

Abstract
It has been demonstrated that a chimeric antigen receptor (CAR) can directly recognize the CD19 molecule expressed on the cell surface of B-cell malignancies independent of major histocompatibility complex (MHC). Although T-cell therapy of tumors using CD19-specific CAR is promising, this approach relies on using expression vectors that stably integrate the CAR into T-cell chromosomes. To circumvent the potential genotoxicity that may occur from expressing integrating transgenes, we have expressed the CD19-specific CAR transgene from mRNA using a high throughput microelectroporation device. This research was accomplished using a microelectroporator to achieve efficient and high throughput non-viral gene transfer of in vitro transcribed CAR mRNA into human T cells that had been numerically expanded ex vivo. Electro-transfer of mRNA avoids the potential genotoxicity associated with vector and transgene integration and the high throughput capacity overcomes the expected transient CAR expression, as repeated rounds of electroporation can replace T cells that have lost transgene expression. We fabricated and tested a high throughput microelectroporator that can electroporate a stream of 2×108 primary T cells within 10 min. After electroporation, up to 80% of the passaged T cells expressed the CD19-specific CAR. Video time-lapse microscopy (VTLM) demonstrated the redirected effector function of the genetically manipulated T cells to specifically lyse CD19+ tumor cells. Our biomedical micro-device, in which T cells are transiently and safely modified to be tumor-specific and then can be re-infused, offers a method for redirecting T-cell specificity, that has implications for the development of adoptive immunotherapy.
Keywords: Electroporation, Cancer, High throughput, mRNA, Chimeric antigen receptor, T cells
1 Introduction
One promising approach to cancer treatment is combining gene therapy with T-cell therapy to redirect specificity to eliminate malignant cells. However, the ability of an endogenous T cell to engage a tumor cell depends on the presence of an αβ T-cell receptor that can be used to specifically recognize a cell target in the context of the major histocompatibility complex (MHC). Unfortunately, for many B-lineage leukemias and lymphomas, the resident immune system of patients with cancer remains incapable of controlling tumor growth, since autologous T cells lack expression of the required receptors and tumor cells have adapted to avoid immunological recognition. Figure 1 shows our approach to overcoming this barrier to immunotherapy. Gene therapy is used to introduce a desired immunoreceptor into T cells ex vivo to redirect specificity for tumor in vivo.
Fig. 1.

Outline of the experimental platform. There are three major parts for this research, ex vivo T-cell proliferation, a microelectroporator, and electro-transfer of mRNA to redirect T-cell specificity and function
Previously, we have demonstrated that a chimeric antigen receptor (CAR, designated CD19RCD28) can directly recognize the CD19 molecule expressed on the cell surface of B-cell malignancies independent of MHC (Singh et al. 2008; Singh et al. 2007; Kowolik et al. 2006; Cooper et al. 2003). The prototypical CAR-dependent signaling event integrates antigen binding via a single-chain variable fragment (scFv) region with a CD3-ζ signaling motif to redirect T-cell specificity, such as to CD19. This technology has been translated to clinical trials and recently, an anti-tumor effect has been attributed to T cells transduced to express CARs specific for GD2 on neuroblastoma cells and CD20 on B-lineage lymphoma cells (Pule et al. 2008; O’Reilly 2008; Till et al. 2008; Cooper 2008; Louis and Brenner 2009). Although this application of gene therapy for cancer is promising, this approach relies on using expression vectors that stably integrate the CAR into T-cell chromosomes. To circumvent the potential genotoxicity that may occur from expressing transgenes from recombinant retrovirus, we have now expressed the CD19-specific CAR transgene from mRNA which cannot integrate and avoids the need to introduce constitutive promoters. Recognizing that CAR expression will be temporary from mRNA, we have combined microelectroporator technology with immunotherapy to develop a new device to electro-transfer mRNA into primary T cells with the expectation that multiple infusions of T cells transiently expressing CAR may be needed to achieve a therapeutic effect (Krassowska and Filev 2007; Li and Harrison 1997; DeBruin and Krassowska 1999). To generate the large numbers of T cells for repeated rounds of electro-transfer and adoptive immunotherapy we have numerically expanded these lymphocytes ex vivo using artificial antigen presenting cells (aAPC).
