

Abstract
Background
Despite reducing pneumonia and other infections, antibody replacement does not appear to treat pulmonary lymphoid hyperplasia (PLH) in patients with common variable immunodeficiency (CVID). The pathogenesis and optimal treatments remain to be clarified.
Objective
We aimed to better understand the pathology of CVID-associated lung disease. Tertiary lymphoneogenesis, although a component of interstitial lung disease associated with autoimmune diseases, has not previously been explored in patients with CVID.
Methods
We examined the clinical characteristics and pathologic findings of 6 patients with CVID with nodular/infiltrative lung disease who had biopsy specimens demonstrating PLH.
Results
In these subjects regions of PLH contained distinct Band T-cell zones, with B-cell predominance in 1 patient and T-cell predominance in the others. Colocalization of Ki67, Bcl6, and CD23 within this ectopic lymphoid architecture demonstrated tertiary lymphoneogenesis with active centers of cellular proliferation. One patient received rituximab with improved pulmonary radiologic findings.
Conclusion
Ectopic lymphoid tissue forming germinal centers suggest tertiary lymphoneogenesis in CVID-associated lung disease. B cell–targeted therapy might disrupt CVID-associated lymphoid hyperplasia.
Keywords: Common variable immunodeficiency, lymphoid neogenesis, chronic lung disease, pulmonary hyperplasia, germinal center, ectopic follicle
Common variable immunodeficiency (CVID) is the most common symptomatic primary immunodeficiency and is characterized by markedly reduced levels of immunoglobulins coupled with a failure to mount functional antibody responses to immunization or infection.1,2 The genetic basis for CVID is known only in a minority of cases, and the biological mechanisms leading to predisposition and progression remain poorly understood.3 Aside from reducing the incidence of pneumonia and other infections,4–6 antibody replacement has minimal effect on the course of noninfectious complications. These have emerged as major causes of both morbidity and mortality in patients with CVID.1–3
Lymphoid infiltrates, granulomatous lung disease, or both are relatively common complications affecting 28.5% to 58% of patients with CVID, depending on the population studied.7–10 High-resolution computed tomography (CT) in these cases demonstrates bronchiectasis, bronchial wall thickening, air trapping, parenchymal consolidation, emphysema, scarring/fibrosis, and/or nodular changes.11–13 In general, the lung pathology in patients with CVID reflects interstitial lung disease, including lymphocytic interstitial pneumonia (LIP), follicular bronchiolitis, granulomatous lung disease, and organizing pneumonia.14 Follicular bronchiolitis, nodular lymphoid hyperplasia, reactive lymphoid infiltrates, and LIP are all forms of pulmonary lymphoid hyperplasia (PLH), within which poorly formed granulomas can also be found.15–17 PLH is included within the umbrella term granulomatous-lymphocytic interstitial lung disease (GLILD), which is used for the pathologic combination of granulomas and lymphoid hyperplasia in CVID-associated lung disease.7,15 However, some limit the use of GLILD to patients in whom both granulomas and lymphocytic infiltrates have been documented.18 In addition to the poorly formed granulomas of PLH, disseminated granulomatous disease in patients with CVID can present as a systemic disorder not confined to the lungs, with granulomata in lymph nodes and organs, such as the liver, skin, and spleen.19,20 Although increasing doses of immunoglobulin replacement improve lung function in some patients with CVID,21,22 for most patients, the inflammatory lung disease appears to be progressive.12 Further study of the cellular constituents of CVID-associated PLH can provide needed clues as to more effective treatment.
METHODS
Patients
Six patients with CVID-associated lung disease seen at the Mount Sinai Medical Center in New York, New York, who underwent lung biopsy that demonstrated PLH between January 2002 and June 2012 were included in this study. These patients were given a diagnosis of CVID based on the presence of 2 parameters: (1) quantitative serum immunoglobulin levels of less than the laboratory reference range of IgG (<500 mg/dL) and very low IgA levels, IgM levels, or both and (2) demonstrated absence of protective levels of antibody for previous immunizations. Clinical and laboratory information was collected from patients’ medical records. This study was approved by the Institutional Review Board at the Mount Sinai School of Medicine.
