

Abstract
Focal adhesions (FAs) are dynamic subcellular structures crucial for cell adhesion, migration and differentiation. It remains an enigma how enzymatic activities in these local complexes regulate their structural remodeling in live cells. Utilizing biosensors based on fluorescence resonance energy transfer (FRET), we developed a correlative FRET imaging microscopy (CFIM) approach to quantitatively analyze the subcellular coordination between the enzymatic Src activation and the structural FA disassembly. CFIM reveals that the Src kinase activity only within the microdomain of lipid rafts at the plasma membrane is coupled with FA dynamics. FA disassembly at cell periphery was linearly dependent on this raft-localized Src activity, although cells displayed heterogeneous levels of response to stimulation. Within lipid rafts, the time delay between Src activation and FA disassembly was 1.2 min in cells seeded on low fibronectin concentration ([FN]) and 4.3 min in cells on high [FN]. CFIM further showed that the level of Src-FA coupling, as well as the time delay, was regulated by cell-matrix interactions, as a tight enzyme-structure coupling occurred in FA populations mediated by integrin αvβ3, but not in those by integrin α5β1. Therefore, different FA subpopulations have distinctive regulation mechanisms between their local kinase activity and structural FA dynamics.
Focal adhesions (FAs) are dynamic subcellular structures connecting the actin cytoskeleton with the extracellular matrix, allowing the cells to sense and respond to the mechanical and chemical cues from the environment1,2,3. FAs contain hundreds of residential and associated molecules with enzymatic activities or adapter functions4. These FA proteins form a three-dimensional nano-structure of slippery clutches within a narrow depth between the actin cytoskeleton and the substrate matrix5,6. As such, the dynamic assembly and disassembly processes of FAs play central roles in cell adhesion, migration and differentiation5,7,8.
The structural regulation of FAs in migrating cells is considered to be driven by local molecular biochemical activities. Epidermal growth factors have been shown to activate the growth factor receptor kinase and cause FA disassembly during migration and invasion9,10. Protein tyrosine kinase Src and focal adhesion kinase (FAK) have been reported to regulate the turnover of FA structures8,11. Meanwhile, Src also interacts with the small GTPase Rac1 and transmembrane integrin receptors to regulate FA assembly12,13. However, the quantitative and spatiotemporal coordination between the enzymatic Src activity and the structural FA dynamics remain elusive and has not been previously investigated, mainly due to the heterogeneous and transient nature of these signals in different subcellular compartments of live cells. Only through single-cell imaging approaches, it has become possible to quantitatively capture these signals and assess the relationship between the local molecular activities and the FA dynamics.
Fluorescence resonance energy transfer (FRET)-based biosensors have been widely used to monitor spatiotemporal molecular activities with high resolution in single live cells14,15. We have previously developed biosensors for monitoring the Src kinase activity at different subcellular locations16. The membrane-tethered Lyn-Src and KRas-Src biosensors have been utilized to show differentially regulated Src activation mechanisms in and outside the rafts microdomains at the plasma membrane16,17,18. In the current study, the Lyn-Src and KRas-Src biosensors based on ECFP and YPet19, a highly sensitive FRET pair, were used to monitor Src activity at different sub-membrane compartments. As such, the enzymatic Src activity visualized by the membrane-targeted Src biosensors, and the structural FA dynamics highlighted by mCherry-paxillin20,21, can be simultaneously monitored to elucidate their dynamic coordination at subcellular levels in the same cell.
Single-cell FRET imaging can provide a panoramic view of the heterogeneous and dynamics processes of molecular activities in a population of cells22,23. However, this invaluable information and the underlying regulation parameters are often lost when only the representative or averaged FRET signals are studied. To decipher the regulation mechanism underlying the heterogeneous and dynamic signals from single cells, we developed a novel correlative FRET imaging microscopy (CFIM) framework for the quantitative analysis of the coordination between a pair of molecular signals in single live cells. The innate cell-cell heterogeneity of the signals is used to evaluate the causality-related parameters without the need of specific pharmacological inhibitors22,24. The dynamics of the signals can be applied to interpret the sequential kinetic parameters of the molecular events25,26. Indeed, our results utilizing CFIM revealed that cell-matrix interactions govern the Src-FA interaction at subcellular levels, via specific integrin subtypes. The results demonstrated that noisy and complex signaling events observed in single live cells can be quantitatively deciphered by CFIM to shed new light on the regulation mechanism of the enzyme-structure coordination.
Results
PDGF induced a concomitant Src kinase activation and FA disassembly
Mouse embryonic fibroblasts (MEFs) were co-transfected with the Lyn-Src biosensor and mCherry-paxillin, and seeded on fibronectin-coated cover glass. Upon PDGF stimulation, an immediate and significant increase of the Lyn-Src ECFP/FRET ratio (representing Src kinase activity in lipid-rafts) was observed concurrently with a decrease of the mCherry-paxillin fluorescence intensity representing FAs (Fig. 1a and Supplementary Video 1). The average Src ECFP/FRET ratio and total fluorescence intensity of mCherry-paxillin within the computationally detected whole-cell contour were quantified and normalized to their levels before stimulation (Fig. 1b). The results revealed that PDGF induced an 80% increase of Src ECFP/FRET ratio, accompanied by a 14% reduction of mCherry-paxillin intensity in the whole cell (Fig. 1c), suggesting a possible coupling between Src activation and FA disassembly at the whole cell level under PDGF stimulation.
Figure 1. The quantification of PDGF-induced Src activation and focal adhesion disassembly.