We report our first-generation high throughput microelectroporation device (HiTMeD) that can process a large number of propagated human T cells within a short time period. The data demonstrate that we can electroporate propagated T cells using HiTMeD such that 2×108 cells can be genetically manipulated within 10 min to express a CD19-specific CAR from in vitro transcribed mRNA and that the CAR+ T cells exhibit redirected killing for CD19+ tumor cells.
2 Materials and methods
2.1 Cell lines and primary human T cells
Human T cells were isolated by density gradient centrifugation over Ficoll-Paque-Plus (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) from buffy coats containing peripheral blood mononuclear cells (PBMC) obtained from Gulf Coast Regional Blood Center (Houston, TX) after consent. We developed an effective ex vivo culture system to rapidly expand large numbers of primary T cells based on aAPC provided in collaboration with Dr. Carl June (University of Pennsylvania). The aAPC were derived from K562 with enforced expression of T-cell costimulatory molecules, 41BBL, CD86, and membrane-bound IL-15 (mIL-15, co-expressed with EGFP). K562-aAPC could be pre-loaded with OKT3 (CD3-specific mAb, 1 mg/mL in phosphate buffered saline (PBS)) at a concentration of 1 μL/106cells, gamma-irradiated at 100 Gy, and co-cultured with T cells typically at a ratio of 1:1 in culture media (CM, RPMI 1640, GIBCO, Grand Island, NY) supplemented with 5% heat-inactivated human serum (Valley Biomedical Inc, Winchester, VA) with 50 units/mL of soluble IL-2 added every 2 days. The OKT3-loaded K562 were added to the culture every 7 days. Adherent U251T glioma cells expressing green fluorescent protein (kindly provided by Dr. Waldemar Debinski, Wake Forest University, NC) were genetically modified to express the truncated CD19 (Cooper et al. 2005; Mahmoud et al. 1999). The GFP+U251T and CD19+GFP+U251T cells were cultured in Dubelco modified Eagle medium (CM2, Hyclone, Logan, UT), supplemented with 10% heat-inactivated fetal calf serum (FCS, Hyclone, Logan, UT).
2.2 In vitro transcription of mRNA species
The mRNA to express a second-generation CD19-specific CAR was prepared from CD19RCD28 cDNA (Singh et al. 2008). CD19RCD28 was cloned into the plasmid pCR4-TOPO (Invitrogen, Carlsbad, CA) to generate CD19RCD28/pCR4-TOPO (Fig. 2(a)), that was linearized with a restriction enzyme, SpeI, downstream of the CAR insert to be transcribed. The in vitro transcription was accomplished using Ribomax Large Scale RNA Production Kit-T7 (Promega, Madison, WI). Capping of the 5′ end of RNA was achieved by assembling anti-reverse cap analog (Ambion, Austin, TX) into the transcription reaction (Mockey et al. 2006; Stepinski et al. 2001). The product was then treated with DNase Q (Promega, Madison, WI) followed by addition of Escherichia Coli poly(A) polymerase in the presence of ATP and manganese (Poly-A-tailing kit, Ambion, Austin, TX) (Elango et al. 2005). The final product was purified with Qiagen RNeasy kit (Qiagen, Valencia, CA). RNA quantity was verified by spectrophotometer (Bio-Rad SmartSpec Plus, Hercules, CA) with a yield of ~400 μg of mRNA per ~30 μg of input DNA plasmid. The same procedure for in vitro transcription was used to make EGFP mRNA using the DNA plasmid pCMV3tag8-EGFP (Stratagene, La Jolla, CA) using NotI to linearize this plasmid (Fig. 2(b)).
Fig. 2.
