

Abstract
Antigen cross presentation is an important mechanism for CD8+ T cell activation by antigen presenting cells (APC). We have investigated mechanisms involved in Hsp90 chaperone mediated cross presentation of ovalbumin (Ova) derived antigens. Hsp90-Ova peptide complexes bound to scavenger receptor expressed by endothelial cells (SREC-I) on the surface of APC. SREC-I then mediated internalization of Hsp90-Ova polypeptide complexes through a Cdc42 regulated, dynamin independent endocytic pathway known as the GPI-AP enriched early endosomal compartment (GEEC) to recycling endosomes. Peptides that did not require processing could then be loaded directly onto MHC-I in endosomes, whereas longer peptides underwent endosomal and cytosomal processing by aminopeptidases and proteases. Cross presentation of Hsp90 chaperoned peptides through this pathway to CD8+ T-cells was highly efficient compared with processing of free polypeptides. In addition, Hsp90 also activated c-src kinase associated with SREC-I, an activity that we determined to be required for effective cross presentation. Extracellular Hsp90 can thus convey antigenic peptides through an efficient endocytosis pathway in APC and facilitate cross presentation in a highly regulated manner.
Keywords: antigen cross presentation, dendritic cell, Hsp90, SREC-I
INTRODUCTION
Heat shock proteins (HSP) are stress-inducible proteins whose main intracellular functions are as molecular chaperones (1, 2). However HSP can also exit cells, gain access to the extracellular environment and interact with the surfaces of immune cells (3–7). HSP are highly qualified to be endogenous danger signals, as proteins that are inducible by stress and can flood their local milieu and signal to inflammatory cells (5, 8–10). These molecular chaperones bind to receptors expressed on antigen presenting cells (APC), chaperone the antigenic peptides through the endocytic pathways and promote antigen cross presentation (3, 11, 12).
It had been shown that extracellular antigens taken up by APC such as dendritic cells (DC) and macrophages can access the intracellular pathways for antigen processing and subsequently elicit CD8+ T cell responses (10, 13–15). This process, known as antigen cross presentation utilizes some of the basic mechanisms involved in the presentation of endogenous antigens. Endogenous antigen processing and presentation requires polypeptide digestion by the proteasome in the cytosol, recruitment of peptides by transporter associated with antigen processing (TAP) in the ER and delivery to the cell surface bound to MHC-I through ER-Golgi-Cell surface anterograde vesicular trafficking (16–20). In the cytosolic pathway of antigen cross presentation, exogenous antigens must penetrate this intracellular pathway. Such exogenous antigens enter this pathway after being taken up into phagosomes, transported out of the phagosomes by Sec61 and then delivered to the cytosol for proteasomal processing (21, 22). Peptides are then re-imported by TAP within the phagosomes and bound to MHC-I in this compartment (21, 22). An alternative cross presentation pathway also exists- the phagosome/endosome pathway- catalyzed by such proteases as cathepsin S, within phagosomes and endosomes (21, 22). Processed antigens then encounter MHC-I in recycling endosomes or phagosomes and can be loaded in this compartment. This pathway excludes a requirement for antigens to cross membrane-bounded vesicles and is thus potentially more rapid.
Although HSP binding to APC may lead to immune consequences through a range of mechanisms, ability to mediate antigen cross presentation is a key factor in the effectiveness of HSP vaccines. Tumor derived Hsp70, Hsp90, GRP94 (gp96) bind to APC and enter cells through receptor-mediated or pinocytic pathways and then may be released from the chaperones within endosomes (3, 7, 23–26). While opsonized antigens enter cells through the Fc receptor, roles for mannose receptor (CD206), langerin (CD207), DC-SIGN (CD209a), Dec-205 (CD205), D-CIR2, CLEC-9a and Dectin-1 in cross presentation of soluble and particulate antigens have been suggested for non-opsonized antigens (27). These receptors are efficient in cross presenting protein antigens to MHC-I molecules. Receptors for molecular chaperones are beginning to be characterized and we have shown avid association of HSP with scavenger receptors (SR) LOX-1, FEEL-1 and SREC-I and recent studies show that SREC-I is important for Hsp70-peptide complex mediated immunity (28–30). Hsp90 was shown to possess exceptional ability to present antigens to CD4+ T cells (23, 31). Factors that may influence the fate of the Hsp90 bound peptides may include the nature of the HSP receptor encountered and the structure of the peptide. For instance, the presence of C-terminal or N-terminal extension of the Ova peptide SIINFEKL bound to Hsp70 appeared to determine whether the peptide is processed by proteasomal or lysosomal proteases (32).
We have investigated the internalization and antigen cross presentation pathways utilized by Hsp90 conjugated peptides (Hsp90.PC) via SREC-I, both in cells expressing SREC-I endogenously (bone marrow derived dendritic cells) and CHO cells engineered to stably express SREC-I. Our experiments indicate a novel pathway of Hsp90.PC internalization by SREC-I. This pathway requires Cdc42, is also known as GEEC (GPI- enriched endosomal compartment) (33) and is regulated by cell surface tyrosine kinase c-src. This process differs from the fluid phase uptake pathway often taken by unconjugated Ova. In addition, Ova presentation was more efficient when polypeptides were bound to Hsp90, compared to Ova. Our experiments suggest that Hsp90 binding to SREC-I permits chaperone-associated antigens to access both endosomal and endosomal-cytosolic pathways of antigen cross presentation.
MATERIALS AND METHODS
Mice
Mice, C57BL/6 (H2b) and Balbc (H2d) and TAP−/− (C57BL/6 background) used in this study were obtained from The Jackson Laboratory. All mice were kept in a specific pathogenfree mouse facility. Studies were done according to institutional guidelines for animal use and care.
Reagents and Antibodies
Ova and Clostridium difficile toxin B were purchased from Sigma-Aldrich. SIINFEKL peptide was custom synthesized (Anaspec) and purified to greater than 95% by HPLC. To prepare maleylated BSA, purified BSA was incubated with maleic anhydride (Sigma-Aldrich) at alkaline pH. Mouse monoclonal human Anti-SREC-I ab was a gift from H. Adachi, Rabbit monoclonal mouse anti SREC-I ab was custom synthesized by GenScript Corporation against specific peptide sequence (TQGTQGSTLDPAGQC). Anti-MHC-I ab (W6/32) from ATCC, FITC-anti-MHC-I ab was from Chemicon. Goat anti-mouse LOX1 blocking antibody was purchased from R&D systems. Monoclonal anti-HA antibody was from Covance, Rat anti-mouse IFNγ was from BD Pharmingen. The drugs and inhibitors, Primaquine, Chloroquine, Leupeptin, Lactacystin, Cytochalsin D, Latrunculin B, DMA (dimethyl amiloride hydrochloride, Poly I (Poly Inosinic acid), MβCD (methyl β-cyclodextrin), PP1 were purchased from Sigma-Aldrich. Rat anti-mouse CD16/32 (Fcγ Receptor) was from Beckman Coulter. GMCSF was purchased from PeproTech Inc. Cathepsin S was from EMD biosciences. The vibrant lipid raft labeling kit and Alexa-labeled secondary antibodies were from Molecular Probes. Cy3-transferrin and all other secondary fluorescent antibodies were from Jackson Immunoresearch. Anti-src and anti-phospho (Y416) src antibodies were from Cell Signaling. The β-galactosidase assay kit was from Promega.
Construction of Baculovirus
Full length human Hsp90 alpha DNA was prepared by PCR amplification from the pET23 plasmid. The full-length cDNA was then cloned into pDEST™10. Overexpression of Hsp90 alpha in Sf9 cells was achieved according to the BAC-to-BAC Transfection kit protocol of Invitrogen. Transfer vector were transformed into DH10BAC competent cells containing bacmid DNA. Later, colonies containing recombinant bacmid were identified and prepared. The bacmid DNA was then transfected into/Sf9cells using CellFECTIN (Invitrogen) to make recombinant baculovirus according to manufacturer’s protocol.
Expression of Hsp90 in Sf9 cells
Sf9 cells were grown in Sf900II serum-free medium (Invitrogen) supplemented with 100 U/ml penicillin-streptomycin and 2mM of L-glutamine in suspension cultures with continuous shaking at 150 rpm at 27°C in a non-humidified environment. The insect cultures were infected in the log phase of growth with recombinant baculovirus. Cells were harvested 48 hr post infection, washed with Hank’s buffered saline solution and protein was purified using the Ni-NTA purification system according to manufacturer’s protocol. AcTEV protease was used to cleave the 6X His tag from the fusion protein and generate unmodified Hsp90.
Cells and Culture conditions
Bone marrow-derived dendritic cells (BMDCs) were generated from the femur and tibiae of C57BL/6, Balbc and TAP−/− mice. The bone marrow was flushed out and cultured in RPMI 1640 supplemented with 10% heat inactivated fetal bovine serum (FBS) and 40ng/ml GMCSF for 6 days. On day 3, a third of the medium was replaced by fresh growth medium. On day 6 the immature BMDC were used for experiment.
Wild type CHO-K1 and HeLa cells were transfected with Human full length SREC-I in pcDNA3 for stable expression of SREC-I. Both cell lines were maintained in Ham’s F12K medium supplemented with 10% heat inactivated FBS. For generation of stable SREC-I expressing cell lines, cells were selected and maintained in the same medium plus 400 µg/ml G418. HeLa and Raw 264.7 cell lines were maintained in DMEM supplemented with 10% heat inactivated FBS.