Biopsy
Five patients underwent open lung biopsy through video-assisted thoracic surgery, and 1 patient, patient 2, had an endoscopic transbronchial biopsy. All biopsies were conducted as part of evaluation of patients with CVID with clinical symptoms and CT findings suggestive of significant lung disease. Pathologic diagnoses were provided by board-certified pathologists and reviewed retrospectively. Patients included in the study had pathology consistent with PLH, including diagnoses of LIP, follicular bronchiolitis, nodular lymphoid hyperplasia, and reactive lymphoid infiltrates. Consistent with PLH,15–17 ill-defined and poorly formed granulomatous inflammation was noted in 4 of the patients, but frank granulomatous disease or well-circumscribed granulomas were not present in any of the pathology samples examined. Handling and processing of samples was done per typical institutional clinical protocols.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections from pulmonary biopsy specimens were used for immunohistochemical analysis. Staining with hematoxylin and eosin, as well as the immunologic markers CD3, CD20, CD23, CD45, Bcl6, and Ki67, was done as part of routine pathologic evaluation or requested as additional studies if paraffin-embedded tissue was available. Appropriate positive and negative controls were reviewed. Requests for pathology slides and embedded tissue from outside institutions were conducted in accordance with medical release authorization protocols.
RESULTS
Clinical characteristics of patients with CVID-associated PLH
Six patients were identified for this study on the basis of a lung biopsy done to further define the radiologic changes observed. Age at lung biopsy, quantitative immunoglobulin levels, method of biopsy, and pathologic diagnosis were obtained (Table I). Patients ranged in age from 28 to 52 years at the time of biopsy and had quantitative IgG levels of 39 to 400 mg/dL, IgA levels of 5 to 44 mg/dL, and IgM levels of less than the level of detection to 168 mg/dL. Slides were evaluated by 2 pathologists, and patients were given a pathologic diagnosis compatible with PLH, including diagnoses of LIP, follicular bronchiolitis, nodular lymphoid hyperplasia, and reactive lymphoid infiltrates.
TABLE I.
Clinical characteristics: Part 1
| Patient no. | Age (y) | Sex | IgG (mg/dL) | IgA (mg/dL) | IgM (mg/dL) | Method of biopsy | Diagnosis |
|---|---|---|---|---|---|---|---|
| 1 | 35 | F | 322 | 7 | 168 | VATS | LIP/follicular bronchiolitis |
| 2 | 29 | M | 400 | 7 | 61 | Transbronchial | Lymphoid interstitial pneumonia |
| 3 | 52 | F | 149 | 7 | 21 | VATS | Bronchiocentric interstitial pneumonia with lymphoid hyperplasia |
| 4 | 41 | M | 39 | 44 | 21 | Open | LIP |
| 5 | 41 | M | 57 | 5 | <1 | VATS | Atypical reactive lymphoid infiltrate with organizing pneumonia |
| 6 | 28 | F | 312 | 16 | 17 | VATS | Atypical nodular lymphoid infiltrate |
Age listed is age at biopsy. Immunoglobulin levels provided were measured at diagnosis before initiating immunoglobulin replacement therapy. Open biopsies were performed with video-assisted thoracoscopic surgery. Diagnoses provided were as reported by the attending pathologists reviewing the case.
F, Female; M, male; VATS, video-assisted thoracoscopic surgery.
Patients underwent pulmonary function testing before lung biopsy. FEV1 and FEV1/forced vital capacity ratio were variable between subjects: 1 patient had mild obstruction (patient 4), 2 patients had restrictive lung disease (patients 3 and 5), and the remaining patients had no evidence of obstruction or restriction (patients 1, 2, and 6; Table II). Measurement of diffusing capacity of the lung for carbon monoxide (DLCO) was more consistent among subjects than FEV1 or FEV1/forced vital capacity ratio, with 4 of 5 patients demonstrating compromised diffusion. Patient 6 did not have DLCO measured before lung biopsy. Retrospective review of radiologic reports from chest CT scans conducted before lung biopsy revealed consistent findings: all 6 patients had reported nodular opacities, 5 of 6 had mediastinal adenopathy, 2 had areas of ground-glass opacity, and 2 had reported evidence of bronchiectasis. Four of 6 patients had a history of pneumonia. Five of the 6 patients had a history of immune thrombocytopenic purpura, 1 had a history of autoimmune hemolytic anemia, and 1 had a history of CVID-associated enteropathy.
TABLE II.