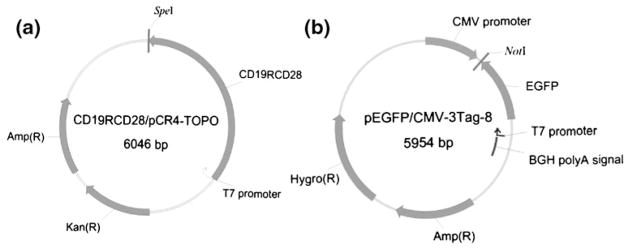
DNA plasmids for in vitro transcription. (a) The second generation CAR (CD19RCD28) cDNA under control of T7 promoter. (b) EGFP cDNA under control of T7 promoter. Amp(R): Ampicillin resistance, Kan(R): Kanamycin resistance, Hygro(R): Hygromycin resistance, BGH: bovine growth hormone
2.3 High throughput microelectroporation device (HiTMeD)
First generation HiTMeD (HiTMeD1) consists of 40× 45 mm electrodes with a 400 μm gap. Figure 3 shows HiTMeD1 which has four inlet ports and the system configured to electroporate cells in continuous flow. We fabricated the HiTMeD1 device by sandwiching the gold-coated glass with a spacer consisting of either polyimide film or SU-8 (MicroChem, Newton, MA). Ports were placed between the electrodes to introduce cells. Adhesive transfer tape (cat # 9461P, 3 M™, St. Paul, MN) was used to assemble spacers and electrodes and sealed with epoxy glue. For electrodes, gold was coated on glass (Mark Optics, Santa Ana, CA) using E-beam evaporator (Sharon Vacuum, Brockton, MA). We chose a commercially available pulse generator (ECM830, BTX Harvard Apparatus, Holliston, MA) which has a multiple pulsing mode to deliver a user-defined electrical pulse to the HiTMeD1 with 10 second intervals. The electrical strength was set at 1 kV/ cm (40 V for the 400 μm gap). T cells were propelled via a syringe pump (KDS230 Multi Syringe, KD Scientific, Holliston, MA). The syringe pump was stopped after delivering the preset volume of the chamber and ran again after delivering the electrical pulse. It took 3 to 5 s to refresh the electroporated sample with new cells. The pump operated to deliver cells with a 10 s interval so that electroporation occurred every 10 s. 14×106 cells passed the chamber over 10 s. The final concentration of T cells in PBS was 2×107 cells/mL. We premixed mRNA with the T cells in the PBS electroporation buffer as 2 μg of each mRNA species per 106 cells. Every 10 s, T cells and mRNA were transferred to the electroporation chamber of the HiTMeD1 and the ECM830 pulse generator delivered a single electrical pulse. The electroporated cells were collected in a flask containing culture media.
Fig. 3.

Description of HiTMeD1 to electroporate T cells in continuous flow. (a) HiTMeD1 is connected to multi-syringe pump. Multiple pulsing mode (square wave) of BTX ECM830 has been used for electroporation (1 kV/cm, 6 ms). (b) Schematic view of HiTMeD1. Multiple inlet ports and polymer films are used to make electrodes and 400 μm gap
2.4 Commercial electroporation
For comparison purposes, T cells were electroporated using Nucleofector Technology (Amaxa/Lonza, Walkersville, MD) which is being used for clinical trials. The conditions for the cell preparation (T-cell proliferation for 7 days stimulation), the ratio with mRNA (2 μg/106cells), and PBS as an electroporation buffer were the same as for HiTMeD1. When the cell suspension (2×107 cells/mL) for the electroporation with HiTMeD1 was ready, a sample was transferred (100 μL) to a cuvette, and electroporated (Program T-14). After 12 h incubation, the cells were analyzed using flow cytometry.
2.5 Flow cytometry
Fluorochrome-conjugated monoclonal antibodies (mAbs) were obtained from BD Biosciences (San Jose, CA): CD3 PerCp Cy5.5 (cat # 340948), CD3 PE (cat # 347347), CD4 PerCp Cy5.5 (cat # 341654), CD4 PE (cat # 347327), CD8 PerCp Cy5.5 (cat # 341051), CD8 PE (cat # 340046), CD28 PerCp Cy5.5 (cat # 337181), CD28 PE (cat # 348047), CD64 PE (cat # 558592), CD86 PE (cat # 555658), CD137L PE (cat # 559446). Propidium iodide (Cat #537059, Calbiochem, San Diego, CA) was used to gate out the dead cells. Goat F(ab′)2 antimouse IgG R-phycoerythrin conjugate (cat # 115-116-146, Jackson ImmunoResearch Laboratories Inc., West Grove, PA) was used at 1/20 dilution to detect cell surface expression of OKT3. Goat F(ab′)2 antihuman IgG (gamma) R-phycoerythrin conjugate (cat # H10104, Caltag Laboratories, Burlingame, CA) was used at 1/20 dilution to detect cell surface expression of mIL-15. Goat F(ab′)2 antihuman IgG (gamma) FITC-conjugate (cat # 109-096-170, Jackson ImmunoResearch Laboratories Inc., West Grove, PA) or Goat F(ab′)2 antihuman IgG (gamma) R-phycoerythrin conjugate (cat # H10104, Caltag Laboratories, Burlingame, CA) were used at 1/20 dilution to detect cell-surface expression of the CD19-specific CAR, CD19RCD28. Blocking of nonspecific antibody binding was achieved using 2% FBS in PBS. Data acquisition was on a FACSCalibur (BD Biosciences, San Jose, CA) using CellQuest version 3.3 (BD Biosciences, San Jose, CA).