Plasmids
The green fluorescent dominant negative constructs derived from Dynamin-2 were from M. McNiven (Mayo Clinic College of Medicine, Rochester, MN) and the YFP-dn Eps15 (lacking amino acids 95–295) was provided by Alexandre Benmerah (Institut Cochin, Université Paris Descartes, Paris, France). The pcDNA3.1-SREC-I (human) was a generous gift from H. Adachi. FLAG-SREC-I construct was made in 3xFLAG-CMV vector. The RhoA, Rac1, Cdc42, Arf6 (Q67L) and Arf1 GFP constructs were from Addgene Inc. (Cambridge, MA). The dominant negative constructs for Rho, Rac, Cdc42 and Arf1 were made using the quick-change site directed mutagenesis kit (Stratagene) according to manufacturer’s protocol.
Hsp90-peptide/protein complex and Anti-SREC-I ab-Ova complex formation
Ova was dialyzed for 36 hours at 4°C with several changes of the buffer (PBS) to remove degradation products as well as possible contaminating peptides from the solution. Small peptides, S8LC (SIINFEKLTEWTS) and S8L (SIINFEKL) were loaded onto Hsp90 in solution at a molar ratio of 50:1 in PBS, incubated for 10 minutes at 45–50°C and cooled at room temperature for 30–40 minutes. For Hsp90-Ova conjugate preparation, soluble Hsp90 and excess Ova (1:2 ratio) were mixed and incubated for 10 minutes at 45°C. The solution mixtures were then incubated at room temperature for 30 minutes. Free Ova was removed using Microcon YM-100 (Millipore) with a 100 kDa cut-off. Alexa 488 and Alexa 555 labeled Hsp90-peptide/Ova (Hsp90.PC) conjugates were made according to the manufacturer’s protocol (Invitrogen). For anti-SREC-I ab-ovalbumin (Ova) preparation, Ova was coupled to anti-SREC-I ab using bis (sulfosuccinimidyl) suberate (BS3) as described by the manufacturer (Thermo Scientific) and labeled with Alexa fluorochromes as described for Hsp90 and purified using Microcon YM-100 ultrafiltration.
Hsp90 Binding Experiments
Cells were pre-incubated without or with mBSA (50µg/ml) or anti-SREC-I ab (100µg/ml) before incubating with Alexa labeled Hsp90 at 4°C in FACS buffer (PBS containing 0.1% BSA and 0.05% NaN3). The cells were then washed with FACS buffer twice, fixed with 4% para-formaldehyde for 10 minutes and analyzed for binding of Alexa-Hsp90 binding by FACScan (BD Biosciences).
Immunofluorescence and Microscopic analysis of Hsp90 Internalization
CHO-K1, CHO-SREC-I, and HeLa-SREC-I cells were labeled with Alexa fluor conjugated Hsp90 complexes for 20–30 minutes on ice. The ice-cold medium was then replaced by warm medium and incubated at 37°C for different periods. Cells were later washed with ice cold stripping buffer (50 mM Sodium Citrate, 280 mM sucrose, pH 4.6) to remove unbound Hsp90.PC. Later the cells were fixed with 4% para-formaldehyde and permeabilized with 0.1% TritonX-100. Cells were stained with different antibodies and later analyzed using a Zeiss 510 confocal microscope (Carl Zeiss GmbH, Jena, Germany). For analysis of binding to BMDC, Fc receptors were pre-blocked. For blocking FcR mediated nonspecific binding immature BMDCs, cells were treated with anti-FcγR antibody (CD16/32 specific for FcγRIII. FcγRII) for 10 minutes at 4°C at a concentration of 1µg/ml per million cells (Kurlander et al., 1984; Lindell et al., 2008). To prevent complication of experiments by Hsp90 interaction with HSP receptor, LOX-1, BMDC were also treated with Goat anti-mouse LOX1 ab (10 µg/ml) for 10–15 minutes on ice.
Fluorophores were visualized using the following filter sets: 488nm excitation and band pass 505–530 emission filter for Alexa 488; 543 nm excitation and BP 560–615 for Cy3; 633 excitation and long pass (LP) 650 for Cy5. Images were taken using a 63x numerical aperture (NA) 1.4 oil immersion objective lens. Figures were made using Adobe photoshop 7.0 (Adobe Systems Inc., San Jose, CA, USA) with little or no contrast adjustments without altering original images.
In vitro cross presentation assay
Immature BMDCs (104) from C57BL/6, Balbc and TAP−/− mice were plated in 96 well plates after purification. Cells were then pulsed with 10µg/ml of Hsp90 and its Ova conjugates (Hsp90-S8L/S8LC/Ova) 10µg/ml free Ova and also with 10µg/ml of anti-SREC-I-Ova complexes for 5 minutes to 2 hours. In all antigen cross presentation experiments a preparation of the BMDC was pulsed with 100ng/ml of SIINFEKL (S8L) as positive control. For inhibitors and drug treatment, cells were incubated with the drugs before they were pulsed with different antigen preparations. The ligands were then removed and cells fixed with para-formaldehyde for 10 minutes at room temperature. Later, peptide-specific T cell hybridoma (B3Z, 105, a generous gift from Dr. N. Shastri) was added to the fixed cells at 37°C for 20 hours. Culture supernatants were harvested and tested for activation of the IL-2 promoter from the stably incorporated Il-2 promoter-LacZ construct and we also measured the amount of IFNγ release by ELISA. Induction of IL-2-LacZ was assayed in cell extracts by measuring activity of LacZ product β-galactosidase. The assay involves cleavage of substrate, ONPG (o-nitrophenyl-β-D-galactopyranoside) by β-galactosidase and measuring the absorbance of cleaved product at 420 nm according to manufacturer’s instruction (Promega). For cross presentation assay in the CHO model system, CHO-SREC-I cells were transfected with murine H2kb (MHC-I) expression vector for 22 hours then chased with Ova or Hsp90 conjugated Ova for 2 hr. Cells were then fixed with 4% para-formaldehyde for 10 minutes at room temperature. Later B3Z cells were added to the wells containing fixed CHO-SREC-I (H2kb) cells for 20 hours at 37°C and assayed as above. In all antigen cross presentation assays immature BMDC were pulsed with anti-FcγR antibody for 10 minutes at 4°C at a concentration of 1µg/ml per million cells and with Goat anti-mouse LOX1 ab (10 µg/ml) for 10–15 minutes on ice to block non-specific ligand uptake through FcR and LOX1.
RESULTS
Hsp90-Ova complexes bind avidly to SREC-I, a member of the class F family of scavenger receptors and mediate antigen cross presentation
We aimed to examine the processing of Hsp90 bound polypeptides (Hsp90.PC) derived from Ovalbumin (Ova) and their presentation to T cells in the context of MHC-I molecule H2Kb. Our experiments utilized synthetic Ova peptides: (i) S8LC (a C-terminally extended Ova peptide S8L, 13 mer) which requires C terminal processing to be loaded onto MHC-I molecule, (ii) S8L (an Ova peptide of 8 mer) that could be readily loaded onto MHC-I directly without processing and (iii) full length Ova. We next examined the ability of such Hsp90.PC to bind SREC-I and showed that Hsp90 binds to SREC-I both in CHO cells stably expressing SREC-I (CHO-SREC-I), and cells with endogenous SREC-I expression including Raw 264.7 cells and the KG1 dendritic cell-like cell line) (Figure 1, supplementary Figure 1). CHO-SREC-I cells incubated with Alexa 488 labeled Hsp90-peptide complexes at 4°C showed efficient binding compared to control CHO-K1 cells (as determined by confocal microscopy) (Figure 1A). We then analyzed internalization of the receptor bound Hsp90 ligand by incubating Alexa 488-Hsp90.PC with CHO-SREC-I on ice followed by warming to 37°C. Hsp90-peptide complexes were rapidly internalized in CHO-SREC-I but not in wild type CHO-K1 cells (Figure 1A). We also examined Hsp90.S8LC uptake by SREC-I in APC. As these cells contain other HSP receptors in addition to SREC-I we examined internalization of fluorescently labeled anti-SREC-I Ab cross-linked to Ova (Supplementary Figure 2). These reagents were also used to confirm that SREC-I can specifically mediate antigen cross presentation in subsequent studies. To avoid anti-SREC-I Ab uptake by the Fc pathway, we incubated cells with Fc receptor blocking antibodies prior to labeling with anti-SREC-I Ab (Supplementary Figure 2). Therefore we conclude that SREC-I is capable of internalizing antigenic peptides bound either to Hsp90 or anti-SREC-I antibodies. We also showed binding of Alexa Hsp90.PC to CHO-SREC-I cells but not to control CHO-K1 cells by FACS analysis (Figure 1B). In addition, to further confirm that Hsp90.PC binds to SREC-I, we incubated Alexa 488-Hsp90.PC with the scavenger receptor ligand, maleylated BSA (mBSA) (Figure 1C). Hsp90.PC binding to SREC-I was reduced in the presence of mBSA as well as when incubated with anti-SREC-I ab (Figure 1C) confirming specific binding to SREC-I. Incubation with unmodified BSA did not affect Hsp90 binding (data not shown).
Figure 1. Hsp90.PC binds to SREC-I and mediates Ova antigen cross presented toB3Z cells after uptake.