(a) The ECFP/FRET ratio images of the Src biosensor (top panels) in MEFs co-transfected with mCherry-paxillin (lower panels) before and after PDGF stimulation. The cold and hot colors in the color bar on the top panel represent the low and high Src activities, respectively. (b) A ratio image (left) and a paxillin intensity image (right) each outlined by an automatically detected cell edge (red). (c) The time courses of the normalized ECFP/FRET ratio (red solid) and the total mCherry fluorescence intensity (blue dotted) over a whole cell body. (d) The mCherry-paxillin intensity image of a representative cell shown in pseudo color before (left) and after (right) high-pass filtering. (e) The pseudo-colored intensity image overlaid with the automatically detected cell edge (solid white), the dividing line between the outer layer and the rest of the cell mask (dotted white), and the boundaries of individual FAs (solid black). The region of interest (ROI) contains the detected FAs located within the cell periphery region between the dotted and solid white lines. An inlet of the panel is shown at the lower right corner with the boundaries of FAs outlined in dashed black. (f) The time courses of the averaged ECFP/FRET ratio (solid red) and the total mCherry intensity (dotted blue) of the FAs in the ROI as shown in (e). (g) The percentage changes of the Src biosensor ECFP/FRET ratio (Src activation) and the paxillin total intensity (FA disassembly) were compared among the whole cell, the FAs in the whole cell (cell FA), and the FAs in ROI (Lam-FA) as shown in (e). Scale bars: 10 μm.
Quantitatively analyze the subcellular coordination between Src activation and FA disassembly by CFIM
We further quantified the mCherry-paxillin dynamics of FA sites at different subcellular regions. The water algorithm27 was modified to detect and segment the FAs in the mCherry-paxillin images with high computation efficiency (Figs. 1d–e, Supplementary Figs. 1a–b and Supplementary Video 2, see Methods for details). A mask image of the cell body was calculated and divided into five layers according to their normalized distance to the centroid. The Src ECFP/FRET ratio and the mCherry-paxillin intensity of the detected FAs within the cell outer layer were then computed via this automated program to represent the local signals at the lamellipodium that are crucial for cell adhesion and migration3,28 (Fig. 1e, Supplementary Fig. 1 and Supplementary Video 2). The quantified results showed a 70% increase of Src activity and a 75% decrease of total FAs at the lamellipodium (Fig. 1f). When the PDGF-induced changes of Src and paxillin signals were compared among three levels: whole cell, FA sites in the whole cell (Cell-FA) and in the lamellipodium (Lam-FA), significantly more paxillin dissolution was detected in the Cell-FA and Lam-FA regions than the whole cell, clearly suggesting differential FA dynamics at different subcellular regions (Fig. 1g).
After the time courses of Src activation and Lam-FA disassembly were quantified, we designed the CFIM analysis procedure to evaluate the correlation between Src activation and Lam-FA disassembly at the single-cell level (Fig. 2). Specifically, the CFIM methods was designed to address two logically independent questions: (1) How are these two signals correlated in magnitude at single-cell level? (2) What are the kinetic similarity and time difference between these two signals? To quantitatively evaluate parameters describing these two kinds of correlation, we performed linear regression on the maximal magnitudes of Src activation and FA disassembly and cross-correlation analysis on the time courses (Fig. 2).
Figure 2. The schematics of CFIM procedure.

CFIM consists of three main components: (1) simultaneous FRET and phenotypic imaging of dynamic molecular events in a live cell, (2) the automatic detection and quantification of these video images, (3) the linear regression analysis for magnitude coupling and the cross-correlation analysis for kinetic coupling.
Lam-FA disassembly is coordinated with Src activation within lipid rafts in magnitude and kinetics
When the quantified time courses of local Src activity and Lam-FA intensity in multiple cells cultured on 2.5 μg/ml fibronectin (FN) coated slides were normalized and plotted together, the cell-cell heterogeneity is apparent with the magnitude of individual curves varying among different cells, although the curves averaged from all the cells demonstrated a consistent trend of Src activation and Lam-FA disassembly (Fig. 3a). Since the cell cycles were synchronized by starvation before imaging, the observed variation in single-cell responses is likely due to the innate cell-cell heterogeneity in structure and function, such as the molecular wiring inside each individual cell.
Figure 3. Lam-FA disassembly is coordinated with Src activation in magnitude and kinetics.

(a) The time courses of normalized Src biosensor ECFP/FRET ratio (pink circles) and the normalized total paxillin intensity (light blue circles) from different individual MEFs, and their average curves of Src ECFP/FRET ratio (red solid line) and paxillin intensity (blue solid line). (b) The time courses of Src activation from different individual cells color-coded by the level of Lam-FA disassembly in the same cell. (c) The max-max plot of maximal Lam-FA disassembly against maximal Src activation. Each dot represents the data from an individual cell. The slope and the correlation coefficient, R, were calculated based on the data. (d) Left Panel: The Src-paxillin CC curves (light blue circles) from different individual cells overlaid with the average CC curve (solid blue) and its ± standard error (SEM, dashed blue lines). The time delay, T, and the maximal cross-correlation value, K, were estimated based on the data. Right panel: the histogram showing the distribution of the time delay values from single cells. (e) The mCherry-paxillin intensity image of a representative SYF-/- cell before and after PDGF stimulation. (f) The time courses of normalized Src ECFP/FRET ratio (pink circles) and the normalized total paxillin intensity (light blue circles) for different individual SYF cells, and their corresponding average curves (red and blue solid lines). Note here the time courses of normalized Src ECFP/FRET ratio were almost identical with the value 1, so the pink circles coincide with the average curve in solid red.
To investigate the spatiotemporal coordination of local Src activation and Lam-FA disassembly in each individual cell, the curve of Src activation (the normalized Src ECFP/FRET ratio −1) was color-coded by the corresponding Lam-FA disassembly (1- the normalized total paxillin intensity) in the same cell. These plots showed a strong correlation between the PDGF-induced Lam-FA disassembly and Src activation within each individual cell (Fig. 3b), as evidenced by higher Src activation associated with stronger Lam-FA disassembly (hotter colors). This observation was further demonstrated by the max-max plot, with each data point obtained from a single cell representing the maximal Lam-FA disassembly against the maximal Src activation (Fig. 3c, Supplementary Fig. 2a, and Methods). The max-max plot revealed a clear linear correlation between the magnitude of Lam-FA disassembly and Src activation at single cell levels, with the slope of this plot representing the relative amount of Lam-FA disassembly per unit of Src activation and hence the capacity of Src activation in causing Lam-FA disassembly (coordination capacity).