2.6 Video time-lapse microscopy after T cells were processed by HiTMeD1
To directly visualize killing of tumor targets by electro-porated CD19-specific T cells, we undertook imaging by VTLM, using the BioStation IM Cell-S1/Cell-S1-P system (Nikon Instruments Inc., Melville, NY). U251T cells were used as targets based on an ability to identify living versus dying/dead cells by phase contrast dynamic morphology (Serrano et al. 2006). GFP+U251T cells were used as CD19neg targets while transfected CD19+GFP+U251T cells were stained with the PKH-26 red fluorescent dye (Cat # MINI26, Sigma, St Louis, MO) according to the manufacturer’s protocol which fluoresces orange (green and red) in our imaging system. The CD19neg and CD19+ GFP+U251T targets were mixed in a 1:1 ratio and plated overnight on a T-35 mm glass bottom plate (Fisher scientific, Pittsburgh, PA) in CM2. CD19RCD28+ T cells were added to the adherent U251T targets and were immediately imaged every 200 s at 37°C. Each image was recorded at 1600×1200 pixels via a 20X objective using phase contrast and fluorescent channels with an exposure time at 1/125 and 1/5 s, respectively.
3 Results
3.1 Propagation of human T cells on aAPC
The human T cells were activated for sustained proliferation from PBMC using the CD3-specific monoclonal antibody (mAb) OKT3 that was pre-bound to γ-irradiated artificial antigen presenting cells derived from K562. These aAPC were genetically modified to enforce expression of CD64, CD86, CD137L, and mIL-15, and were then loaded with OKT3. Figure 4(a) shows the surface expression of these co-stimulatory molecules. After 7 days in co-culture with γ-irradiated OKT3-aAPC the T cells expanded by approximately 50-fold and after 14 days the numeric expansion was almost 1,000-fold (Fig. 4(b)). At day 7 the outgrowth was 74% CD4+ and 26% CD8+ T cells and after an additional 7 days of culture the percentage of CD8+ T cells had increased to 54% (Fig. 4(c)).
Fig. 4.

Phenotypic characteristics of aAPC and propagation of human T cells on aAPC. (a) Surface expression of co-stimulatory molecules and adsorbed OKT3 on K562-aAPC. Open histograms show the expression on K562 parental cells and filled histograms show the expression on genetically modified K562 which function as aAPC. Expression of CD64, CD86, CD137L, membrane-bound IL-15, and OKT3 were detected with antibodies. (b) Propagation of primary T cells on aAPC. Irradiated OKT3-loaded K562-aAPC were added to the T-cell culture for every 7 days at two different ratios. (c) T-cell subsets after 7 and 14 days of numeric expansion on OKT3-loaded aAPC at 1 : 1 (T cells : aAPC) ratio
3.2 Electro-transfer of CAR into numerically expanded T cells using HiTMeD1
The HiTMeD1 was developed to electro-transfer mRNA into flowing T cells. An electric field of 1 kV/cm was used for each electroporation experiment. Ex vivo numerically expanded and activated T cells were pre-mixed in PBS with one or two mRNA species, and delivered via a controlled-rate syringe pump to the electroporation chamber every 10 s where they received a single square pulse (6 millisecond duration) (Supplementary material 1 demonstrating the electroporation process is provided). After 12 h, expression of EGFP and/or CAR was evaluated by flow cytometry. Figure 5 demonstrates that the CD19-specific CAR coded from a 2,000 bp mRNA species and/or EGFP coded from an 800 bp mRNA species could be electro-transferred into the ex vivo propagated T cells. The expression of CAR and EGFP mRNA transgenes was 80% and 91%, respectively as determined by flow cytometry analysis. The chosen mRNA input concentration was 2 μg/106cells. Greater than 2 μg/106cells of mRNA did not increase the transfection efficiency for a given concentration of T cells (2×107 cells/ mL). The viability of T cells measured by exclusion of PI using flow cytometry was 50 to 60%, 12 h after electro-poration. This compared favorably to electro-transfer of same mRNA into propagated human T cells using the commercial cuvette-based device (Fig. 5(a)). The HiTMeD1 electro-transferred mRNA into 20-fold more T cells 2×108 cells, compared to 107 T cells for the commercial cuvette-based devices. Since mRNA can be rapidly degraded and is not integrated, the CAR expression decreased over time as expected. Nevertheless, the CAR expression was still 70% at 48 h after electroporation (Fig. 5(b)). To determine whether HiTMeD1 could electro-transfer more than one mRNA species, we pre-mixed the mRNAs coding for both CAR and EGFP and 86% of the processed T cells achieved dual expression 12 h after electroporation (Fig. 5(c)).