(A) CHO-K1 and CHO-SREC-I cells were incubated with Alexa 488-Hsp90/Hsp90-S8LC (green) at 4°C for 30 min and counterstained with DAPI to examine cell surface binding. Next, we assayed for internalization of ligands after chase with fresh medium at 37°C for 5–10 min followed by fixation and fluorescence microscopy. (B) CHO-K1 and CHO-SREC-I cells were incubated with Alexa 488 labeled Hsp90 as above and binding at the cell surface quantitated by flow cytometry. (C) CHO-SREC-I cells were incubated with Alexa 488-labeled Hsp90 with or without scavenger receptor agonist mBSA or anti-SREC-I ab. Binding of Hsp90 to the CHO-SREC-I cells was then assayed by flow cytometry. (D) Immature BMDC from either Balbc or C57BL/6J mice were incubated with Hsp90, free Ova, Ova peptide (S8L), Hsp90.Ova, Hsp90.S8LC, Ova peptide complexes and anti SREC-I Ab-Ova complexes for 2 hours at 37°C. Cells were then fixed, washed with 1X PBS and cultured with B3Z overnight before assaying secreted IFNγ by ELISA. As a control, cross presentation assays were carried out using Balbc derived BMDC which lack Ova-specific H2kb (lane 1). (E) CHO-K1 cells were transfected with SREC-I and H2Kb (murine MHC-I) for 22 hours and then chased with Hsp90, free Ova, Ova peptide and Hsp90.PC for 2 hours. Cells were then fixed, washed and cultured with B3Z overnight. The IFNγ secreted by B3Z was assayed by ELISA. (F) In a control experiment for (E) CHO-K1 cells were transfected with H2Kb alone for 22 hrs, and chased with free or Hsp90 conjugated Ova antigens as in (E) for 2 hrs and assayed for antigen cross presentation. (G) Untransfected CHO-K1 cells were chased with ligands as E and F and IFNγ secreted by B3Z assayed for antigen cross presentation. Data shown in D-F are the mean +SD of triplicate wells. The results shown (A–F) are representative of at least three independent experiments. (H) Immature BMDC from C57BL/6J mice were incubated with Hsp90, S8L, free Ova and Hsp90.Ova for 2 hours at 37°C with or without incubation with anti-LOX-1 blocking ab or anti-SREC-I blocking ab. Cells were then fixed, washed with 1X PBS and cultured with B3Z overnight before assaying secreted IFNγ by ELISA.
Hsp90-chaperoned Ova is presented to MHC-I in BMDC
We next evaluated the role of SREC-I in cross presentation of Ova and Hsp90-Ova complexes. To address complications due to the presence of multiple Hsp90 receptors in APC we used anti SREC-I Ab cross-linked to Ova as ligand, as described above as well as Hsp90-Ova complexes. We employed immature BMDC as these are efficient antigen presenting cells (34). These cells were incubated at 37°C, with free Ova, free S8L peptide, Hsp90-S8LC complexes, Hsp90-Ova complexes or anti SREC-I antibody cross-linked to Ova. Cells were then fixed, washed and cultured with Ova-specific B3Z CD8+ T cell hybridoma. We then assayed interferon γ (IFNγ) secretion as a measure of the rate of antigen cross presentation and T cell activation by BMDC. Hsp90-Ova complexes induced abundant IFNγ secretion in CD8+ cells, which greatly exceeded effects of free Hsp90 (Figure 1D). Incubation with free Ova induced a low level of cross presentation likely due to uptake by fluid phase endocytosis or by mannose receptor (MR) (Figure 1D). In addition, anti-SREC-I conjugated Ova strongly activated IFNγ production to a similar degree compared to Hsp90-Ova, indicating that SREC-I can specifically mediate access to the antigen cross presentation pathways in DC. As a control, cross presentation assays were carried out using Balbc derived BMDC which lack Ova-specific H2kb. Cells were incubated with anti-FcR antibody for blocking FcR-mediated nonspecific uptake of Hsp90.PC. Hsp90-Ova failed to activate cross presentation in BMDC from Balbc mice (Figure 1D)
To further establish the roles of SREC-I and Hsp90.PC in antigen cross presentation we examined whether CHO-K1 cells could be engineered to mediate cross presentation by forced expression of the MHC class I molecule H2Kb required for recognition of the S8L sequence. Cresswell et al have shown recently that non-APC can be engineered to cross present antigens and we have employed a similar approach here (35). CHO-K1 cells transfected with murine H2Kb were able to cross present free Ova when in large excess but were incapable of cross priming Ova antigens bound to Hsp90 or anti-SREC-I (Figure 1F). However, expression of SREC-I in the CHO cells expressing H2Kb rendered them capable of cross presenting Hsp90-chaperoned Ova or S8LC (Figure 1E). SREC-I expression is thus sufficient to mediate Hsp90.PC uptake and cross presentation of peptides (Figure 1E). As a further control, wild type CHO-K1 cells were incubated with Hsp90.PC and peptides alone as described above and B3Z cells were cultured overnight with minimal IFNγ production (Figure 1G). Although these experiments prove the principal that SREC-I can mediate cross presentation of antigens bound to Hsp90, we examined whether IFNγ production in BMDC might also involve other scavenger receptors such as LOX-1. LOX-1 was shown to mediate cross presentation of Ova antigens complexed with Hsp70 (Delneste et al., 2002). Indeed blocking antibodies to LOX-1 reduced Hsp90-Ova induced IFNγ production by B3Z cells by approximately 50% (as did anti-SREC-I ab) (Figure 1H). Therefore all subsequent cross presentation experiments involving Hsp90-Ova included LOX-1 blocking antibodies. We observed minimal effects in such experiments of blocking FEEL-1, receptor also reported to bind HSPs (data not shown). We did not detect significant expression of HSP receptors CD91/LRP1 or FEEL-1 in BMDC (B Zhu and SK Calderwood, in preparation). In addition an excess of CD91/LRP1 agonist α2-macroglobulin did not reduce Hsp90-Ova induced binding to BMDC. We therefore concluded that Hsp90-Ova cross presentation in BMDC is largely mediated by SREC-I and LOX-1. Interestingly, LOX-1 blocking antibodies did not markedly decrease Hsp90 uptake by KG1 or Raw264.7 cells (Supp. Fig. 1).
Extracellular Hsp90-Ova complexes are internalized by a receptor-mediated, clathrin- and dynamin-independent pathway and are regulated by the actin cytoskeleton
We next addressed the nature of the internalization pathway for Hsp90.PC in cells expressing SREC-I in order to examine potential mechanisms for chaperone mediated antigen cross presentation. We aimed to characterize this process in detail as it might indicate sites for interaction of chaperoned peptides and MHC class I. Immature BMDC and CHO-SREC-I cells were incubated with Alexa 488 labeled Hsp90-S8LC at 4°C and then warmed to 37°C. The cells showed an intracellular Alexa labeled punctuate distribution characteristic of endosomes (supplementary Figure 3, Figure 1A). To further investigate the mode of endocytosis of Hsp90.PC we next inhibited clathrin-mediated endocytosis, the dominant endocytic pathway in eukaryotic cells. We employed two strategies including: (i) overexpression of EH29-YFP (Eps15 (Δ95/295)-YFP) (36) which creates aggregates in clathrin-coated pits and (ii) overexpression of a dominant-negative dynamin 2 (K44A)-GFP (37–39) in CHO-SREC-I cells. Transfected cells (both KG1 and CHO-SREC-I) were incubated with Alexa 555 -Hsp90.PC as described above. We observed minimal inhibition of Hsp90.PC uptake in cells transfected with either dynamin 2(K44A)-GFP or Eps15 (Δ95/295)-YFP consistent with a clathrin and caveolin independent endocytic route, as both pathways require dynamin (Figure 2). Quantitative analysis of endocytosis in cells expressing either dynamin 2 (K44A) or Eps15 (Δ95/295) confirmed this observation (supplementary Figure 4).
Figure 2. Hsp90.PC is internalized through a dynamin and clathrin independent endocytosis pathway.