Clinical characteristics: Part 2
| Patient no. | FEV1 (% predicted) | FEV1/FVC ratio (%) | DLCO (% predict) | Complications | CT findings |
|---|---|---|---|---|---|
| 1 | 113 | 108 | 70 | ITP | Nodular opacities Ground-glass opacity Mediastinal lymphadenopathy |
| 2 | 88 | 76 | 58 | ITP AIHA |
Nodular opacities |
| 3 | 61 (before) 62 (after) |
98 | 51 | PNA Enteropathy |
Nodular opacities Mediastinal lymphadenopathy Bronchiectasis |
| 4 | 71 | 75 | 109 | PNA ITP |
Nodular opacities Mediastinal lymphadenopathy |
| 5 | 71 (before) 72 (after) |
104 | 77 | PNA ITP |
Nodular opacities Ground-glass opacity Mediastinal lymphadenopathy |
| 6 | 91 | 107 | NA | PNA ITP |
Nodular opacities Mediastinal lymphadenopathy Bronchiectasis |
Pulmonary function testing was conducted before biopsy. One patient, patient 6, did not have DLCO measured as part of preoperative evaluation. CT findings are provided retrospectively as diagnosed by attending radiologists.
AIHA, Autoimmune hemolytic anemia; FVC, forced vital capacity; ITP, immune thrombocytopenic purpura; PNA, pneumonia.
CVID-associated PLH is lymphocytic inflammation with a variable ratio of T and B cells
For immunohistochemical analysis, we used CD45 as a marker of all leukocytes, CD3 as a marker of T cells, and CD20 as a marker of B cells. In concordance with previously reported studies, 5 patients with CVID and PLH had a predominance of T cells in the pulmonary lymphocyte infiltrate (Fig 1, patients 1 and 3, and Table III), with CD4+ lymphocytes as the predominant T cells (Fig 1, B, and Table III). However, the B-cell/T-cell ratio was found to be variable in patients with CVID-associated PLH because patient 5 had a B-cell predominance, with the majority of CD45+ leukocytes also being CD20+ (Fig 1, A, patient 5). Interestingly, patient 5 had been initially given a diagnosis of a pulmonary mucosa-associated lymphoid tissue lymphoma based on the presence of a monoclonal B-cell population based on PCR gene rearrangement studies, perhaps reflecting the dominance of large proliferating B-cell follicles in this case. The diagnosis was subsequently revised to atypical reactive lymphoid infiltrate with organizing pneumonia; flow cytometry demonstrated mixed κ and λ chain expression, suggesting polyclonal B-lymphocyte expansion consistent with PLH rather than monoclonal B-cell malignancy. In all cases examined, the vast majority of CD45+ leukocytes in the lungs are CD33− lymphocytes (88.3%), either CD3+ T cells (77.39%) or CD19+ B cells (12.42%; Fig 1, B, and Table III). Although EBV has been associated with PLH,16 results of immunohistochemical staining for EBV-encoded small RNAs were negative in all subjects.
FIG 1.

The B-cell/T-cell ratio is variable in patients with CVID-associated PLH. A, Immunohistochemical staining of lungs for 2 representative patients demonstrating that the B-cell/T-cell ratio is variable among patients with CVID-associated PLH (original magnification ×40). B, Flow cytometric analysis of lungs from a representative patient with CVID-associated PLH.
TABLE III.
Summary of pathologic characteristics
| Patient no. | T cells | B cells | Lymphoid neogenesis |
|---|---|---|---|
| 1 | CD3+, CD4+ 63.2%, CD3+, CD8+: 12.8% | CD19+: 12.4% | Formation of reactive follicular centers (BCL2−, BCL6+) and intact follicular dendritic cell networks (CD23+) |
| 2 | CD3+, CD4+ 51%, CD3+, CD8+: 41% | CD19+: 3% | Presence of ectopic B-cell follicles |
| 3 | Majority of CD45+ cells are also CD3+ | B cells are confined to follicular aggregates | Extensive lymphocytic infiltrates in both diffuse and follicular arrangements with germinal centers |
| 4 | Diffuse CD3+ T cells are numerous and most prominent leukocyte population | CD20+ B cells are limited to follicular structures | Lymphoid follicles of CD20+ B cells and CD23+ follicular dendritic cells containing BCL6+ germinal centers |
| 5 | CD3+ T cells diffusely distributed in the background of B-cell nodules | Large nodules of CD20+ B cells are prominent | Focal small germinal centers within follicles highlighted as CD20+, CD10+, and Ki67+ |
| 6 | Slight predominance of T cells relative to B cells | CD20+ B cells localized within numerous nodules | Ectopic follicles with 5% to 10% CD10+ germinal center B cells |
Pulmonary lymphocytes are organized into B-cell follicles and T-cell zones in patients with CVID-associated PLH
Peribronchial lymphocyte infiltrate of patients with CVID-associated PLH was characterized by distinct segregation into apparent B- and T-cell zones (Fig 2). Consistent among all patients, CD20+B cells were found as components of the ectopic follicles, containing only a few interspersed CD3+ T cells. In contrast, T cells were more diffusely present, with a range overlapping that of B cells in some areas but also existent in areas in which no B cells were found. In patient 5, the only patient with PLH with overall B-cell predominance, T cells were found circumscribing large B-cell follicles. In cases of T cell–predominant PLH, such as patient 4, CD3+ T lymphocytes were found both encircling B-cell follicles and in T cell–predominant zones in the periphery of the follicles.
