC within the thermal ablation zone represent a potential mechanism mediating recurrence and tumor progression to thermal ablative therapies for HCC. These studies provide experimental evidence supporting diverse roles for HCC cells expressing putative stem cell markers CD44 and CD90 in HCC biological response to heat stress. Given the enrichment of CD44+ and CD90+ cells after sublethal heat stress and the preferential localization of CD44+ cells to tumor margin, these data may suggest that sublethal heat stress at the tumor ablation margin leaves behind residual CD44+ and/or CD90+ cells that can repopulate the tumor, thereby resulting in tumor relapse and progression. Of translational relevance, these data suggest that PI3K-mTOR may be a potential therapeutic target in CD44+ and CD90+ cells and support further studies investigating inhibition of the PI3K-AKT-mTOR pathway in combination with thermal ablation as a mechanism to sensitize potentially more refractory HCC subpopulations to heat stress. Although there is much more to be learned regarding the roles of CD44+ and CD90+ cells in HCC biological response to thermal ablation, these experiments suggest that the CSC hypothesis may be biologically and clinically relevant to the field of interventional oncology.
Acknowledgments
This publication was supported by CTSA Grant TL1 TR000137 from the National Center for Advancing Translational Science (NCATS). Additional research support provided in part by a SIR Foundation allied scientist training Grant. Infrastructure support was provided by NIH construction grant NIH C06 RR018898.
Footnotes
Conflict of interest Mr. Scott Thompson received a research grant from the SIR Foundation—Allied Scientist Training Grant. Dr. Lewis Roberts received research grants from Bristol Myers-Squibb, Merck, Nordion, and Bayer. Dr. Matthew Callstrom, Ms. Kim Butters, Ms. Shari Sutor, Mr. Bruce Knudsen, Dr. Joseph Grande, and Dr. David Woodrum declare that they have no conflict of interest.
Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Contributor Information
Scott M. Thompson, Email: thompson.scott@mayo.edu, Medical Scientist Training Program, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
Matthew R. Callstrom, Email: callstrom.matthew@mayo.edu, Department of Radiology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
Kim A. Butters, Email: butters.kim@mayo.edu, Department of Radiology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
Shari L. Sutor, Email: sutor.shari@mayo.edu, Genomics Research Center, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
Bruce Knudsen, Email: Knudsen.bruce@mayo.edu, Department of Laboratory Medicine and Pathology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA.
Joseph P. Grande, Email: grande.joseph@mayo.edu, Department of Laboratory Medicine and Pathology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
Lewis R. Roberts, Email: Roberts.lewis@mayo.edu, Division of Gastroenterology and Hepatology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
David A. Woodrum, Email: woodrum.david@mayo.edu, Department of Radiology, College of Medicine, Mayo Clinic, 200 First Street Southwest, Rochester, MN 55905, USA
References
- 1.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 2.Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197(4302):461–463. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 5.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell. 2012;22:699–701. doi: 10.1016/j.ccr.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Park CY, Tseng D, Weissman IL. Cancer stem cell-directed therapies: recent data from the laboratory and clinic. Mol Ther. 2009;17:219–230. doi: 10.1038/mt.2008.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324(5935):1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong CM, Ma S, Guan XY. Biology of hepatic cancer stem cells. J Gastroenterol Hepatol. 2011;26:1229–1237. doi: 10.1111/j.1440-1746.2011.06762.x. [DOI] [PubMed] [Google Scholar]
- 10.Haraguchi N, Ishii H, Mimori K, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Investig. 2010;120:3326–3339. doi: 10.1172/JCI42550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry JC, Park JK, Jiang J, et al. miR-199a-3p targets CD44 and reduces proliferation of CD44 positive hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2010;403:120–125. doi: 10.1016/j.bbrc.2010.10.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang ZF, Ho DW, Ng MN, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Ma S, Lee TK, Zheng BJ, et al. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 14.Ma S, Chan KW, Lee TK, et al. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol Cancer Res. 2008;6:1146–1153. doi: 10.1158/1541-7786.MCR-08-0035. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita T, Forgues M, Wang W, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 16.Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52:280–281. doi: 10.1016/j.jhep.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Yoon SK. The biology of cancer stem cells and its clinical implication in hepatocellular carcinoma. Gut Liver. 2012;6:29–40. doi: 10.5009/gnl.2012.6.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji J, Wang XW. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin Oncol. 2012;39:461–472. doi: 10.1053/j.seminoncol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rountree CB, Mishra L, Willenbring H. Stem cells in liver diseases and cancer: recent advances on the path to new therapies. Hepatology. 2012;55:298–306. doi: 10.1002/hep.24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colombo F, Baldan F, Mazzucchelli S, et al. Evidence of distinct tumour-propagating cell populations with different properties in primary human hepatocellular carcinoma. PLoS One. 2011;6:e21369. doi: 10.1371/journal.pone.0021369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquardt JU, Raggi C, Andersen JB, et al. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology. 2011;54:1031–1042. doi: 10.1002/hep.24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song K, Wu J, Jiang C. Dysregulation of signaling pathways and putative biomarkers in liver cancer stem cells [review] Oncol Rep. 2013;29:3–12. doi: 10.3892/or.2012.2082. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Lingala S, Khoobyari S, et al. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838–845. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee TK, Castilho A, Ma S, Ng IO. Liver cancer stem cells: implications for a new therapeutic target. Liver Int. 2009;29:955–965. doi: 10.1111/j.1478-3231.2009.02040.x. [DOI] [PubMed] [Google Scholar]