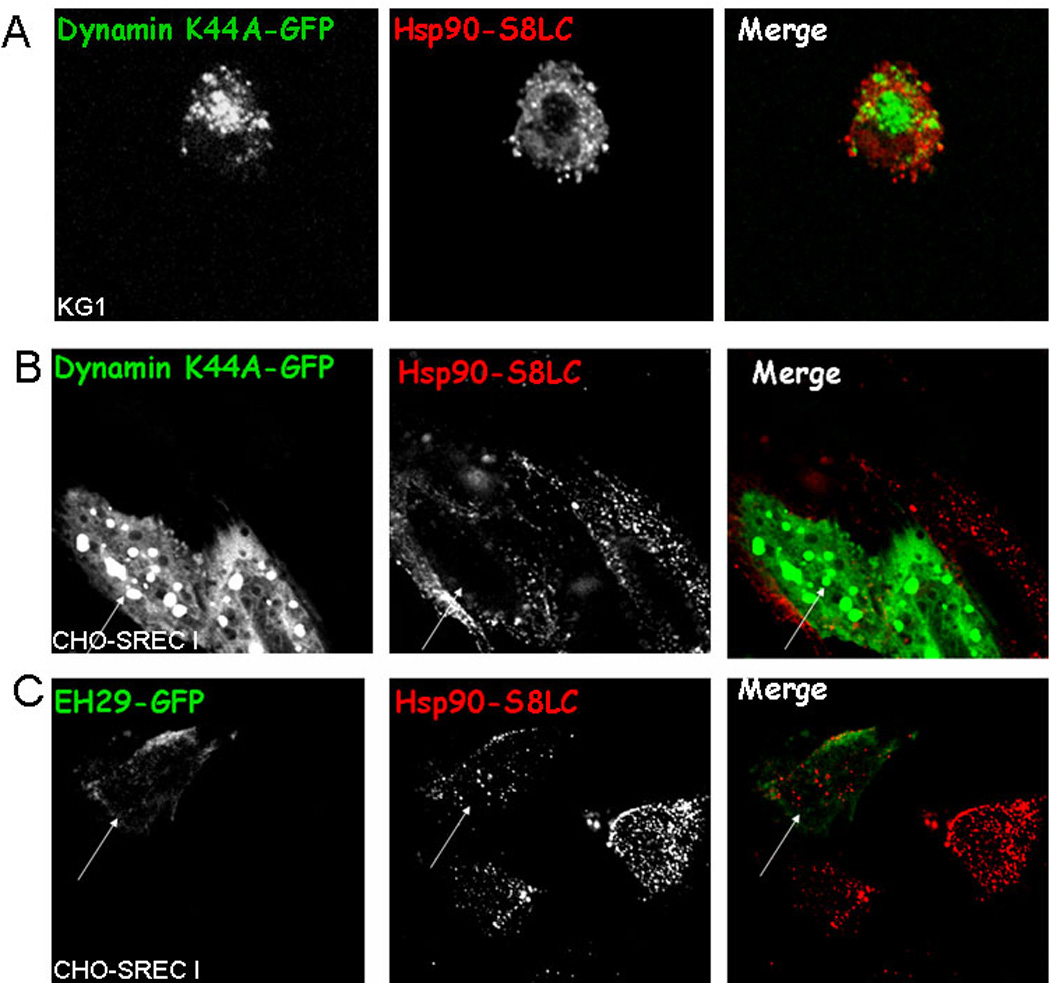
(A) KG1 cells were transfected with dynamin (K44A)-GFP (green) for 22 hours and cells then incubated with Alexa 555-Hsp90.PC (red). (B) CHO-SREC-I cells were transfected with dynamin (K44A)-GFP (green) and chased with Alexa 555-Hsp90.PC (red). (C) CHO-SREC-I cells were transfected with EH29-YFP (green) and labeled and chased with Alexa 555-Hsp90.PC (red). Experiments were carried out three times with reproducible results. More than 50 cells were examined in each experiment and representative samples presented.
Dynamin-independent receptor mediated endocytosis has been shown to involve Rhodependent macropinocytosis (40, 41), Arf6-regulated endocytosis (42), and Cdc42-dependent internalization of GPI-anchored proteins (GPI-AP) into uncoated tubular structures. Cdc42 regulated internalization of ligand-receptor complexes occurs through a class of newly characterized endocytic vesicles known as GEEC (GPI-AP enriched endosomal compartment), which later fuse with EEA1 or Rab5-positive early endosomes (Sabharanjak et al., 2002). Each of these clathrin and dynamin independent uptake processes requires actin but differs in further requirements for specific Rho GTPases such as Rho, Rac, Cdc42 and Arf1, Arf6 that regulate actin dynamics.
Extracellular anti-SREC-I ab-Ova complexes are internalized by a receptor-mediated pathway regulated by Rho-GTPases activity in BMDC
We next examined whether internalization of anti-SREC-I ab in immature BMDC follows a similar internalization pathway to Hsp90. Cells were treated with Rho-GTPase inhibitor, Toxin B (2ng/ml) for 2 hours prior to incubation with Alexa 555-anti-SREC-I antibody. We found that anti-SREC-I antibody internalization was perturbed in cells treated with the drug (Figure 3A, D). To confirm our finding of scavenger receptor mediated uptake of anti-SREC-I ab in these cells, cells were treated with non-selective scavenger receptor inhibitor, Poly I (50µg/ml) (Figure B) and DMA (500 µM) along with anti-SREC-I ab. Cells treated with Poly I showed little or no fluorescence suggesting SREC-I receptor mediated uptake of Alex 555 labeled anti-SREC-I ab (Figure B, E). On the other hand, in the presence of a specific inhibitor of macropinocytosis, DMA, cells showed somewhat similar levels of internalized anti-SREC-I ab punctate as control cells (Figure C, F), suggesting role for receptor (SREC-1) mediated uptake of anti-SREC-I ab as opposed to non-specific pinocytosis.
Figure 3. Anti-SREC-I antibody is internalized in a Rho-GTPase regulated pathway.

(A, D) Immature BMDC were incubated without (D) or with (A) Toxin B (2ng/ml). Cells were then pulsed with Alexa 555 labeled anti-SREC-I ab (red) at 4°C then incubated at 37°C for 10 min. Cells were fixed and analyzed using fluorescence microscopy. (B, E) Immature BMDC were treated with (B) or without (E) Poly I (50µg/ml) along with Alexa 555 labeled anti-SREC-I ab and antibody internalization examined under fluorescence microscope. (C, F) Immature BMDC were treated as in A, D, B and E and with (C) or without (F) DMA (500µM) along with Alexa labeled anti-SREC-I ab. Cells were later fixed and analyzed as described in A. Experiments were carried out after pre-blocking with FcR blocking antibodies as described in Materials and Methods. In these experiments BMDC were pre-treated with LPS (10µg/ml) for 2 h to boost expression of SREC-I. Experiments were carried out twice, reproducibly.
Rho-GTPase activity is required for Hsp90-peptide uptake via SREC-I
We next investigated the role of the actin cytoskeleton in uptake of Hsp90.PC in CHO-SREC-I and BMDC cells using actin depolymerizing agents Latrunculin B and Cytochalasin D. These agents caused marked inhibition of Hsp90.PC uptake, suggesting a requirement for the actin cytoskeleton in endocytosis of Hsp90.PC-SREC-I complexes in CHO-SREC-1 (Figure 4 A , C) and BDMC (data not shown). (In each case the position of the cells can be visualized by the DAPI stained nuclei). We then investigated the role of the Rho GTPases, initially employing Clostridium difficile Toxin B, a molecule that inhibits Rac, Rho and Cdc42 GTPases (43). Hsp90.PC endocytosis was severely blocked by Toxin B although the toxin had minimal effects on internalization of transferrin (Tf) through receptors that use the clathrin-dependent uptake pathway (Supplementary Figure 5).
Figure 4. Hsp90.PC internalization is actin and Rho GTPase dependent.

(A) CHO-SREC-I cells were treated with Cytochalasin D (10µM) before incubating with Alexa 555-Hsp90.PC. at 4°C for 20 minutes, then chasing with medium for 10 minutes at 37°C. (B) CHO-SREC-1 cells were incubated without Cytochalasin D and with Alexa 555-Hsp90.PC at 4°C for 20 minutes, then at 37°C in growth media for 10 minutes. (C) CHO-SREC-I cells were treated with latrunculin B (1µM) and then labeled with Alexa 555-Hsp90.PC (red) at 4°C for 20 minutes, then incubated at 37°C with media for 10 minutes. (D) Control CHO-SREC-I cells were incubated without Lat B and with Alexa 555-Hsp90.PC at 4°C for 20 minutes, then at 37°C as described in (A) (E) Human myeloid DC were transfected with Cdc42-GFP (green) for 22 hours and then incubated with Alexa 555 anti SREC-I ab (red) in the presence of Hsp90.PC. (F–H) CHO-SREC-I cells were transfected with (F) Cdc42 (N17)-GFP (green) (G) RhoA (N19)-GFP (green) and (H) Rac1 (N17)-GFP (green) for 22 hours and then labeled with Alexa 555-Hsp90.PC (red) for 20 minutes at 4°C. Later the cells were chased with normal growth media for 10 minutes at 37°C. Experiments were carried out twice with reproducible results. More than 50 cells were examined in each experiment and representative samples shown.
We then tested the requirement for individual Rho GTPases in Hsp90.PC internalization. Individual Rho GTPase were initially investigated using overexpressed wild type, GFP-tagged mammalian expression constructs of RhoA, Rac1 and Cdc42. Cdc42-GFP was expressed in human myeloid DC and CHO-SREC-I (data not shown) and cells were incubated with Alexa 555-anti-SREC-I Ab for 20 minutes on ice. The Cdc42-GFP was closely localized with labeled anti SREC-I Ab (Figure 4E) on the plasma membrane although there was minimal colocalization of SREC-1 with either Rac 1-GFP or Rho A-GFP (data not shown), suggesting a specific role for Cdc42. Similar findings were observed with Hsp90.PC uptake in CHO-SREC-1 (data not shown). To examine a causal role for these GTPases, we constructed dominant negative forms of RhoA (N19), Rac1 (N17), and Cdc42 (N17) shown previously to inhibit actindependent Rho GTPase mediated endocytosis (44). We then overexpressed these dominant negative constructs in cells for 22 hours and assayed for uptake of fluorescently labeled Hsp90.PC. Overexpression of RhoA (N19) had a minimal effect on Hsp90.S8LC uptake (Figure 4G) whereas Cdc42 (N17) expression blocked internalization of Hsp90.PC quantitatively (Figure 4F). Overexpression of Rac1 (N17) had an intermediate effect and reduced the level of internalized Hsp90-SREC-I complexes (Figure 4H). These experiments suggest a specific regulatory role for Cdc42 as well as less pronounced participation of Rac1 in dynamin-independent endocytosis of Hsp90.PC. The experiments further suggest that Hsp90.PC may be internalized through a pathway utilized by GPI-anchored proteins (GPI-AP) such as CD59 (Naslavsky et al., 2004). SREC-I has been shown to contain potential N-linked glycosylation motifs in its extracellular domain (Ishii et al., 2002). GPI-AP are targeted to specialized endosomes (GEEC) by a process dependent on actin, Cdc42, and plasma membrane sphingolipids (45). GEECs are distinct endocytic organelles which are acidic in nature and are compartments of the major pinocytic pathway (46).