Fig. 5.

Electro-transfer of mRNA into T cells. (a) Flow cytometry analysis of CD19-specific CAR and EGFP transgenes in gated CD3+ T cells 12 h after electroporation. (b) CD19-specific CAR expression on CD3+ T cells by flow cytometry 12 and 48 h after electro-transfer of mRNA. (c) Four experiments with HiTMeD1 in which T cells were electroporated without mRNA, with CAR mRNA, with EGFP mRNA, and with both CAR and EGFP mRNA species
3.3 Redirected killing of tumor targets by CAR+ T cells after electro-transfer
We undertook VTLM to directly assess the ability of the genetically manipulated T cells to lyse CD19+ tumor targets. To mimic a tumor microenvironment we pre-mixed GFP+U251T cells at a ratio of 1:1 that did and did not express CD19. To distinguish these two populations we pre-labeled CD19+GFP+U251T target cells using the fluorescent dye PKH-26, which resulted in cells appearing orange around the nuclei. The CD19neg parental and CD19+ transfected GFP+U251T targets (84% expressed CD19 by flow cytometry) were adhered to a glass slide prior to the addition of electroporated T cells. The killing events were directly visualized over 7 h to reveal the engagement/disengagement of T cells (small, darkgrey, irregular bodies) to adherent spindle-shaped green/orange U251T cells (Fig. 6). After contact with the genetically manipulated T cells, the CD19+U251T cells rounded and imploded while the CD19negU251T cells generally did not. Adherent live cells continue to express GFP and remain flat and spread out. (Supplementary material 2 demonstrating the redirected killing of tumor targets is provided). The killing of U251T cells over time was also analyzed for 14 h by Nikon imaging software (NIS-Elements AR v3.00). CAR+ T cells, CD19+U251T tumor cells, and CD19negU251T tumor cells were co-cultured at a ratio of 10:1:1 (4×105, 4×104, and 4×104 cells). 50% killing of CD19+U251T target cells was observed at approximately 4 h after the beginning of the assay (Fig. 7). CD19negU251T cells were not targeted by CD19-specific T cells and cell division was observed which resulted in cell viability over 110% after 14 h. No cell division among the target CD19+U251T tumor cells was observed. We assume that the specific killing of the target cells by CAR+ T cells precluded the proliferation of these target cells.
Fig. 6.

Redirected specificity of electroporated T cells. Killing of CD19+GFP+U251T target cells within 7 h of co-culture with CD19-specific T cells (12 h after processing through HiTMeD1). Orange+ adherent CD19+GFP+U251T cells can be differentiated from CD19negGFP+U251T cells. T cells (approximately 10 μm diameter) can be seen as black (mobile) structures. See supplementary material 2 (Movie 2) for the original data used to perform this analysis
Fig. 7.

CD19-specific time-dependent killing of tumor cells by CAR+ T cells. The killing of U251T cells over time was analyzed for 14 h by Nikon imaging software (NIS-Elements AR v3.00). 50% killing of CD19+U251T target cells was observed at approximately 4 h after the beginning of the assay. CD19negU251T cells were not targeted by CD19-specific T cells and cell division was observed which resulted in cell viability over 110%
4 Discussion and conclusions
Other investigators have also electro-transferred mRNA expressing immunoreceptors into human T cells, but their approach relies on using cuvettes (Cohen et al. 2007; Holtkamp et al. 2006; Mitchell et al. 2008). This commercially-based electroporation methodology limits the numbers of cells that can be electroporated and the need to manipulate multiple cuvettes with attendant risks of loss of sterility and cross-contamination.