The linear correlation coefficient, R, measures the coordination strength between the magnitude of Src activation and Lam-FA disassembly among different cells17, with R2 representing the coefficient of determination, quantifying the portion of variability in the Lam-FA signal which is determined by the variability in Src activity17. R = 0 indicates no correlation between two signals and R = 1 indicates that the variability in the Lam-FA signal is completely determined by that of Src activation. Hence, the R value of 0.74 in this case clearly indicates a strong correlation between the Src activation and Lam-FA disassembly in magnitude, which is drastically higher than that of unrelated events generated by randomization tests (p-value < 1.0e-3). The cell group with high Src activations (Src activation in each cell is above its median value from the whole cell population) also had significantly more Lam-FA disassembly than cells with below-median Src activations (Supplementary Fig. 2b), confirming the coupling between the magnitudes of Src activation and Lam-FA disassembly.
Since the time courses of the normalized Src FRET ratio and normalized paxillin intensity both leveled off around 20 min after stimulation (Figs. 3a–3b), it is expected that final levels of the normalized FRET ratio and paxillin intensity are also coupled. Indeed, linear regression analysis confirmed this correlation (R: −0.72 ± 0.14, 95% CI), with a relatively broad distribution of both the normalized FRET ratio and paxillin intensity broad (Supplementary Figs. 2c–2d). In contrast, without normalization, the final levels of FRET ratio and paxillin intensity are uncoupled with R: −0.05 ± 0.67 (95% CI, Supplementary Fig. 2d), with distributions quite different from their normalized counterparts (Supplementary Figs. 2e–2f). These results underscore the importance of the normalization processes in revealing the underlying coupling between different dynamic processes24,29.
Thus, the innate cell-cell heterogeneity can be utilized by CFIM to verify the Src-FA magnitude coupling, without the perturbation of signaling using Src inhibitors. Two important parameters “slope” and “R-value” can also be quantified by CFIM to characterize the capacity of Src enzymatic activity in causing the FA disassembly as well as the strength of the Src-FA magnitude coupling, respectively.
The colored curves in Figure 3b also showed a gradual increase of Lam-FA disassembly during the time course of Src activity elevation, suggesting a temporal coordination between the kinetics of Src activation and Lam-FA disassembly. This dynamic coordination was quantified by the Src-Lam-FA cross-correlation (CC) functions in each single cell to reveal a maximum of K = 0.84 (representing the kinetic similarity between two signals) at T = 1.2 min (representing the time delay between two signals) on the average CC curve (Fig. 3d). The parameter K denotes the maximal value of the CC function, which measures the similarity between two time courses. K = 0 indicates no similarity between the time courses, while K = 1 indicates that the time courses are identical. Therefore, these results suggest that the PDGF-induced Lam-FA disassembly was dynamically coupled with Src activation in lipid rafts, as Src activation leading and acting upstream of Lam-FA disassembly with an average time delay of 1.2 min.
Control experiment results indicate that the expression of Src FRET biosensor did not perturb the natural FA disassembly process, since the mCherry-paxillin intensity at Lam-FAs decreased with similar kinetics upon PDGF stimulation with or without the co-expression of the Lyn-Src biosensor (Supplementary Fig. 3a). Interestingly, Lam-FA increased to 2.5 folds of the basal level upon PDGF stimulation in the Src/Yes/Fyn triple knockout (SYF-/-) fibroblasts30 while the Src ECFP/FRET ratio remained unchanged during the time course, suggesting possibly positive regulations of FAs by other signaling molecules in the absence of Src family kinases (Figs. 3e–f). The PDGF-induced Src activation and FA disassembly in SYF-/- fibroblasts can be rescued by expressing the wild-type Src gene, but not the kinase dead Src mutant (Supplementary Figs. 3b–c). These perturbation experiments confirmed that the PDGF-stimulated Src kinase activity is essential in causing Lam-FA disassembly as suggested by the CFIM results based on the innate heterogeneous signals (Fig. 3c). These results are also consistent with previous reports that constitutively active Src can cause the turnover of focal adhesion during cell migration, although these earlier reports cannot differentiate the importance of lipid-rafts Src activity from other subcellular locations11,31. Experiments in cells co-transfected with Lyn-Src and another FA marker protein vinculin conjugated with mCherry further showed that PDGF induced a Src activation with concomitant vinculin disassembly (Supplementary Fig. 3d), confirming a coordination between lipid rafts Src activation and general FA protein disassembly in response to PDGF stimulation.
The magnitude of Lam-FA disassembly is regulated by FN concentration
The FN concentration ([FN]) is known to regulate migration speed via cell-ECM adhesion and FAs32,33,34. We therefore investigated the effect of variable [FN] on the coordination between Src activation and Lam-FA disassembly. As in the case of 2.5 μg/ml [FN], PDGF induced Src activation and Lam-FA disassembly in cells seeded on 10 μg/ml and 20 μg/ml [FN] (Figs. 4a–b, Supplementary Fig. 4). Lam-FA disassembly upon PDGF stimulation was statistically more significant in cells seeded on 2.5 μg/ml [FN] than that on 10 or 20 μg/ml [FN], although their average Src activation was not significantly different (Figs. 4c–d). Interestingly, there were no significant difference in FA disassembly or Src activation between the groups of cells on 10 and 20 μg/ml [FN].
Figure 4. Fibronectin concentration affects the magnitude of Lam-FA disassembly.

(a–b) Quantified results for MEFs seeded on (a) 10 μg/ml and (b) 20 μg/ml FN: the time courses of normalized Src ECFP/FRET ratio (pink circles) and the normalized total paxillin intensity (light blue circles) from different individual cells, and their corresponding average curves (red and blue solid lines); (c–d) show the statistics of (c) the maximal Src activation and (d) Lam-FA disassembly by whisker plots. (*) indicates statistically significant difference between the data distribution using the Kolmogorov-Smirnov (KS) test, n = 20, 23 and 33, p ≤ 6.1e-4.