Hsp90-peptide-SREC-I and CD59 share a similar clathrin and dynamin independent endocytic pathway that is regulated by Arf1 and requires membrane cholesterol
To test whether internalization of Hsp90.PC by SREC-I follows this GPI-AP endocytic pathway regulated by Cdc42, we labeled HeLa (SREC-I) cells with fluorescently labeled anti-SREC-I Ab and fluorescently labeled anti CD59 Ab for 20–30 minutes on ice. We used anti SREC-I Ab as ligand to avoid non-specific binding of Hsp90 to other scavenger receptors expressed on HeLa cells and employed HeLa cells as they express detectable levels of CD59. Indeed, CD59 and SREC-I cross-linked antibodies (anti SREC-I-Ab-Ova) became co-localized on the plasma membrane upon binding (Figure 5A). We also found that SREC-I and CD59 traffic to the GEEC compartment (Figure 5B) shown previously to be a specific immediate destination for internalized GPI-AP such as CD59 (Naslavsky et al., 2004). In addition MHC class I molecules were observed co-localizing with anti SREC-I Ab in this compartment identified by GPI-AP marker CD59, suggesting a role for GEEC as a potential location for Hsp90.PC and MHC class I interaction (Figure 5C). We then tested the localization of MHC-I and anti-SREC-I ab in CHO-SREC-I cells. In 2 minutes of internalization, anti-SREC-I ab and MHC-I were seen in the compartment marked by FITC-CD59 (Figure 5A, B and C).
Figure 5. Anti SREC-I Ab uptake into the CD59-marked GEEC compartment requires cholesterol and is regulated by ARF 1 GTPase.

(A) HeLa -SREC-I cells were labeled with Alexa 555 labeled anti SREC-I Ab (red) and FITC-CD59 (green) at 4°C for 20 minutes. (B) HeLa-SREC-I cells were labeled with anti MHC-I Ab (red) and FITC-CD59 (green) at 4°C for 20 minutes and chased at 37°C for 2 minutes. (C) HeLa -SREC-I cells were labeled with Alexa 555 labeled anti SREC-I Ab (red) at 4°C for 20 minutes and then chased at 37°C as in B. Cells were then stained for MHC-I with anti-MHC-I ab (green). (D) CHO-SREC-I cells were transfected with ARF 1 (T31N)-HA (green) for 22 hours, labeled with Hsp90.PC at 4°C for 20 minutes then chased at 37°C for 10 minutes. (E) HeLa-SREC-I cells were treated with MβCD (10 mM) and then incubated with Alexa 555-anti SREC-I Ab (red). (F) Control HeLa-SREC-I cells were incubated without MβCD and with Alexa 555-anti SREC-I Ab (red) as in E. (G–H) HeLa-SREC-I cells were incubated with CTXB (green) and Alexa 555- anti SREC-I Ab (red) (G) at 4°C for 20 minutes and (H) later chased for 2 minutes at 37°C with fresh growth media. Experiments were carried out twice with reproducible results. More than 50 cells were examined in each experiment.
To confirm the internalization of SREC-I Ab into GEEC by the Cdc42 regulated pinocytic pathway, we examined the requirement for another class of small GTPases. As ARF1 GTPase is involved in regulating this pathway we determined the effect of expressing dominant negative ARF1 in CHO-SREC-I cells on Hsp90-S8LC uptake. Indeed, overexpression of this construct inhibited endocytosis of Hsp90.PC (Figure 5D). We also tested the role of, ARF6 as activated ARF6 can also mediate GPI-AP uptake in mammalian cells (Naslavsky et al., 2004). However overexpression of ARF6 (T27N)-HA only slightly decreased the uptake of Hsp90.PC indicating that that ARF6 may not be crucial for Hsp90.PC uptake via SREC-I in those cells (data not shown). Cdc42 recruitment and activation in the plasma membrane and cytosolic actin dynamics also require membrane cholesterol. We therefore examined the role of mild cholesterol depletion in endocytosis of anti-SREC-I antibodies in HeLa cells via the GEEC pathway using cholesterol depleting/sequestering drug cyclodextrin (MβCD) (Figure 5E, F). MβCD treatment decreased the number of fluorescent internalized vesicles compared to control cells demonstrating a role for membrane cholesterol in uptake of SREC-I (Figure 5E). Similar results were seen when we examined endocytosis of Hsp90.PC in CHO-SREC-1 cells (data not shown). Thus plasma membrane cholesterol plays a role in endocytosis of ligand-bound SREC-I through the GEEC pathway. Cholesterol depletion from the plasma membrane may inhibit cortical actindependent SREC-I uptake in cells, thereby inhibiting Hsp90.PC uptake as suggested by studies in other systems (47, 48).
SREC-I is localized to lipid raft domains in the plasma membrane
Lipid rafts are cholesterol-rich membrane microdomains containing abundant cell signaling molecules (49). Ligand-induced reorganization of signaling molecules is often observed in lipid raft domains within the plasma membranes of immune cells (50). To investigate whether SREC-I is localized to such lipid raft domains upon ligand binding, we labeled HeLa (SREC-I) with fluorescently labeled anti-SREC-I Ab. We used cholera toxin-B (CTX-B), which binds specifically to the ganglioside GM1 as a marker for lipid raft domains. Indeed the ligands were found to be co-localized on the plasma membrane (Figure 5 G). We observed however only partial colocalization of CTX-B and anti-SREC-I ab after a 2 minute chase at 37°C, indicating differing trafficking routes after internalization of these molecules despite prior residence in similar domains on the cell surface (Figure 5H). Thus Hsp90-peptide-SREC-I complexes appear to be localized to lipid rafts, where they may potentially encounter molecules that could mediate signaling pathway activation and internalization.
c-src phosphorylation and Hsp90.PC internalization via SREC-I
The activation of non-receptor tyrosine kinases such as c-src has been shown to modify many receptors anchored in the lipid raft domains of the plasma membrane through phosphorylation of motifs in the intracellular domain (51, 52). We therefore investigated interactions between c-src and SREC-I after Hsp90 binding in CHO-SREC-I. SREC-I and c-src were visualized after Hsp90 binding using confocal microscopy and the molecules were strongly co-localized on plasma membrane domains (Figure 6A). Treatment of CHO-SREC-I cells with PP1, a Src kinase specific inhibitor reduced Hsp90.PC uptake (Figure 6B) compared with uptake in untreated control cells (Fig 1A) (Hanke et al., 1996) but did not affect the binding of ligand at 4°C (Figure 6B). Similar results were seen when anti-SREC-I ab was used as ligand. PP1 inhibited internalization of the antibody suggesting a specific role for c-src in endocytosis of SREC-I bound ligands (Figure 6 C, D). We next tested the phosphorylation status of c-src using phosphospecific antibodies directed against activating tyrosine residue Y-416. c-src became phosphorylated on this residue in both CHO-SREC-1 and Hela–SREC-I cells in the presence of Hsp90.PC whereas minimal c-src-Y416 phosphorylation was found in untreated controls (Figure 6E). These experiments therefore further suggest a role for c-src activity in Hsp90.PC internalization via SREC-I.
Figure 6. Hsp90.PC internalization via SREC-I requires Src kinase activity.

(A) CHO-SREC-I cells were stained with anti-c-src antibody (green) and Alexa 555 labeled anti-SREC-I Ab (red) at 4°C for 20 minutes and analyzed by confocal microscopy as in Materials and Methods. (B) CHO-SREC-I cells were not treated or treated with c-src kinase inhibitor PP1 (10 µM), then incubated with Alexa 555 Hsp90.S8LC complexes (red) at 4°C or 37°C as indicated. (C–D) Immature BMDC were not treated (C) or treated with PP1 (10 µM) (D), then pulsed with Alexa 555 labeled anti-SREC-I ab (red) for 20 minutes at 4°C and later chased with fresh medium for 10 minutes at 37°C. Cells were later fixed and stained with nuclear dye, DAPI (blue). (E) CHO-SREC-I and HeLa-SREC-I cells were incubated with Hsp90.S8LC and assayed for c-src phosphorylation. Levels of total c-src and c-src-phospho-Y416 were examined in the lysates from control cells and cells incubated with Hsp90.S8LC by immunoblot. Experiments were done twice with reproducible outcomes.
These investigations therefore indicate a novel, highly-regulated pathway for uptake of Hsp90 peptide complexes after binding to SREC-I. We next investigated the role of this complex pathway in regulating chaperone-mediated antigen cross presentation.