Using mRNA to express a desired transgene as an approach to gene therapy avoids the need to develop integrating vectors with strong promoters which can cause genotoxicity by deleterious site-directed integration events (Baum et al. 2003; Edelstein et al. 2004; Hacein-Bey-Abina et al. 2003). However, the expression of transgene(s) from introduced mRNA is transient and a methodology is required to sustain transgene expression after infusion. What is needed, and is provided here, is a cost-effective approach to rapidly modify a large number of clinical-grade T cells so that more than one infusion can be undertaken to replace lost transgene expression in vivo. Indeed, to sustain a therapeutic anti-tumor effect, large numbers of CAR+ mRNA-loaded T cells will likely be required for more than one infusion. Assuming an upper limit of the number of T cells that can typically be electroporated in a cuvette to be 107, 20 cuvettes would be needed to electro-transfer 2×108 cells, which when undertaken in compliance with current good manufacturing practices for Phase I/II trials exposes the methodology to contamination and tedium. The HiTMeD1 uses continuous flow to avoid the need for multiple cuvettes and since we can adapt our technology to an unbreached system, we can minimize the potential for contamination (Craiu and Scadden 2008; Fratantoni et al. 2003; Wang et al. 2006).
In addition to generating a device for non-viral gene transfer, we also developed an approach to manufacturing large number of T cells using aAPC loaded via a CD64 transgene with OKT3. This is a clinically-appealing approach as (i) the aAPC have been manufactured as a master cell bank by the Production Assistance for Cellular Therapies (PACT) under the auspices of NHLBI, and (ii) OKT3 is available as clinical-grade material. Some electro-poration buffers are reported to have higher transfection results (Li 2004; Miller et al. 1988; Nathwani et al. 1994). However, we used PBS to reduce cost and improve biocompatibility since this buffer can be adapted for clinical use. Ideally, the electroporation buffer and electroporated T cells will be infused directly into humans without any additional procedure to save processing time and cost.
In this work, by combining bioengineering with gene transfer into human T cells, we have developed a new approach with the potential for rapid treatment of tumors recognized by an introduced CAR. We fabricated and tested HiTMeD1 that can electroporate a stream of 2×108 primary T cells within 10 min. HiTMeD1 can synchronously electro-transfer one or more mRNA species into a large number of activated primary human T cells, and these genetically manipulated T cells can exhibit desired effector function, such as redirected killing of tumor cells. After electroporation, up to 80% of the primary T cells expressed the CD19-specific CAR. VTLM demonstrated the redirected effector function of the genetically manipulated T cells to specifically lyse CD19+ tumor cells. Our biomedical microdevice, in which T cells are transiently and safely modified to be tumor-specific, offers a method for modifying T cells that has implications for the development of adoptive immunotherapy. We foresee the implementation of the HiTMeD1 to introduce mRNA to express desired transgenes such as CAR into T cells for infusion and re-infusion into patients for cancer therapy.
Supplementary Material
Acknowledgments
Support from CCSG Grant (CA16672), RO1 (CA124782, CA120956), R21 (CA129390, CA116127), DOD (PR064229), The Alex Lemonade Stand Foundation, The Alliance for NanoHealth Competitive Research, The Burroughs Wellcome Fund, The Gillson Longenbaugh Foundation, The Leukemia and Lymphoma Society, The Lymphoma Research Foundation, The Miller Foundation, The National Foundation for Cancer Research, The Pediatric Cancer Research Foundation, The National Marrow Donor Program, The William Lawrence and Blanche Hughes Foundation, Longenbaugh Foundation. We thank Rice University for access to the cleanroom to make the micro devices.
Footnotes
Electronic supplementary material. The online version of this article (doi:10.1007/s10544-010-9440-3) contains supplementary material, which is available to authorized users.
Contributor Information
Yoonsu Cho, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Carrie Yuen, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Sourindra N. Maiti, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
Simon Olivares, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Hillary Gibbons, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Helen Huls, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Robert Raphael, Department of Bioengineering, Rice University, Houston, TX, USA.
Thomas C. Killian, Department of Physics and Astronomy, Rice University, Houston, TX, USA
Daniel J. Stark, Department of Physics and Astronomy, Rice University, Houston, TX, USA
Dean A. Lee, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
Hiroki Torikai, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Daniel Monticello, InCellerate Inc., Houston, TX, USA.