To quantitatively evaluate the effect of [FN] on the magnitude and kinetic Src-FA coordination in single cells, we applied CFIM to compare two distinct sets of [FN] (low: 2.5 μg/ml and high: 10 μg/ml). CFIM revealed that the linear correlation coefficient R and the slope of the magnitude Src-FA coordination were significantly lower in cells on high [FN] than those in cells on low [FN], with the coordination strength, R, decreased from 0.74 to 0.28, and the coordination capacity “slope” decreased from 0.84 to 0.32 (Figs. 5a–b). Furthermore, the peak value of the average CC-curve, K, representing the kinetic similarity, was significantly lower on high [FN], decreased from 0.84 to 0.74 (Fig. 5c). Consistently, the time delay between Src activation and Lam-FA disassembly, T, increased from 1.2 min to 4.3 min when [FN] was raised from 2.5 to 10 μg/ml (Fig. 5d). These results suggest that Src activation and Lam-FA disassembly are more closely coordinated when cells are seeded on low [FN]. Surprisingly, this tight coordination apparently only occurs at the rafts micro-domains, as the dominant Src activity reported by the general membrane-tagged KRas-Src biosensor outside of lipid rafts is uncoupled from the Lam-FA disassembly (Figs. 5e–f)18.
Figure 5. Fibronectin concentration affects the Src-FA coordination in magnitude and kinetics.

(a–b) The bar graphs represent parameters in Src-FA magnitude coordination: (a) the correlation coefficients and (b) the slopes of the linear models on different [FN]. (*) indicates significant difference based on the randomized test. Correlation coefficient: p = 0.02; Slope: p = 0.04. (c–d) show the parameters for the Src-paxillin kinetic coordination. Bar graphs represent (c) the maximal value (CC peak) and (d) the time delay of the Src-FA CC curves for cells seeded on different [FN], respectively. (*) indicates significant difference, p ≤ 0.04. (e–f) The comparison of (e) the correlation coefficients and (f) the slopes of the linear model between the Lyn-Src/Lam-FA and KRas-Src/Lam-FA coordinations. (*) indicates significant difference with p < 2.0e-4.
FAs mediated by different integrin subtypes have distinct Src-FA coordination
MEFs express integrin receptors including αvβ3 and α5β1 subtypes which can bind to FN35. Since a tighter Src-FA coordination was observed at lower [FN], we reasoned that integrin engagement at high [FN] may antagonize the Src-FA coupling. To investigate the contribution of different integrin subtypes, we examined the effect of antibodies blocking the interactions between integrin and FN, i.e. LM609 specific for integrin αvβ336 and MAB2514 for α5β137, when cells are seeded on 10 μg/ml [FN].
As shown in Supplementary Figure 5 and Supplementary Videos 3–4, the pre-treatment of blocking antibodies did not affect the basal morphology of the Lam-FAs before PDGF. Neither did the pre-treatment significantly affect the average magnitude of PDGF-induced Src activation, suggesting that this PDGF-induced Src activation was relatively independent of specific integrin ligation (Fig. 6a). When examining the averaged Lam-FA responses, the pre-treatment with any of the antibodies in blocking the nascent integrin ligation significantly enhanced the Lam-FA disassembly upon PDGF stimulation (Fig. 6b and Supplementary Videos 3–4). These results suggest that the overall PDGF-induced disassembly of Lam-FA populations is affected by integrins and their interactions with ECM proteins.
Figure 6. Integrins α5β1 and αvβ3 mediate FA populations with distinct Src-FA coordinations.

MEFs were seeded on 10 μg/ml [FN] with or without antibodies blocking different integrin receptors. (a–b) show the statistics of (a) the maximal Src activation and (b) maximal LAM-FA disassembly by whisker plots. (*) indicates significant difference by Kolmogorov-Smirnov test, n = 27, 26, 23 p ≤ 0.03. (c–f) The bar graphs with 95% confidence intervals compare the parameters of the Src-FA coordination: (c) the correlation coefficient; (d) the slope; (e) the maximal value (CC peak); (f) the time delay of the Src-FA CC curve. (**) indicates significant difference from all the other groups in the panel, (*) indicates significant difference between indicated groups: Correlation coefficients, p ≤ 0.02; Slope, p ≤ 0.05; CC Peak, p ≤ 0.02; Time delay, p ≤ 0.005, n = 24, 26, 20.
Single cell analysis by CFIM further showed that blocking integrin α5β1 function by MAB2514 significantly enhanced the linear correlation coefficient R and the slope of the max-max plot, clearly indicating an increased level of Src-FA coupling under PDGF stimulation upon the integrin α5β1 inhibition (Figs. 6c–d). The coordination strength and capacity increased from 0.28 to 0.45 and from 0.32 to 1.37, respectively. In contrast, the blockage of integrin αvβ3 by LM609 significantly inhibited this coordination, essentially reduced the coordination strength and capacity to near zero (Figs. 6c–d). These results suggest that the Src-driven disassembly mainly occurs in the αvβ3-mediated, but not α5β1-mediated, Lam-FA populations at the single-cell level.
Consistently, blocking integrin α5β1 functions, but not integrin αvβ3, significantly enhanced the kinetic Src-FA similarity and eliminated almost all the time delay between Src activation and Lam-FA disassembly (Figs. 6e–f). The Src-FA kinetic similarity increased from 0.74 to 0.81, and the Src-FA time lag decreased from 4.3 min to 0.8 min. Additional experiments confirmed that another rat antibody against integrin α5β1 (MAB16), also reduced the time delay between Src activation and Lam-FA disassembly (Supplementary Fig. 6). These results suggest that the relatively large time delay and uncoordinated action between Src activation and Lam-FA disassembly observed on the high [FN] group is mainly attributed to the integrin α5β1 ligation. Therefore, the Lam-FA populations mediated by integrin α5β1 are relatively independent of the control of Src activity, whereas those associated with integrin αvβ3 are closely coupled with Src activation upon PDGF stimulation.