Cross presentation of Hsp90-chaperoned Ova peptides requires actin, Rho GTPases and c-src activity
To determine the significance of the GEEC pathway in Hsp90/SREC-I mediated antigen cross presentation, we next evaluated the ability of the actin and Rho GTPase inhibitors used in the earlier experiments to block cross presentation. Dendritic cells were treated with cytochalasin D (Figure 7A, D) or Toxin B before chasing with either Hsp90-Ova complexes or anti SREC-I Ab-Ova conjugate and assaying for cross presentation by either IFN-γ release or IL-2 reporter assay (Figure 7B, E). As in the earlier experiments, Ova polypeptides non-covalently coupled to Hsp90 or chemically cross-linked to anti SREC-I Ab activated cross presentation in DC (Figure 7). Cytochalasin D inhibited cross presentation by Hsp90 conjugated to full length Ova or S8LC as well as anti-SREC-I Ova, indicating a requirement for actin dynamics in Hsp90 / SREC-I mediated cross presentation pathway (Figure 7A, D). Likewise, Toxin B was effective in reducing Hsp90.PC mediated antigen cross presentation, confirming a key role for the Rho GTPases (Figure 7B, E). The experiments in Figure 7 A, D and 7 B, E therefore suggest a role for the GEEC pathway of endocytosis in Hsp90 mediated cross presentation assayed by either IFN-γ release or IL-2 reporter assay.
Figure 7. Cross presentation of Hsp90 chaperoned peptides after binding SREC-I requires actin, Rho GTPase activity and c-src activation.

(A–C) Immature BMDC were treated with (A, D) cytochalasin D (10 µM) (B, E) Toxin B (2 ng/ml), and (C, F) PP1 (10µM) and incubated at 37°C. Cells were then pulsed with Ova polypeptides, Hsp90.PC complexes and anti SREC-I Ab-Ova for 2 hours at 37°C, fixed and washed then cultured with B3Z T cell hybridoma cells overnight. As a control, immature BMDC from Balbc mice were pulsed with Hsp90-Ova in each experiment. IFNγ secreted by B3Z was measured by sandwich ELISA using anti IFNγ Ab (A, B, C). IL-2 promoter induction in B3Z cells was assayed using an endogenous IL-2 promoter–LacZ reporter construct, with intracellular β-galactosidase activity employed as readout. Data shown are the means +SD of triplicate assays. The results shown are representative of at least three independent experiments.
As our studies shown in Figure 5 indicate that c-src regulates Hsp90.PC internalization through the GEEC pathway, we next treated BMDC with c-src inhibitor PP1 and examined the cross presentation capacity of immature BMDC. C-src inhibition blocked cross presentation through the Hsp90-SREC-I pathway (by Hsp90-S8LC, Hsp90-Ova and anti-SREC-I Ova) but not of free Ova or free S8L. (Figure 7C, F). Cross presentation of Hsp90 chaperoned peptides through SREC-I thus appears to involve the GEEC pathway and is subject to c-src regulation (Figure 7 A-F).
Hsp90-chaperoned peptides are transferred from GEEC to recycling endosomes, processed by endosomal proteases and cross presented by BMDC
We next investigated the intracellular compartments involved in Hsp90-mediated antigen cross presentation. To assess the trafficking route of Hsp90.PC following internalization through the GEEC pathway, we incubated KG1 cells with Hsp90.PC for 20 minutes on ice and later chased for 2–30 minutes with or without human Cy3-Tf. Excess surface Cy3-Tf was removed by washing cells with buffer (50 mM Sodium-citrate, 280 mM Sucrose, 0.01 mM Deferoxamine Mesylates, pH 4.6) for 2 minutes on ice before fixation. We used KG1 cells rather than CHO-SREC-I as Cy3-Tf only recognizes transferrin receptors expressed in human cells. Cy3-Tf binds to transferrin receptors expressed on the surface and is recycled back to the plasma membrane after releasing iron in the intracellular compartment. We found that after 10 minutes Hsp90.PC became localized in recycling endosomes marked with Cy3-Tf (Figure 8A). We also stained for EEA1 (early endosome antigen 1) and found that after 5 minutes of internalization, Hsp90.PC became localized in early endosomes marked with EEA1 (Supplementary Figure 3 C). Our experiments therefore indicate that ligand-bound Hsp90 traffics through the GEEC, early endosome and recycling endosome compartments. (It had been shown before that Hsp90 chaperoned peptides can be transferred to recycling endosomes containing MHC-I (Kurotaki et al., 2007)). Next, to investigate a causal role for the early or recycling endosomes in Hsp90-mediated antigen cross presentation, immature BMDC were treated with primaquine, an inhibitor which blocks the membrane recycling pathway, and chloroquine, a known inhibitor of vacuolar acidification of endosomal compartments, before chasing with Hsp90 conjugated Ova peptides or anti-SREC-I antibody cross linked to Ova. BMDC incubated with these drugs exhibited a reduced level of cross presentation compared to control BMDC (Figure 8B, F, C, G). These experiments suggest that SREC-I complexes, after trafficking to recycling endosomes release Ova peptides (S8L, S8LC) that can be loaded onto recycling MHC-I, thus mediating presentation to the B3Z T cell hybridoma cells. Both primaquine and chloroquine treatment resulted in the inhibition of Hsp90 mediated S8LC presentation indicating that recycling endosomes and vacuolar acidification are required for S8LC processing and uploading to MHC-I molecules found into these compartments (Figure 8B, F, C and G).
Figure 8. Hsp90-mediated Ova peptide cross presentation requires endosomal protease, cathepsin S and is inhibited by leupeptin.

(A) KG1 cells were labeled with Alexa-555-Hsp90.S8LC complexes and Cy3 Tfn at 37°C. (B–C) Immature BMDC were treated with either (B, F) primaquine (20 µM) or (C, G) chloroquine (20 µM) at 37°C for 2 hours before incubation with Hsp90-Ova peptide complexes. Cells were then fixed, washed and cultured with B3Z T cells overnight and assayed for IFNγ release. (D–E) Immature BMDC were treated with (D, H) cathepsin S inhibitor (200 µM) or (E, I) (leupeptin 5 µM) at 37°C for 2 hours and then incubated with Ova peptides, free Ova, Hsp90-Ova complexes and anti SREC-I-Ab-Ova. As a control, for non-specific activation, immature BMDC from Balbc mice were pulsed with Hsp90-Ova in each experiment. Cells were then fixed, washed and cultured with B3Z T cell. As control in each experiment, we also examined BMDC activation by Hsp90 or the activity of B3Z cells alone (in 7, 8, 9). B3Z activation was assayed either by IFNγ secretion or IL-2 promoter activity as in Fig. 7. Data shown are the mean +SD of triplicate wells. The results shown are representative of at least three independent experiments.
We also examined the effect of cysteine protease inhibitor leupeptin on Hsp90- and SREC-I-mediated antigen cross presentation. Indeed leupeptin strongly inhibited Ova peptide cross presentation (Figure 8E and I). Leupeptin reduced presentation mediated by Hsp90-S8LC, Hsp90-Ova, anti-SREC-I Ova and free Ova. Cross presentation of free S8L was not affected. We next studied the effect on cross presentation of the endosomal protease, cathepsin S (22). Hsp90/SREC-I-mediated cross presentation of polypeptides that require proteolytic processing was clearly inhibited by the cathepsin S inhibitor further suggesting a requirement for endosomal proteases in such antigen cross presentation (Figure 8D and H). It is however not clear whether endosomal acidification is required for antigen processing by cathepsin S. Cathepsin S has a broad tolerance for pH and can function at neutral pH. Presentation of fully processed peptide S8L is not inhibited by chloroquine as might be predicted. It is notable that in this experiment and subsequent experiments of antigen cross presentation similar results were obtained whether IL-2 promoter activity or IFNγ release by B3Z were assayed.
Hsp90.Ova complexes require proteasomal activity and TAP expression for efficient cross presentation of Ova peptides
For cross presentation by APC, antigens must be fully processed to the 8-mer SIINFEKL peptide and loaded onto MHC class I. We therefore next examined whether components of the endosome-cytosol pathway are also required for chaperone-mediated cross presentation of Ova. To probe a role for the proteasome, BMDC were treated with proteasome inhibitor, lactacystin (20µM) (Figure 9). Lactacystin treatment for 30 min had minimal inhibitory effect on Hsp90 mediated S8LC peptide cross presentation but affected the cross presentation of full length Ova bound to Hsp90 or anti-SREC-I Ab, suggesting a role for the proteasomal system for processing the larger Ova polypeptides (in addition to digestion by cysteine proteases in the endosomes shown in Figure 8D and H). BMDC were also incubated with Hsp90.PC for 2 hours after treatment with lactacystin. In this case, lactacystin had profound inhibitory effects on this more gradual component of cross presentation, particularly with full length Ova complexed to Hsp90 or bound to anti-SREC-I (Figure 9). These findings may suggest essential, initial Ova processing in the endosome followed by transfer of peptides to the cytosol and processing by the proteasome. The latter process seems essential for antigen presentation in the context of larger polypeptides but dispensable for smaller peptides such as S8LC. The more gradual processing through this compartment may reflect the extra steps needed for polypeptides to cross endosomal membranes and enter the proteasome. In addition smaller peptides may lack the signals for polyubiquitinylation required for efficient targeting to the proteasome.
Figure 9. Role of proteasome activity in cross presentation of hsp90-chaperoned peptides.

(A–B) Immature BMDC were treated with lactacystin (20µM) before incubating with Ova peptides, free Ova, Hsp90-Ova complexes and anti SREC-I-ab-Ova for (A, C) 30 minutes and (B, D) 2 hours. Cells were then fixed, washed and cultured with B3Z T cells overnight. For control, immature BMDC from Balbc mice were pulsed with Hsp90-Ova in all cases. B3Z activation was assayed either by IFNγ secretion or IL-2 promoter activity as in Fig. 7. Data are represented as the mean +SD of triplicate assays. The results shown are representative of at least three independent experiments.