Susan S. Kelly, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
Partow Kebriaei, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA.
Richard E. Champlin, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
Sibani L. Biswal, Department of Chemical and Biomolecular Engineering, Rice University, Houston, TX, USA
Laurence J. N. Cooper, Email: ljncooper@mdanderson.org, Division of Pediatrics, Children’s Cancer Hospital, The University of Texas Graduate School of Biomedical Sciences at Houston, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
References
- Baum C, Dullmann J, Li Z, Fehse B, Meyer J, Williams DA. Blood. 2003;101:2099. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Cancer Res. 2007;67:3898. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LJN. Blood. 2008;112:2172. doi: 10.1182/blood-2008-06-163006. [DOI] [PubMed] [Google Scholar]
- Cooper LJN, Topp MS, Serrano LM, Gonzalez S, Chang W, Naranjo A. Blood. 2003;101:1637. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- Cooper LJN, Al-Kadhimi Z, Serrano LM, Pfeiffer T, Olivares S, Castro A. Blood. 2005;105:1622. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- Craiu A, Scadden D. Methods Mol Biol. 2008;423:301. doi: 10.1007/978-1-59745-194-9_22. [DOI] [PubMed] [Google Scholar]
- DeBruin KA, Krassowska W. Biophys J. 1999;77:1213. doi: 10.1016/S0006-3495(99)76973-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein ML, Abedi MR, Wixon J, Edelstein RM. J Gene Med. 2004;6:597. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- Elango N, Elango S, Shivshankar P, Katz MS. Biochem Biophys Res Commun. 2005;330:958. doi: 10.1016/j.bbrc.2005.03.067. [DOI] [PubMed] [Google Scholar]
- Fratantoni JC, Dzekunov S, Singh V, Liu LN. Cytotherapy. 2003;5:208. doi: 10.1080/14653240310001479. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E. N Engl J Med. 2003;348:255. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C. Blood. 2006;108:4009. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N. Cancer Res. 2006;66:10995. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- Krassowska W, Filev PD. Biophys J. 2007;92:404. doi: 10.1529/biophysj.106.094235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Curr Gene Ther. 2004;4:309. doi: 10.2174/1566523043346336. [DOI] [PubMed] [Google Scholar]
- Li PCH, Harrison DJ. Anal Chem. 1997;69:1564. doi: 10.1021/ac9606564. [DOI] [PubMed] [Google Scholar]
- Louis CU, Brenner MK. Curr Pharm Des. 2009;15:424. doi: 10.2174/138161209787315765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud MS, Fujii R, Ishikawa H, Kawano MM. Blood. 1999;94:3551. [PubMed] [Google Scholar]
- Miller JF, Dower WJ, Tompkins LS. Proc Natl Acad Sci USA. 1988;85:856. doi: 10.1073/pnas.85.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Karikari I, Cui X, Xie W, Schmittling R, Sampson JH. Hum Gene Ther. 2008;19:511. doi: 10.1089/hum.2007.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockey M, Goncalves C, Dupuy FP, Lemoine FM, Pichon C, Midoux P. Biochem Biophys Res Commun. 2006;340:1062. doi: 10.1016/j.bbrc.2005.12.105. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Gale KM, Pemberton KD, Crossman DC, Tuddenham EDG, McVey JH. Br J Haematol. 1994;88:122. doi: 10.1111/j.1365-2141.1994.tb04987.x. [DOI] [PubMed] [Google Scholar]
- O’Reilly RJ. Nat Med. 2008;14:1148. doi: 10.1038/nm1108-1148. [DOI] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G. Nat Med. 2008;14:1264. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Kim D. Blood. 2006;107:2643. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Serrano LM, Pfeiffer T, Olivares S, McNamara G, Smith DD. Cancer Res. 2007;67:2872. doi: 10.1158/0008-5472.CAN-06-2283. [DOI] [PubMed] [Google Scholar]
- Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H. Cancer Res. 2008;68:2961. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepinski J, Waddell C, Stolarski R, Darzynkiewicz E, Rhoads RE. RNA. 2001;7:1486. [PMC free article] [PubMed] [Google Scholar]
- Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA. Blood. 2008;112:2261. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bhunia AK, Lu C. Biosens Bioelectron. 2006;22:582. doi: 10.1016/j.bios.2006.01.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.