FIG 2.

Pulmonary lymphocytes are organized into B- and T-cell zones in patients with CVID-associated PLH. Hematoxylin-eosin and immunohistochemical staining for CD3 (T cells) and CD20 (B cells) is shown for 5 patients. Areas of B- and T-cell predominance are labeled as B and T, respectively. Magnification of images is ×40.
B-cell follicles in patients with CVID-associated PLH have characteristics of ectopic germinal centers
Immunohistochemical staining of lungs for 2 representative patients demonstrated CD20+ B-cell follicles containing Ki67+ proliferating cells23 within Bcl6+ germinal centers24,25 colocalized with CD23+ follicular dendritic cells,26,27 whereas a third representative patient demonstrates B-cell follicles (CD20+) containing germinal centers (CD10+) with follicular T cells (CD3+) interspersed (Fig 3). Ectopic B-cell follicles were described in the pathology reports of all 6 patients, with germinal centers noted in all 5 patients who underwent open lung biopsy or video-assisted thoracoscopic surgery and were evaluated for this finding (Table III). Taken together, these findings demonstrate ectopic lymphoid follicles with actively proliferating germinal centers and follicular dendritic cell networks within the lungs of patients with CVID with PLH.
FIG 3.

Ectopic follicles and germinal centers with localized proliferation in patients with CVID-associated PLH. Magnification is ×100 in patient 1, ×40 in patient 4, and ×200 in patient 5.
Use of rituximab in patients with CVID-associated PLH
Patient 1 received B cell–depletive therapy with rituximab as a treatment for CVID-associated PLH. As described above, the patient was given a diagnosis of LIP with tertiary lymphoid follicles and localized cellular proliferation within the pulmonary parenchyma. It was the identification of these ectopic B-cell follicles that inspired the use of rituximab. Of note, her PLH had a T-cell predominance (Fig 1, B). She received 750 mg of rituximab intravenously on a weekly basis with 40 mg of dexamethasone, acetaminophen, and diphenhydramine pretreatment for a total of 6 weeks, with eradication of B cells in the blood confirmed by means of flow cytometry. The patient had no adverse events as a result of the therapy. Treatment with rituximab was added to a baseline treatment of chronic lung disease with 250 mg of azithromycin 3 times weekly and inhaled 80/4.5 μg budesonide/formoterol twice daily.
Two months after the completion of therapy, the clinical response to rituximab was assessed by means of high-resolution CT of the chest and pulmonary function testing. The CT scan demonstrated amelioration of radiologic pathology compared with a study done 3 months before rituximab treatment, with significant interval improvement of ground-glass and solid opacities, as well as hilar and mediastinal adenopathy, and few persistent scattered nodules that were stable or smaller than in the prior study (Fig 4). Posttreatment pulmonary function testing conducted 3 months after completion of rituximab revealed no change in her diminished DLCO (70% of predicted value before treatment vs 69% of predicted value after treatment). Of note, the patient reported subjective improvement in her exercise tolerance and respiratory capacity after rituximab treatment.
FIG 4.
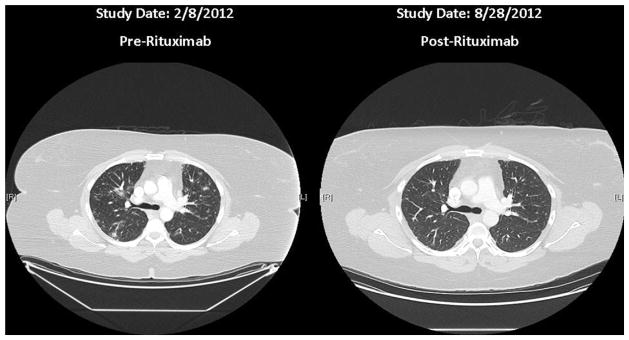
CT scan of the chest of patient 1 before and 2 months after rituximab treatment. The CT scan demonstrated significant interval improvement after rituximab treatment, with resolution of ground-glass and solid opacities, as well as hilar and mediastinal adenopathy, with few persistent scattered nodules that were stable or smaller than those in the prior study.