Peptides processed in the cytosol by the proteasome are taken up by TAP in the ER or TAP containing endosomes for loading onto MHC-I. Therefore we asked whether TAP is required for chaperone-mediated antigen cross presentation by comparing the efficiency of Hsp90.Ova cross presentation in BMDC from TAP−/− mice and wild type mice. TAP−/− and control BDMC were incubated with antigens for 15 min and 2 hr. In accordance with the lactacystin experiments, TAP−/− DC were able to present Hsp90 / SREC-I associated Ova antigens quite efficiently when incubated for 15 min (Fig. 10). However, cross presentation observed after 120 min incubation with Ova or anti-SREC-I-Ova was inhibited in TAP−/− DC. This further suggest that at least two kinetically distinct pathways of antigen cross presentation may be accessed by Hsp90.PC, a rapid pathway that is sufficient for peptides requiring minimal processing and inhibited by antagonists of endosomal proteases and a more slowly developing, proteasome- and TAP-dependent route especially significant for larger polypeptides.
Figure 10. Role of TAP in Ova antigen cross presentation via the Hsp90-peptide-SREC-1 pathway.

Immature BMDC from TAP−/− mice and C57 BL/6 (H-2b) mice were incubated with Hsp90, free precursor peptide (S8L), free Ova, Hsp90.Ova and anti SREC-I Ab. Ova complexes at 37°C for (A, C) 15mins and (B, D) 2 hours. Cells were then fixed, washed and cultured with B3Z T cell hybridoma overnight. As a control, immature BMDC from Balbc mice were pulsed with Hsp90-Ova in each experiment. B3Z activation was assayed either by IFNγ secretion or IL-2 promoter activity as in Fig. 7. Mean IFNγ levels +SD of triplicate assays are indicated. The results shown are representative of at least three independent experiments.
DISCUSSION
Our experiments show that antigens complexed to Hsp90 can be specifically bound by SREC-I, internalized and cross presented by MHC-I (Figure 1). In BMDC, this process is complicated by the presence of multiple HSP receptors. Our studies suggest that SREC-I plays a major role in BMDC, in tandem with LOX-1. Other cell surface proteins characterized as HSP-receptors do not seem to play a major role in BMDC. The other receptors may carry out additional roles in immunity. Hsp90.PC-induced SREC-I uptake involves the GEEC pathway characterized previously for GPI anchored proteins such as CD59 and the folate receptor (53). One function of peptide association with Hsp90 may thus be to direct the antigen along the GEEC pathway of endocytosis initiated by binding SREC-I (Fig. 2). Another function of Hsp90 is to chaperone peptides and deter proteolysis and the studies of Shastri et al show efficient chaperoning of Ova polypeptides by intracellular Hsp90 in the conventional pathway of antigen presentation (54).
Cell surface SREC-I appeared to be localized to lipid raft domains upon ligand binding. While the full significance of these findings is not clear, these domains contain a number of GPI anchored proteins and tyrosine kinases important in cell regulation. Hsp90 may thus carry out an additional function in antigen cross presentation involving the triggering of signaling pathways that promote peptide internalization and other processes. Lipid raft localization suggested to us that src-family kinases could play a role in recruitment of SREC-I into the clathrin-independent pathway. Indeed Hsp90.PC was shown to activate c-src and lead to its colocalization with SREC-I (Fig. 5), mechanisms required for SREC-I mediated antigen cross presentation. Further interactions of Hsp90-SREC-I complexes with lipid raft domain regulatory proteins are under investigation. It is tempting to speculate that SREC-I mediates rapid formation of a signaling platform upon Hsp90.PC binding which may increase the docking sites for other signaling molecules. It may be significant that MHC-I and SREC-I were both localized in this same lipid raft domain compartment marked by CD59.
Although Hsp90.PC-SREC-I complexes are internalized through the GEEC pathway, we did not detect antigen cross presentation at the earlier period (two to 5 minutes) when the complexes are confined to GEEC (Data not shown). This finding may reflect the finite nature of peptide processing, presentation and IFNγ transcription and release required to detect antigen cross presentation, compared to kinetically rapid uptake into GEEC. The mechanistic role of GEEC in cross presentation is therefore yet to be determined. Hsp90-Ova complexes, after internalization through the GEEC pathway next trafficked to early or recycling endosomes, where peptides could be loaded onto recycling MHC-I molecules (Figure 7, Supplementary Figure 3, Figure 11). Recent reports suggest that early/recycling endosomes can participate in peptide loading (23). Our studies indicate that early endosomes and / or recycling endosomes are important sites for Hsp90 / SREC-I mediated cross presentation, as the endosomal cysteine protease cathepsin S was required for maximal Ova peptide generation and subsequent cross presentation (Fig 8). Our results also indicate that while proteasomal function and TAP are also required for efficient cross presentation of full length Ova bound to Hsp90, smaller Ova peptides could be presented in a TAP-independent and lactacystin insensitive manner suggesting that generation of immune responses can occur directly within the endosome (Figures 8, 9, 10). Our findings thus suggest the possibility of complex antigen cross presentation pathways for Hsp90 chaperoned Ova and Ova peptides that may depend on the size of the chaperoned polypeptide and other factors (Figure 10).
Figure 11. Schematic diagram of Hsp90-mediated antigen cross presentation.

(A) Endosomal pathway of cross presentation. Hsp90.Ova/Ova peptide complexes are internalized by BMDC in a SREC-I receptor-mediated, Cdc42-regulated pinocytic pathway into the GEEC compartment which is regulated by the activity of Arf1 and c-src. Ova peptides then traverse early endosomes complexed to Hsp90 and SREC-I and can be processed and loaded onto MHC-I molecules in recycling endosomes. MHC-I-Ova peptide complexes can then recycle to the plasma membrane and encounter CD8+ T cells. (B) Endosome-to-cytosolic pathway of cross presentation. In an additional pathway, observed with Hsp90 complexes containing full length Ova, the Ova traverses the SREC-I/GEEC pathway defined in this study and is partially processed at an endosomal site as in (A). However, a proportion of the peptides evidently enter the cytosol, are processed by the proteasome and enter compartments containing TAP such as endosomes and loaded onto MHC-I or the ER where they may be further processed by ERAP (Endoplasmic Reticulum amino peptidases) and loaded onto newly synthesized MHC-I molecules. MHC-I-processed Ova can then travel from the ER (or other TAP-containing compartments) to the plasma membrane for activation of CD8+ T cells.
Under in vivo conditions, Hsp90.PC would likely interact with DC expressing a range of HSP receptors (25, 29, 55, 56). The outcome, in terms of immunity may thus be an integral of a number of such interactions. One of the HSP receptors, SRA-1 appears to be an inhibitor of immune processes by blocking TLR4 activation, although its role in antigen cross presentation is not known (57). In addition, we have found that FEEL-1 (a class H SR) is capable of binding to Hsp90 but fails to cross present Ova peptide in CHO-K1 cells expressing FEEL-1 and murine MHC-I molecule H2Kb (A Murshid & SK Calderwood, unpublished). LOX-1 was shown previously to be capable of cross presentation initiated by Hsp70.Ova complexes (56). Indeed blocking of LOX-1 significantly reduced Hsp90-Ova mediated cross presentation indicating roles for both LOX-1 and SREC-I. SREC-I and LOX-1 appear to function in tandem in a number of immune-related functions (Jeannin et al., 2005). We also failed to demonstrate a role for the receptor CD91 in responses to Hsp90.Ova complexes (data not shown) although this receptor seems important in antigen cross presentation of some other HSP-peptide complexes (3). In addition, SREC-I also possesses other properties in addition to cross presenting Hsp90 chaperoned peptides and our previous studies suggest that it may facilitate uptake of HSP-bound peptides through the Class II pathway (30).
This work therefore suggests a specific pathway for molecular chaperone-mediated antigen cross presentation that differs from those observed in cross presentation of antigens in other forms and requires the scavenger receptor SREC-I.
Supplementary Material
Acknowledgments
# SKC is supported by NIH research grants R01CA047407; R01CA094397 and R01CA119045. JG is a recipient of the US Department of Defense Breast Cancer Research programs and Susan G. Komen Breast Cancer Foundation. We thank the Department of Radiation Oncology, BIDMC for their support.