Discussion
The seamless integration of advanced imaging and analysis technologies is essential for visualizing and quantifying the molecular interactions in single live cells with high spatiotemporal accuracy. Here we have integrated FRET imaging, automated computational method, and correlation analysis to develop a CFIM method for the quantitative evaluation of the spatiotemporal coordination between an enzymatic activity and a structural dynamics at subcellular regions (Fig. 2). CFIM utilizes the heterogeneity among different cells and the dynamic signals reported by live cell imaging to quantify the cause-effect characteristics including the coordination strength, capacity, the kinetic similarity and time delay between two molecular events. Growth factor stimulation was further employed to expose important signaling networks hidden at the basal state, enhancing the sensitivity and precision of our approach24,29,38.
Because of the ratiometric nature of FRET imaging and the normalization process employed in CFIM, the FRET signals and total paxillin intensity should be independent of the fluorescence protein expression level and the instability of optics39. Indeed, our results show that the final levels of Src FRET ratio are not correlated with those of total paxillin intensity, although the expression levels of the reporters may be correlated in cells (Supplementary Fig. 2). Only after further normalization with the basal-level signals, the responses of Src activity and Lam-FA induced by the growth factor become correlated at the single-cell level (Fig. 3 and Supplementary Fig. 2). Thus, these results highlight the accuracy and specificity of the CFIM approach in single live-cell imaging.
Our results indicate that the raft-domain Src activation is linearly correlated in magnitude with FA disassembly at lamellipodia regions, and that Src activation precedes FA disassembly in minutes. This linear relationship between two distinctive biochemical and biophysical quantities of Src activation and FA disassembly revealed from single cells is exciting and may serve as a general rule in governing the FA dynamics in live cells. The likelihood of Src-FA causality was quantitatively evaluated by coordination strength and kinetic similarity, and further confirmed by traditional experiments and analysis in SYF-/- cells expressing Src mutants. Thus, CFIM allows the quantitative evaluation of the coordination between molecular events in the innate single-cell system without the need for specific inhibitors.
It is intriguing that only the Src activity at lipid rafts (Lyn-Src), but not that occurring at general membrane regions (KRas-Src), is coordinated with the FA disassembly upon PDGF stimulation (Figs. 4e–f). This is consistent with the fact that FAs belong to highly ordered membrane micro-domains and lipid rafts40. Although the majority of Src activity is concentrated at the non-raft regions of the plasma membrane18,41, a fraction of Src can associate with FAK at FA sites and lipid rafts to modulate the FA regulation42. Therefore, the subcellular localization of Src is crucial for its roles in regulating FA dynamics. This concept can be extendable to other functions of Src in general. In fact, Src inhibits Rho GTPase at the FA sites43, whereas it activates Rho GTPase at podosomes44. These results clearly highlight the importance of subcellular localization of molecules in governing their functions.
CFIM also revealed that the Src-FA coordination can be modulated by the concentration of the matrix protein, fibronectin, and possibly its ligation to the corresponding integrin receptors, with Src activation tightly coupled with the disassembly of integrin αvβ3-mediated Lam-FA population but poorly coordinated with that mediated by integrin α5β1 (Fig. 7). As such, CFIM can measure the control parameters with high accuracy and statistical significance, providing powerful tools to unravel and quantify the underlying molecular mechanism governing the spatiotemporal molecular activity-structure relationship in live cells. This approach can be generally applicable to the investigation of, in principle, any pair of molecular activity/structure coordination in space and time. Similar to previously reported correlative or multiplexing analysis methods25, our approach is also readily applicable for studying the spatiotemporal relationship among a large number of signaling/structural events measured from different individual cells, by assigning one common molecular event as a reference to align the different molecular signals.
Figure 7. The proposed biological model.

The PDGF induced Src activation is directly coordinated with the disassembly of FA populations mediated by integrin αvβ3, but is relatively separated from the FAs mediated by integrin α5β1.
It has been proposed that the paxillin-containing FAs at the cell periphery consists of a continuum combination of classical FAs, fibrillar adhesions and focal complexes45. Our results suggest that different populations of Lam-FAs exist in a single cell, whose regulation routes are distinctively wired to govern their dynamics. Specifically, the disassembly of Lam-FAs mediated by integrin αvβ3 is tightly controlled by Src activity upon PDGF stimulation. In fact, the blockage of integrin αvβ3 significantly inhibited the coordination between Src activation and Lam-FA disassembly. Consistently, when integrin α5β1 was blocked, both magnitude and kinetic correlations were significantly enhanced, and the time lag between Src activation and Lam-FA disassembly was eliminated (Figs. 6c–f). It is possible that integrin αvβ3-associated Lam-FAs can be directly controlled by the Src-mediated phosphorylation events of paxillin, FAK, or ERK to cause protein cleavage and FA disassembly8,46. In contrast, the regulation of integrin α5β1-associated Lam-FAs may largely rely on events relatively independent of Src activity, such as the re-arrangement of the actin cytoskeleton or different endocytosis routes adopted by integrins47. These notes are consistent with previous reports that PDGFR co-localizes and can in fact physically associate with integrin αvβ3 and Src tyrosine kinase48,49,50,51, and that integrins β3 and β1 had distinctive dynamics inside focal adhesions52. Therefore, our work highlights the power of CFIM in quantitatively deciphering the complex and noisy signaling network distinctively wired at different subcellular locations in live cells.
Methods
Cell Culture, Transient Transfection and Reagents
Mouse embryonic fibroblasts (MEFs) (from ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 unit/ml penicillin, 100 μg/ml streptomycin, and 1 mM sodium pyruvate (GIBCO BRL) in a humidified 95% air, 5% CO2 incubator at 37°C before transfection. Different DNA plasmids were transfected into cells using Lipofectamine 2000 according to the protocol from the vendor (Invitrogen). The transfection condition was optimized for MEFs (60%–80% confluent), with 3.5 μg of DNA plasmids for each 35 mm cell culture dish. The amount of DNA transfected varies in this range between different experiments with Lyn-Src-YPet (2.5 μg) and mCherry-paxillin (1 μg). The Src biosensor was engineered in the PCDNA3.1 vector with the CMV promoter; mCherry-paxillin was engineered in the mCherry-C1 vector with the same promoter (CMV).