DISCUSSION
All patients had diagnoses consistent with PLH, including LIP, follicular bronchiolitis, nodular lymphoid hyperplasia, and reactive lymphoid infiltrate.16,28–30 PLH represents a clinically relevant proportion of CVID-associated lung disease because nearly 40% of patients with CVID with respiratory symptoms and radiographic findings had PLH in a large study.7 CVID-associated lung disease can have either obstructive31 or restrictive32 respiratory phenotypes, and we similarly found pulmonary function testing to be variable among our patients. In contrast, diminished DLCO was a more consistent finding that was noted in 4 of the 5 patients tested. This finding supports DLCO as the most reliable parameter of pulmonary function testing to assist in the diagnosis of CVID-associated lung disease. Nodular opacities were noted on CT scans for all patients, suggesting this might be a defining feature of CVID-associated PLH. Ground-glass opacity and bronchiectasis were each noted in one third of patients, a similar proportion to that previously reported.12,13
All patients included in this study had extrapulmonary noninfectious complications associated with CVID, including immune thrombocytopenic purpura, autoimmune hemolytic anemia, and enteropathy. This finding highlights the overlapping consequences of dysregulated immune responses and the association of CVID-associated lung disease with autoimmunity and lymphoproliferative disorders.7,11 Two thirds of the patients with PLH had a history of pneumonia. Supporting the idea of stratified immune dysregulation among CVID syndromes, the degree of disruption in memory B-cell differentiation correlates with the risk for lung disease.33 Similarly, we speculate that immune dysregulation imparting greater risk of pneumonia in patients with CVID also leads to lymphoid hyperplasia because increased B-cell expansion might be a failed attempt to compensate for a lack of protective affinity-matured, class-switched cells or another manifestation of exaggerated lymphoproliferation that underlies the benign and malignant complications of CVID.34 Studies have linked infectious agents, such as EBV and human herpesvirus 8, to PLH.7,16 We did not test for human herpesvirus 8, but EBV was not present in any of the pulmonary specimens examined.
In all patients we found distinct B-cell follicles and T-cell zones, demonstrating lymphoneogenesis as a feature of CVID-associated PLH. Accordingly, this lymphoid architecture allows for B- and T-cell interactions locally within the lung, mimicking those that occur in secondary lymphoid tissue. Germinal centers are sites of B-cell affinity maturation and isotype class-switching within lymphoid tissue as a result of T-cell coactivation and stimulation.35 We noted ectopic Bcl6+ or CD10+ germinal centers within networks of CD23+ follicular dendritic cells and CD3+ T cells in patients with CVID-associated PLH. This finding demonstrates that despite deficient maturation of antibody responses inherent to CVID, tertiary lymphoneogenesis defined by ectopic germinal centers occurs within patients with PLH. Additionally, we identified Ki67+ proliferating cells within the ectopic B-cell follicles, providing evidence that these are sites of localized cellular division within the lung.
Tertiary lymphoid neogenesis has been described as a feature of many chronic inflammatory states,36 including those affecting the lung.37 B cell–targeted therapy has demonstrated potential in multiple chronic inflammatory conditions in which tertiary lymphoid neogenesis has been identified, including multiple sclerosis, rheumatoid arthritis, and Sjögren syndrome.38–43 Notably, lung diseases associated with such conditions have also been successfully treated with rituximab, including pulmonary complications of antisynthetase syndrome, connective tissue disease, rheumatoid arthritis, and Sjögren syndrome.44–47 Similarly implicating B lymphocytes in the pathogenesis of pulmonary complications associated with humoral immunodeficiency, it is salient that chronic lung disease is less common in X-linked agammaglobulinemia,48 an immune defect characterized by the absence of B cells. Thus B cells, by acting as antigen-presenting cells, producing proinflammatory chemokines and cytokines, or both, might perpetuate the leukocyte accumulation within the lung in patients with CVID. Emphasizing the antibody-independent role of B cells in perpetuating chronic inflammation, B cell–depletive therapy in patients with other diseases has demonstrated efficacy as a treatment of autoimmunity, even without significantly reducing autoantibody levels.49
In 5 of the 6 cases examined, T cells were the predominant lymphocyte found in biopsy samples, whereas in one case B cells were the dominant lymphocyte. T cell–dominant LIP has been previously reported in patients with CVID,50 but B cell–dominant PLH has not. All prior reports of immunomodulatory treatment for GLILD have used T cell–targeted therapy,51–53 and one might postulate that B cell–dominant disease might be more responsive to rituximab or other B cell–targeted therapies. In our case the patient receiving rituximab demonstrated significant radiologic improvement, despite the presence of only 12% B cells versus 77% T cells in the pulmonary infiltrate, suggesting that T-cell inflammation was at least partially ameliorated. Thus B cells might be providing local antigen presentation, cellular expansion/recruitment signals, or both that promote PLH, regardless of whether it is B- or T-cell dominant. Additionally, nodules noted by using CT scans might be representative of ectopic B-cell follicles, thus explaining the resolution of the radiographic finding after B-cell depletion. Rituximab monotherapy might be a useful treatment option for patients with CVID-associated PLH, particularly in patients who might not tolerate chronic T cell–targeted therapy.