- 25.Martelli AM, Evangelisti C, Follo MY, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem. 2011;18:2715–2726. doi: 10.2174/092986711796011201. [DOI] [PubMed] [Google Scholar]
- 26.Dubrovska A, Kim S, Salamone RJ, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci USA. 2009;106:268–273. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunayama J, Matsuda K, Sato A, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28:1930–1939. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- 28.Xu Q, Simpson SE, Scialla TJ, et al. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 29.Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261–4268. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu HS, Lin JH, Huang WC, et al. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer. 2011;117:1516–1528. doi: 10.1002/cncr.25599. [DOI] [PubMed] [Google Scholar]
- 31.Prinsloo E, Setati MM, Longshaw VM, Blatch GL. Chaperoning stem cells: a role for heat shock proteins in the modulation of stem cell self-renewal and differentiation? Bioessays. 2009;31:370–377. doi: 10.1002/bies.200800158. [DOI] [PubMed] [Google Scholar]
- 32.Wright MH, Calcagno AM, Salcido CD, et al. Brca1 breast tumors contain distinct CD44+ /CD24−; and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lencioni R, Crocetti L. Local-regional treatment of hepatocellular carcinoma. Radiology. 2012;262:43–58. doi: 10.1148/radiol.11110144. [DOI] [PubMed] [Google Scholar]
- 34.Tiong L, Maddern GJ. Systematic review and meta-analysis of survival and disease recurrence after radiofrequency ablation for hepatocellular carcinoma. Br J Surg. 2011;98:1210–1224. doi: 10.1002/bjs.7669. [DOI] [PubMed] [Google Scholar]
- 35.Wang JH, Wang CC, Hung CH, et al. Survival comparison between surgical resection and radiofrequency ablation for patients in BCLC very early/early stage hepatocellular carcinoma. J Hepatol. 2012;56:412–418. doi: 10.1016/j.jhep.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 36.Cho YK, Kim JK, Kim MY, et al. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology. 2009;49:453–459. doi: 10.1002/hep.22648. [DOI] [PubMed] [Google Scholar]
- 37.Yin XY, Xie XY, Lu MD, et al. Percutaneous thermal ablation of medium and large hepatocellular carcinoma: long-term outcome and prognostic factors. Cancer. 2009;115:1914–1923. doi: 10.1002/cncr.24196. [DOI] [PubMed] [Google Scholar]
- 38.Kao WY, Chiou YY, Hung HH, et al. Risk factors for long-term prognosis in hepatocellular carcinoma after radiofrequency ablation therapy: the clinical implication of aspartate amino-transferase–platelet ratio index. Eur J Gastroenterol Hepatol. 2011;23:528–536. doi: 10.1097/MEG.0b013e328346d529. [DOI] [PubMed] [Google Scholar]
- 39.Ng KK, Poon RT, Lo CM, et al. Analysis of recurrence pattern and its influence on survival outcome after radiofrequency ablation of hepatocellular carcinoma. J Gastrointest Surg. 2008;12:183–191. doi: 10.1007/s11605-007-0276-y. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Zheng Y, Li S, et al. Percutaneous microwave ablation of larger hepatocellular carcinoma. Clin Radiol. 2013;68:21–26. doi: 10.1016/j.crad.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Thompson SM, Callstrom MR, Knudsen B, et al. Development and preliminary testing of a translational model of hepatocellular carcinoma for MR imaging and interventional oncologic investigations. J Vasc Interv Radiol. 2012;23:385–395. doi: 10.1016/j.jvir.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson SM, Callstrom MR, Knudsen B, et al. AS30D model of hepatocellular carcinoma: tumorigenicity and preliminary characterization by imaging, histopathology, and immunohistochemistry. Cardiovasc Intervent Radiol. 2013;36:198–203. doi: 10.1007/s00270-012-0466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamashita T, Honda M, Nio K, et al. Oncostatin m renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010;70:4687–4697. doi: 10.1158/0008-5472.CAN-09-4210. [DOI] [PubMed] [Google Scholar]
- 44.Wang XQ, Ongkeko WM, Chen L, et al. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528–539. doi: 10.1002/hep.23692. [DOI] [PubMed] [Google Scholar]
- 45.Cheung ST, Cheung PF, Cheng CK, et al. Granulin-epithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology. 2011;140:344–355. doi: 10.1053/j.gastro.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 46.Lingala S, Cui YY, Chen X, et al. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol. 2010;89:27–35. doi: 10.1016/j.yexmp.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hermansen SK, Christensen KG, Jensen SS, Kristensen BW. Inconsistent immunohistochemical expression patterns of four different CD133 antibody clones in glioblastoma. J Histochem Cytochem. 2011;59:391–407. doi: 10.1369/0022155411400867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mather JP. In vitro models. Stem Cells. 2012;30:95–99. doi: 10.1002/stem.774. [DOI] [PubMed] [Google Scholar]