Cells expressing various exogenous proteins were starved in cell culture medium with 0.5% FBS for 36 hr before passing onto fibronectin-coated glass bottom dishes (Cell E&G) overnight prior to imaging. This step also served the purpose of synchronizing the status of cell cycle and reducing its effect on the variation of intracellular protein expression and molecular wiring. FN stock (1 mg/ml) was diluted with PBS to obtain the working solutions with different concentrations. The dishes were incubated with FN solutions at 37°C for 4 hr before usage to ensure sufficient coating. The coated FN density measured by fluorescent intensity has been shown to be linearly dependent on the concentration of FN solution at this range33. To inhibit the cell-matrix interaction, the MEF cells were pretreated with integrin αvβ3 antibody LM609, or integrin α5β1 antibody MAB2514 (10 μg/ml, EMD Millipore) for 2 hr before imaging and the application of PDGF.
Microscope Imaging
During imaging, the cells were maintained in CO2-independent medium (Gibco BRL) without serum at 37°C before stimulation by PDGF (10 ng/ml, Sigma). Images were collected by a Zeiss Axiovert inverted microscope equipped with 100× objective (1.4 NA) and a cooled charge-coupled device camera (Cascade 512 B; Photometrics) using the MetaFluor 6.2 software (Universal Imaging). The parameters of dichroic mirrors, excitation and emission filters for different fluorescence proteins were described previously19,39. In brief, the Lyn-Src biosensor was excited at 420 ± 20 nm, and the emissions collected at 475 ± 40 nm or 535 ± 25 nm for ECFP or FRET images, respectively. The mCherry-paxillin probe was excited at 560 ± 40 nm and the emission collected at 653 ± 95 nm for mCherry images.
Image Analysis
The image analysis was conducted using our customized software fluocell53 developed in MATLAB (version R2008a, The MathWorks Inc., Natick MA). The source code of fluocell is published via Google Code (http://code.google.com/p/fluocell). All the images were background-subtracted and smoothed using a median-filter with a window size of 3 × 3 pixels. A combination of the FRET images of the Lyn-Src biosensor and the mCherry-paxillin images were used to compute the masks of the cells. The mCherry images highlight the intensity value at the location of the FAs and ensure the inclusion of the FAs within the detected cell mask. The masks of the cell were then divided into five layers with the outer layer representing the lamellipodium region. In some cells, the FA sites were only correctly detected at the lamellipodia, but not at the perinuclear regions (Supplementary Fig. 1c). It also happened that some image frames could not encompass the whole-cell body. In these cells where only part of the cell body was imaged, a fan-shaped region was manually selected in the first image of the video sequence (Supplementary Figs. 1c–d). The interception of this fan region and the outer layer of the cell mask were utilized for all the images in the sequence to represent the active lamellipodium region of interest (ROI). As such, we quantified the average Src FRET ratio and total paxillin intensity within the Lam-FA region.
For the detection of FA locations based on the mCherry intensity images, the water algorithm27,54 was modified and employed (Supplementary Methods). This modification reduced the CPU time (on a laptop computer equipped with dual core Intel CPUs at 1.20 GHz and 2 GB of RAM) of the FA detection in one single image from 74.3 sec to 0.22 sec, and achieved a 338-fold increase of computing speed for a representative cell (Supplementary Fig. 1a). In fact, the pixels labeled as FAs using our modified method are the same as those detected by the water algorithm (Supplementary Fig. 1b). As such, this modification allowed us to not only detect the locations of the FAs as mask images with high efficiency but also quantify subcellular Src activity and paxillin dynamics of a large amount of images at these local sites, which is critical for the quantitative analysis of dynamic cellular processes.
The average Src ECFP/FRET ratio within the FAs in the regions of interest was obtained by calculating the average ratio between the ECFP and FRET images at the pixels that lie in both the ROI and FA mask. These Src ECFP/FRET ratios were further normalized by the mean average ratio within these corresponding regions before PDGF to obtain the time courses representing the dynamic and local Src activity as shown in Fig. 3a. The PDGF-induced increase of Src activity, or Src activation, was further quantified by normalized (Src ECFP/FRET ratio-1) (see Fig. 3b). Meanwhile, the total paxillin intensity was obtained by computing the sum of the intensity values from the mCherry image within the same regions and divided by the total paxillin intensity in the whole cell to account for photobleach. Then the time course of total paxillin intensity was normalized by the mean total intensity before PDGF to obtain normalized curves representing the percentage change of paxillin intensity comparing to the basal level (Fig. 3a). Since the normalized paxillin intensity curves usually have values between 0 and 1, the curve, (1-normalized intensity), was used to represent the level of paxillin disassembly (Fig. 3b). As a result, the paxillin disassembly curve measures the percentage of mCherry-paxillin leaving the FAs after stimulation. As such, these normalized and quantified amounts of Src activation and paxillin disassembly induced by PDGF should be independent of specific protein expression in individual cells.
In signal processing, the cross-correlation function is a measurement of similarity between two dynamics waveforms when a time-lag is applied. The discrete cross-correlation function (CC) between the signals A and B (A–B CC curve) were defined by the equation
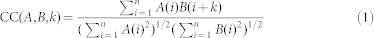 |
where,
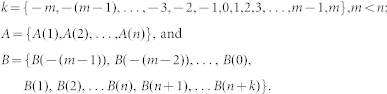 |
Our customized cross-correlation function was used instead of the MATLAB functions xcorr or xcov, which inaccurately extends the Be signals to both sides using the value zero (see Supplementary Methods). In addition, both signals were normalized such that the maximal value of the auto-correlation function is 1. The average CC curves and the standard error can be computed based on the values of CC curves from different cells.