The absence of an improvement in DLCO might be attributable to the relatively high pretreatment DLCO because a study of rituximab in patients with interstitial lung disease associated with connective tissue disease demonstrated improvements in DLCO 9 to 12 months after initiation of therapy in patients in whom DLCO ranged from 16% to 54% before treatment.45 Additionally, improvement in DLCO might be limited by the short-term follow-up of our patient, who only received 1 course of rituximab. Treatment of GLILD with azathioprine (or other T cell–targeted therapy) for 18 weeks in combination with 4 to 6 courses of rituximab resulted in significant improvement in both lung radiographic findings and DLCO in some subjects.52 Larger studies are necessary to determine whether rituximab monotherapy is a useful treatment for patients with CVID-associated PLH.
Multiple treatments for patients with CVID-associated chronic lung disease have been used with success in reports of small sample size, including corticosteroids,50 cyclosporine,53 and infliximab.54 Chase et al52 have published a pilot study demonstrating the efficacy of combination therapy with azathioprine and rituximab in treating CVID-associated GLILD to suppress both T- and B-lymphoid pathways. Notably, the response to treatment of CVID GLILD is variable and often unsatisfactory, with agents such as azathioprine or cyclophosphamide described as efficacious in certain cases but ineffective in others.51 A possible reconciliation of these disparate findings is that the B-cell follicles and germinal centers of patients with CVID-associated PLH are important targets for therapy, and T cell–targeted therapy alone might not be adequate in many cases. Moreover, anti-TNF therapy might be more appropriate for patients with CVID-associated disseminated granulomatous disease, in whom it has shown some success,54,55 although it has not been examined in detail in patients with PLH.
Our work describes the immunopathology of CVID-associated PLH in 6 cases, with the novel identification of tertiary lymphoneogenesis within the lungs. Ki67+ staining demonstrates that cellular expansion occurs locally within B-cell follicles in patients with CVID-associated PLH, disruption of which by rituximab could be essential to effective treatment. This ectopic lymphoid activity might provide a mechanism by which PLH is perpetuated in patients with CVID. More research is needed to determine whether ectopic follicular organization and tertiary lymphoid neogenesis are important mechanisms of pathogenesis. Likewise, further studies will be essential in establishing the most appropriate treatment approach for the spectrum of CVID-associated lung disease.
Clinical implications.
Pulmonary lymphoneogenesis is a novel finding in patients with CVID. Immunomodulatory therapy aiming at disrupting leukocyte accumulation and proliferation within PLH should be considered.
Acknowledgments
Supported by the Thrasher Research Fund Early Career Award, National Institutes of Health grants AI 048693 and AI 061093, the Jeffrey Modell Foundation, and the David S. Gottesman Immunology Chair.
We thank Laureen Ojalvo for critically reading this manuscript and the Department of Pathology at Mount Sinai Medical Center for assistance in preparation of paraffin-embedded sections and immunohistochemical staining. We also thank all health care providers who helped in the management of these patients and the patients for their participation.
Abbreviations used
- CT
Computed tomography
- CVID
Common variable immunodeficiency
- DLCO
Diffusing capacity of the lung for carbon monoxide
- GLILD
Granulomatous-lymphocytic interstitial lung disease
- LIP
Lymphocytic interstitial pneumonia
- PLH
Pulmonary lymphoid hyperplasia
Footnotes
Disclosure of potential conflict of interest: P. J. Maglione has received grants from the Thrasher Research Fund and the Jeffrey Modell Foundation. C. Cunningham-Rundles has received grants from the National Institutes of Health. The rest of the authors declare that they have no relevant conflicts of interest.
References
- 1.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489–502. doi: 10.1016/S0140-6736(08)61199-X. [DOI] [PubMed] [Google Scholar]
- 3.Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 4.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–60.e4. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 5.Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 2010;137:21–30. doi: 10.1016/j.clim.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109:1001–4. doi: 10.1067/mai.2002.124999. [DOI] [PubMed] [Google Scholar]
- 7.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114:415–21. doi: 10.1016/j.jaci.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 8.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 9.Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–16. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 10.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bondioni MP, Soresina A, Lougaris V, Gatta D, Plebani A, Maroldi R. Common variable immunodeficiency: computed tomography evaluation of bronchopulmonary changes including nodular lesions in 40 patients. Correlation with clinical and immunological data. J Comput Assist Tomogr. 2010;34:395–401. doi: 10.1097/RCT.0b013e3181cad9da. [DOI] [PubMed] [Google Scholar]
- 12.Gregersen S, Aalokken TM, Mynarek G, Fevang B, Holm AM, Ueland T, et al. Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol. 2010;104:503–10. doi: 10.1016/j.anai.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Touw CM, van de Ven AA, de Jong PA, Terheggen-Lagro S, Beek E, Sanders EA, et al. Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy Immunol. 2010;21:793–805. doi: 10.1111/j.1399-3038.2009.00963.x. [DOI] [PubMed] [Google Scholar]
- 14.Hampson FA, Chandra A, Screaton NJ, Condliffe A, Kumararatne DS, Exley AR, et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol. 2012;67:587–95. doi: 10.1016/j.crad.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 15.Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID) Clin Immunol. 2010;134:97–103. doi: 10.1016/j.clim.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122:2150–64. doi: 10.1378/chest.122.6.2150. [DOI] [PubMed] [Google Scholar]
- 17.Guinee DG., Jr Update on nonneoplastic pulmonary lymphoproliferative disorders and related entities. Arch Pathol Lab Med. 2010;134:691–701. doi: 10.5858/134.5.691. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez Perez ER. Granulomatous lymphocytic interstitial lung disease. Immunol Allergy Clin North Am. 2012;32:621–32. doi: 10.1016/j.iac.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009;133:198–207. doi: 10.1016/j.clim.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis associated CVID: a case-control versus sarcoidosis study. Eur Respir J. 2013;41:115–22. doi: 10.1183/09031936.00189011. [DOI] [PubMed] [Google Scholar]
- 21.Arish N, Eldor R, Fellig Y, Bogot N, Laxer U, Izhar U, et al. Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax. 2006;61:1096–7. doi: 10.1136/thx.2004.029819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Gracia J, Vendrell M, Alvarez A, Pallisa E, Rodrigo MJ, de la Rosa D, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol. 2004;4:745–53. doi: 10.1016/j.intimp.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol. 2010;105:193–210. doi: 10.1016/S0065-2776(10)05007-8. [DOI] [PubMed] [Google Scholar]
- 25.Okada T, Moriyama S, Kitano M. Differentiation of germinal center B cells and follicular helper T cells as viewed by tracking Bcl6 expression dynamics. Immunol Rev. 2012;247:120–32. doi: 10.1111/j.1600-065X.2012.01120.x. [DOI] [PubMed] [Google Scholar]
- 26.Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. 2012;150:194–206. doi: 10.1016/j.cell.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park CS, Choi YS. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology. 2005;114:2–10. doi: 10.1111/j.1365-2567.2004.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koss MN, Hochholzer L, Langloss JM, Wehunt WD, Lazarus AA. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology. 1987;19:178–85. doi: 10.3109/00313028709077131. [DOI] [PubMed] [Google Scholar]
- 29.Popa V. Lymphocytic interstitial pneumonia of common variable immunodeficiency. Ann Allergy. 1988;60:203–6. [PubMed] [Google Scholar]
- 30.Tian X, Yi ES, Ryu JH. Lymphocytic interstitial pneumonia and other benign lymphoid disorders. Semin Respir Crit Care Med. 2012;33:450–61. doi: 10.1055/s-0032-1325156. [DOI] [PubMed] [Google Scholar]