To ensure the data quality for the cross-correlation analysis, only the cells with more than 10% Src activation and more than 10% FA disassembly were included. In addition, outlier cells are detected and excluded based on a test of the maximal values and the shape of the CC curves. If the maximal value or more than half of the CC curve lies outside of the range encompassing 3 times standard deviation extending from the average of all other CC curves, this particular cell is labeled as an outlier and excluded from the analysis55. This condition was applied and the process repeated until no outliers were detected. In our experiments, less than 20% of the cells were detected as outliers and excluded from the cross-correlation analysis.
Statistical Analysis
The Kolmogorov-Smirnov test (kstest, MATLAB) was used to compare the distribution of samples of maximal Src activation and paxillin disassembly since the KS test does not require the samples to have normal distribution (Supplementary Fig. 2). The statistical methods based on re-sampling of the data such as the bootstrap method and randomized testing were used to calculate the distribution of the parameters in the Src-paxillin magnitude and kinetic correlation, and estimate their statistical properties56,57. Following standard procedures, the distribution and 95% confidence intervals of the parameters were obtained by the bootstrap method for 2000 times allowing replications25,56 (bootstrap and bootci, MATLAB). In this paper, we chose the well-studied and widely used randomization tests to perform the statistical inference57. Randomization with 5000 repeats was generated and used for statistical inference on the parameters including the correlation coefficient, slope, max CC value and time delay.
Author Contributions
Author contributions: S.L., J.S., S.C. and Yingxiao W. designed research; S.L., J.S., SC.C., Yi W., J.E. and M.O. performed research; S.L., Yi W., S.C.C., J.L. and Yingxiao W. analyzed data; S.L., J.S., S.C. and Yingxiao W. wrote the paper.
Supplementary Material
Supplementary Methods, Figure and Video Legends
Movie S1
Movie S2
Movie S3
Movie S4
Acknowledgments
We thank Drs. Roger Y. Tsien and Ryan G. Holzer (University of California, San Diego), Maria Nemethova and Victor Small (Institute of Molecular Biotechnology, Vienna, Austria), Martin A. Schwartz (University of Virginia) for valuable reagents, constructs, and SYF-/- cell lines. We also thank Dr. Gandauz Danuser (Harvard University) for insightful discussion. We thank Jason Fan (University of Toronto, Canada) for critically proofreading this manuscript. This work is supported by grants from NIH HL098472, HL109142, NS063405, NSF CBET0846429 (Yingxiao W. and S. L.), NIH HL121365 (S.C. and Yingxiao W.), the University of Illinois Research Fund (S.L.) and the Wallace H. Coulter Foundation and Beckman Laser Institute, Inc. (Yingxiao W.). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Vicente-Manzanares M., Ma X., Adelstein R. S. & Horwitz A. R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo K. A., Dembo M., Kaverina I., Small J. V. & Wang Y. L. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol 153, 881–888 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel M. L., Schneider I. C., Aratyn-Schaus Y. & Waterman C. M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol 26, 315–333, 10.1146/annurev.cellbio.011209.122036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R., Itzkovitz S., Ma'ayan A., Iyengar R. & Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol 9, 858–867, 10.1038/ncb0807-858 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K., Ji L., Applegate K. T., Danuser G. & Waterman-Storer C. M. Differential transmission of actin motion within focal adhesions. Science 315, 111–115 (2007). [DOI] [PubMed] [Google Scholar]
- Kanchanawong P. et al. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584, 10.1038/nature09621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J. et al. Cell migration: integrating signals from front to back. Science 302, 1704–1709 (2003). [DOI] [PubMed] [Google Scholar]
- Webb D. J. et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol 6, 154–161 (2004). [DOI] [PubMed] [Google Scholar]
- Xie H. et al. EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCgamma signaling pathway. J Cell Sci 111 (Pt 5), 615–624 (1998). [DOI] [PubMed] [Google Scholar]
- Xu Y., Benlimame N., Su J., He Q. & Alaoui-Jamali M. A. Regulation of focal adhesion turnover by ErbB signalling in invasive breast cancer cells. Br J Cancer 100, 633–643 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V. J. & Frame M. C. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. Embo J 17, 81–92 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox E. A., Sastry S. K. & Huttenlocher A. Integrin-mediated adhesion regulates cell polarity and membrane protrusion through the Rho family of GTPases. Mol Biol Cell 12, 265–277 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L. M. & Hall A. Role of actin polymerization and adhesion to extracellular matrix in Rac- and Rho-induced cytoskeletal reorganization. J Cell Biol 138, 913–926 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Campbell R. E., Ting A. Y. & Tsien R. Y. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol 3, 906–918 (2002). [DOI] [PubMed] [Google Scholar]
- Wang Y., Shyy J. Y. & Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng 10, 1–38 (2008). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Visualizing the mechanical activation of Src. Nature 434, 1040–1045 (2005). [DOI] [PubMed] [Google Scholar]
- Lu S. et al. The spatiotemporal pattern of Src activation at lipid rafts revealed by diffusion-corrected FRET imaging. PLoS Comput Biol 4, e1000127 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J. et al. Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol 16, 48–57 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M., Sun J., Chien S. & Wang Y. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc Natl Acad Sci U S A 105, 14353–14358 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A., Wiseman P. W., Choi C., Horwitz A. R. & Gratton E. Stoichiometry of molecular complexes at adhesions in living cells. Proc Natl Acad Sci U S A 106, 2170–2175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A. et al. Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J Cell Sci 121, 196–204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. A. et al. Dynamic proteomics of individual cancer cells in response to a drug. Science 322, 1511–1516, 10.1126/science.1160165 (2008). [DOI] [PubMed] [Google Scholar]