- 31.Martinez Garcia MA, de Rojas MD, Nauffal Manzur MD, Munoz Pamplona MP, Compte Torrero L, Macian V, et al. Respiratory disorders in common variable immunodeficiency. Respir Med. 2001;95:191–5. doi: 10.1053/rmed.2000.1020. [DOI] [PubMed] [Google Scholar]
- 32.Watts WJ, Watts MB, Dai W, Cassidy JT, Grum CM, Weg JG. Respiratory dysfunction in patients with common variable hypogammaglobulinemia. Am Rev Respir Dis. 1986;134:699–703. doi: 10.1164/arrd.1986.134.4.699. [DOI] [PubMed] [Google Scholar]
- 33.Detkova D, de Gracia J, Lopes-da-Silva S, Vendrell M, Alvarez A, Guarner L, et al. Common variable immunodeficiency: association between memory B cells and lung diseases. Chest. 2007;131:1883–9. doi: 10.1378/chest.06-2994. [DOI] [PubMed] [Google Scholar]
- 34.da Silva SP, Resnick E, Lucas M, Lortan J, Patel S, Cunningham-Rundles C, et al. Lymphoid proliferations of indeterminate malignant potential arising in adults with common variable immunodeficiency disorders: unusual case studies and immunohistological review in the light of possible causative events. J Clin Immunol. 2011;31:784–91. doi: 10.1007/s10875-011-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–57. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 36.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–53. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 37.Randall TD. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. 2010;107:187–241. doi: 10.1016/B978-0-12-381300-8.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 39.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 40.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–80. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 42.Pijpe J, van Imhoff GW, Spijkervet FK, Roodenburg JL, Wolbink GJ, Mansour K, et al. Rituximab treatment in patients with primary Sjogren’s syndrome: an open-label phase II study. Arthritis Rheum. 2005;52:2740–50. doi: 10.1002/art.21260. [DOI] [PubMed] [Google Scholar]
- 43.Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, et al. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum. 2003;48:3187–201. doi: 10.1002/art.11311. [DOI] [PubMed] [Google Scholar]
- 44.Glace B, Gottenberg JE, Mariette X, Philippe R, Pereira B, Lequerre T, et al. Efficacy of rituximab in the treatment of pulmonary rheumatoid nodules: findings in 10 patients from the French AutoImmunity and Rituximab/Rheumatoid Arthritis registry (AIR/PR registry) Ann Rheum Dis. 2012;71:1429–31. doi: 10.1136/annrheumdis-2011-200915. [DOI] [PubMed] [Google Scholar]
- 45.Keir GJ, Maher TM, Hansell DM, Denton CP, Ong VH, Singh S, et al. Severe interstitial lung disease in connective tissue disease: rituximab as rescue therapy. Eur Respir J. 2012;40:641–8. doi: 10.1183/09031936.00163911. [DOI] [PubMed] [Google Scholar]
- 46.Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med. 2012;106:581–7. doi: 10.1016/j.rmed.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Swartz MA, Vivino FB. Dramatic reversal of lymphocytic interstitial pneumonitis in Sjogren’s syndrome with rituximab. J Clin Rheumatol. 2011;17:454. doi: 10.1097/RHU.0b013e31823ac199. [DOI] [PubMed] [Google Scholar]
- 48.Aghamohammadi A, Allahverdi A, Abolhassani H, Moazzami K, Alizadeh H, Gharagozlou M, et al. Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology. 2010;15:289–95. doi: 10.1111/j.1440-1843.2009.01679.x. [DOI] [PubMed] [Google Scholar]
- 49.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 50.Kohler PF, Cook RD, Brown WR, Manguso RL. Common variable hypogammaglobulinemia with T-cell nodular lymphoid interstitial pneumonitis and B-cell nodular lymphoid hyperplasia: different lymphocyte populations with a similar response to prednisone therapy. J Allergy Clin Immunol. 1982;70:299–305. doi: 10.1016/0091-6749(82)90066-5. [DOI] [PubMed] [Google Scholar]
- 51.Boursiquot JN, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol. 2013;33:84–95. doi: 10.1007/s10875-012-9778-9. [DOI] [PubMed] [Google Scholar]
- 52.Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID) J Clin Immunol. 2013;33:30–9. doi: 10.1007/s10875-012-9755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax. 2000;55:88–90. doi: 10.1136/thorax.55.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malbran A, Juri MC, Fernandez Romero DS. Common variable immunodeficiency and granulomatosis treated with infliximab. Clin Immunol. 2010;134:359–60. doi: 10.1016/j.clim.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 55.Lin JH, Liebhaber M, Roberts RL, Dyer Z, Stiehm ER. Etanercept treatment of cutaneous granulomas in common variable immunodeficiency. J Allergy Clin Immunol. 2006;117:878–82. doi: 10.1016/j.jaci.2006.01.034. [DOI] [PubMed] [Google Scholar]