- Sun J. et al. Antagonism between binding site affinity and conformational dynamics tunes alternative cis-interactions within Shp2. Nat Commun 4, 2037, 10.1038/ncomms3037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Saidon C., Cohen A. A., Sigal A., Liron Y. & Alon U. Dynamics and variability of ERK2 response to EGF in individual living cells. Mol Cell 36, 885–893, 10.1016/j.molcel.2009.11.025 (2009). [DOI] [PubMed] [Google Scholar]
- Machacek M. et al. Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch C. M., Elliott H., Danuser G. & Hahn K. M. Imaging the coordination of multiple signalling activities in living cells. Nat Rev Mol Cell Biol 12, 749–756, 10.1038/nrm3212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir E. et al. Molecular diversity of cell-matrix adhesions. J Cell Sci 112 (Pt 11), 1655–1669 (1999). [DOI] [PubMed] [Google Scholar]
- Webb D. J., Parsons J. T. & Horwitz A. F. Adhesion assembly, disassembly and turnover in migrating cells -- over and over and over again. Nat Cell Biol 4, E97–100, 10.1038/ncb0402-e97 (2002). [DOI] [PubMed] [Google Scholar]
- Sachs K., Perez O., Pe'er D., Lauffenburger D. A. & Nolan G. P. Causal protein-signaling networks derived from multiparameter single-cell data. Science 308, 523–529, 10.1126/science.1105809 (2005). [DOI] [PubMed] [Google Scholar]
- Klinghoffer R. A., Sachsenmaier C., Cooper J. A. & Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. Embo J 18, 2459–2471 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V. J., Wyke J. A. & Frame M. C. v-Src-induced degradation of focal adhesion kinase during morphological transformation of chicken embryo fibroblasts. Oncogene 10, 2247–2252 (1995). [PubMed] [Google Scholar]
- Palecek S. P., Loftus J. C., Ginsberg M. H., Lauffenburger D. A. & Horwitz A. F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385, 537–540 (1997). [DOI] [PubMed] [Google Scholar]
- Gupton S. L. & Waterman-Storer C. M. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 125, 1361–1374 (2006). [DOI] [PubMed] [Google Scholar]
- DiMilla P. A., Stone J. A., Quinn J. A., Albelda S. M. & Lauffenburger D. A. Maximal migration of human smooth muscle cells on fibronectin and type IV collagen occurs at an intermediate attachment strength. J Cell Biol 122, 729–737 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- Gao B., Saba T. M. & Tsan M. F. Role of alpha(v)beta(3)-integrin in TNF-alpha-induced endothelial cell migration. Am J Physiol Cell Physiol 283, C1196–1205 (2002). [DOI] [PubMed] [Google Scholar]
- Tang C. H., Lu D. Y., Tan T. W., Fu W. M. & Yang R. S. Ultrasound induces hypoxia-inducible factor-1 activation and inducible nitric-oxide synthase expression through the integrin/integrin-linked kinase/Akt/mammalian target of rapamycin pathway in osteoblasts. J Biol Chem 282, 25406–25415, 10.1074/jbc.M701001200 (2007). [DOI] [PubMed] [Google Scholar]
- Pe'er D. & Hacohen N. Principles and strategies for developing network models in cancer. Cell 144, 864–873, 10.1016/j.cell.2011.03.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M. et al. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J Biol Chem 283, 17740–17748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaus K., Le Lay S., Balasubramanian N. & Schwartz M. A. Integrin-mediated adhesion regulates membrane order. J Cell Biol 174, 725–734 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L. A., Klinghoffer R. A., Sachsenmaier C. & Cooper J. A. SRC catalytic but not scaffolding function is needed for integrin-regulated tyrosine phosphorylation, cell migration, and cell spreading. Mol Cell Biol 22, 2427–2440 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S. K. & Schlaepfer D. D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18, 516–523 (2006). [DOI] [PubMed] [Google Scholar]
- Thomas S. M. & Brugge J. S. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 13, 513–609 (1997). [DOI] [PubMed] [Google Scholar]
- Berdeaux R. L., Diaz B., Kim L. & Martin G. S. Active Rho is localized to podosomes induced by oncogenic Src and is required for their assembly and function. J Cell Biol 166, 317–323 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. T., Horwitz A. R. & Schwartz M. A. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–643, 10.1038/nrm2957 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quizi J. L. et al. SLK-mediated phosphorylation of paxillin is required for focal adhesion turnover and cell migration. Oncogene 10.1038/onc.2012.488 (2012). [DOI] [PubMed] [Google Scholar]
- Margadant C., Monsuur H. N., Norman J. C. & Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol 23, 607–614, 10.1016/j.ceb.2011.08.005 (2011). [DOI] [PubMed] [Google Scholar]
- Schneller M., Vuori K. & Ruoslahti E. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. Embo J 16, 5600–5607, 10.1093/emboj/16.18.5600 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Salgado E. G. et al. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U S A 100, 13298–13302, 10.1073/pnas.2336149100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon T. J. & Eck M. J. Structure and regulation of Src family kinases. Oncogene 23, 7918–7927 (2004). [DOI] [PubMed] [Google Scholar]
- Woodard A. S. et al. The synergistic activity of alphavbeta3 integrin and PDGF receptor increases cell migration. J Cell Sci 111 (Pt 4), 469–478 (1998). [DOI] [PubMed] [Google Scholar]
- Rossier O. et al. Integrins beta1 and beta3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat Cell Biol 14, 1057–1067, 10.1038/ncb2588 (2012). [DOI] [PubMed] [Google Scholar]
- Lu S. et al. Computational analysis of the spatiotemporal coordination of polarized PI3K and Rac1 activities in micro-patterned live cells. PLoS ONE 6, e21293, 10.1371/journal.pone.0021293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N. Q. et al. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol 3, 466–472 (2001). [DOI] [PubMed] [Google Scholar]
- Cipra T. et al. Detection and interpolation of outliers in biosignals. Act Nerv Super (Praha) 32, 283–291 (1990). [PubMed] [Google Scholar]
- Efron B. & Tibshirani R. J. An Introduction to the Bootstrap. (Chapman & Hall, 1993). [Google Scholar]
- Manly B. F. J. Randomization, Bootstrap and Monte Carlo Methods in Biology. 2nd edn, (Chapman & Hall, 1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Figure and Video Legends
Movie S1
Movie S2
Movie S3
Movie S4