REFERENCES
- 1.Lindquist S, Craig EA. The heat shock proteins. Ann. Rev. Genet. 1988;22:631–637. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 2.Wegele H, Muller L, Buchner J. Hsp70 and Hsp90--a relay team for protein folding. Rev Physiol Biochem Pharmacol. 2004;151:1–44. doi: 10.1007/s10254-003-0021-1. [DOI] [PubMed] [Google Scholar]
- 3.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 4.Mambula SS, Calderwood SK. Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int J Hyperthermia. 2006;22:575–585. doi: 10.1080/02656730600976042. [DOI] [PubMed] [Google Scholar]
- 5.Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol. 2006;177:7849–7857. doi: 10.4049/jimmunol.177.11.7849. [DOI] [PubMed] [Google Scholar]
- 6.Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000;1:151–155. doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- 7.Berwin B, Hart JP, Rice S, Gass C, Pizzo SV, Post SR, Nicchitta CV. Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. Embo J. 2003;22:6127–6136. doi: 10.1093/emboj/cdg572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vabulas RM, Ahmad-Nejad P, Costa Cda, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–31339. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 9.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 10.Kottke T, Pulido J, Thompson J, Sanchez-Perez L, Chong H, Calderwood SK, Selby P, Harrington K, Strome SE, Melcher A, Vile RG. Antitumor Immunity Can Be Uncoupled from Autoimmunity following Heat Shock Protein 70-Mediated Inflammatory Killing of Normal Pancreas. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pockley AG, Muthana M, Calderwood SK. The dual immunoregulatory roles of stress proteins. Trends Biochem Sci. 2008;33:71–79. doi: 10.1016/j.tibs.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Murshid A, Gong J, Calderwood SK. Heat-shock proteins in cancer vaccines: agents of antigen cross-presentation. Expert Rev Vaccines. 2008;7:1019–1030. doi: 10.1586/14760584.7.7.1019. [DOI] [PubMed] [Google Scholar]
- 13.Arnold D, Faath S, Rammensee H, Schild H. Cross-priming of minor histocompatibility antigen-specific cytotoxic T cells upon immunization with the heat shock protein gp96. J Exp Med. 1995;182:885–889. doi: 10.1084/jem.182.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicchitta CV. Biochemical, cell biological and immunological issues surrounding the endoplasmic reticulum chaperone GRP94/gp96. Curr Opin Immunol. 1998;10:103–109. doi: 10.1016/s0952-7915(98)80039-3. [DOI] [PubMed] [Google Scholar]
- 15.Gong J, Zhu B, Murshid A, Adachi H, Song B, Lee A, Liu C, Calderwood SK. T Cell Activation by Heat Shock Protein 70 Vaccine Requires TLR Signaling and Scavenger Receptor Expressed by Endothelial Cells-1. J Immunol. 2009 doi: 10.4049/jimmunol.0901235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 17.Ortmann B, Copeman J, Lehner PJ, Sadasivan B, Herberg JA, Grandea AG, Riddell SR, Tampe R, Spies T, Trowsdale J, Cresswell P. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science. 1997;277:1306–1309. doi: 10.1126/science.277.5330.1306. [DOI] [PubMed] [Google Scholar]
- 18.Rock KL, York IA, Saric T, Goldberg AL. Protein degradation and the generation of MHC class I-presented peptides. Adv Immunol. 2002;80:1–70. doi: 10.1016/s0065-2776(02)80012-8. [DOI] [PubMed] [Google Scholar]
- 19.Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity. 1996;5:103–114. doi: 10.1016/s1074-7613(00)80487-2. [DOI] [PubMed] [Google Scholar]
- 20.Solheim JC, Carreno BM, Hansen TH. Are transporter associated with antigen processing (TAP) and tapasin class I MHC chaperones? J Immunol. 1997;158:541–543. [PubMed] [Google Scholar]
- 21.Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25:607–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 23.Kurotaki T, Tamura Y, Ueda G, Oura J, Kutomi G, Hirohashi Y, Sahara H, Torigoe T, Hiratsuka H, Sunakawa H, Hirata K, Sato N. Efficient cross-presentation by heat shock protein 90-peptide complex-loaded dendritic cells via an endosomal pathway. J Immunol. 2007;179:1803–1813. doi: 10.4049/jimmunol.179.3.1803. [DOI] [PubMed] [Google Scholar]
- 24.Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, Salle Hde la, Schild H. Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol. 1999;162:3757–3760. [PubMed] [Google Scholar]
- 25.Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. J Biol Chem. 2004;279:51250–51257. doi: 10.1074/jbc.M406202200. [DOI] [PubMed] [Google Scholar]
- 26.Singh-Jasuja H, Toes RE, Spee P, Munz C, Hilf N, Schoenberger SP, Ricciardi-Castagnoli P, Neefjes J, Rammensee HG, Arnold-Schild D, Schild H. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med. 2000;191:1965–1974. doi: 10.1084/jem.191.11.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Calderwood SK, Theriault J, Gray PJ, Gong J. Cell surface receptors for molecular chaperones. Methods. 2007;43:199–206. doi: 10.1016/j.ymeth.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Theriault JR, Adachi H, Calderwood SK. Role of scavenger receptors in the binding and internalization of heat shock protein 70. J Immunol. 2006;177:8604–8611. doi: 10.4049/jimmunol.177.12.8604. [DOI] [PubMed] [Google Scholar]
- 30.Gong J, Koido S, Calderwood SK. Cell fusion: from hybridoma to dendritic cell-based vaccine. Expert Rev Vaccines. 2008;7:1055–1068. doi: 10.1586/14760584.7.7.1055. [DOI] [PubMed] [Google Scholar]
- 31.Kutomi G, Tamura Y, Okuya K, Yamamoto T, Hirohashi Y, Kamiguchi K, Oura J, Saito K, Torigoe T, Ogawa S, Hirata K, Sato N. Targeting to static endosome is required for efficient cross-presentation of endoplasmic reticulum-resident oxygen-regulated protein 150-peptide complexes. J Immunol. 2009;183:5861–5869. doi: 10.4049/jimmunol.0803768. [DOI] [PubMed] [Google Scholar]
- 32.Castellino F, Boucher PE, Eichelberg K, Mayhew M, Rothman JE, Houghton AN, Germain RN. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J Exp Med. 2000;191:1957–1964. doi: 10.1084/jem.191.11.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabharanjak S, Sharma P, Parton RG, Mayo rS. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 34.Lutz MB, Kurts C. Induction of peripheral CD4+ T-cell tolerance and CD8+ T-cell cross-tolerance by dendritic cells. Eur J Immunol. 2009;39:2325–2330. doi: 10.1002/eji.200939548. [DOI] [PubMed] [Google Scholar]
- 35.Giodini A, Rahner C, Cresswell P. Receptor-mediated phagocytosis elicits cross-presentation in nonprofessional antigen-presenting cells. Proc Natl Acad Sci U S A. 2009;106:3324–3329. doi: 10.1073/pnas.0813305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benmerah A, Lamaze C, Begue B, Schmid SL, Dautry-Varsat A, Cerf-Bensussan N. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J Cell Biol. 1998;140:1055–1062. doi: 10.1083/jcb.140.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature. 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- 38.van Dam EM, Broeke T, Jansen K, Spijkers P, Stoorvogel W. Endocytosed transferrin receptors recycle via distinct dynamin and phosphatidylinositol 3-kinase-dependent pathways. J. BIol.Chem. 2002;277:48876–48883. doi: 10.1074/jbc.M206271200. [DOI] [PubMed] [Google Scholar]
- 39.Miwako I, Schroter T, Schmid SL. Clathrin- and dynamin-dependent coated vesicle formation from isolated plasma membranes. Traffic. 2003;4:376–389. [PubMed] [Google Scholar]
- 40.Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15:3309–3319. doi: 10.1091/mbc.E04-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J.Biol Chem. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 42.Naslavsky N, Weigert R, Donaldson JG. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol Biol Cell. 2003;14:417–431. doi: 10.1091/mbc.02-04-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giry M, Popoff MR, Eichel-Streiber Cvon, Boquet P. Transient expression of RhoA, -B, and -C GTPases in HeLa cells potentiates resistance to Clostridium difficile toxins A and B but not to Clostridium sordellii lethal toxin. Infect Immun. 1995;63:4063–4071. doi: 10.1128/iai.63.10.4063-4071.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110(Pt 6):707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 45.Cheng ZJ, Singh RD, Sharma DK, Holicky EL, Hanada K, Marks DL, Pagano RE. Distinct mechanisms of clathrin-independent endocytosis have unique sphingolipid requirements. Mol Biol Cell. 2006;17:3197–3210. doi: 10.1091/mbc.E05-12-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 47.Mayor S, Sabharanjak S, Maxfield FR. Cholesterol-dependent retention of GPI-anchored proteins in endosomes. Embo J. 1998;17:4626–4638. doi: 10.1093/emboj/17.16.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- 49.Le Roy C, Wrana JL. Signaling and endocytosis: a team effort for cell migration. Dev Cell. 2005;9:167–168. doi: 10.1016/j.devcel.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol. 2006;23:49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arcaro A, Aubert M, Hierro MEEspinosa del, Khanzada UK, Angelidou S, Tetley TD, Bittermann AG, Frame MC, Seckl MJ. Critical role for lipid raft-associated Src kinases in activation of PI3K–Akt signalling. Cell Signal. 2007;19:1081–1092. doi: 10.1016/j.cellsig.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Lee H, Park DS, Wang XB, Scherer PE, Schwartz PE, Lisanti MP. Src-induced phosphorylation of caveolin-2 on tyrosine 19. Phospho-caveolin-2 (Tyr(P)19) is localized near focal adhesions, remains associated with lipid rafts/caveolae, but no longer forms a high molecular mass hetero-oligomer with caveolin-1. J Biol Chem. 2002;277:34556–34567. doi: 10.1074/jbc.M204367200. [DOI] [PubMed] [Google Scholar]
- 53.Naslavsky N, Weigert R, Donaldson JG. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell. 2004;15:3542–3552. doi: 10.1091/mbc.E04-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kunisawa J, Shastri N. Hsp90alpha chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24:523–534. doi: 10.1016/j.immuni.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 55.Facciponte JG, Wang XY, Subjeck JR. Hsp110 and Grp170, members of the Hsp70 superfamily, bind to scavenger receptor-A and scavenger receptor expressed by endothelial cells-I. Eur J Immunol. 2007;37:2268–2279. doi: 10.1002/eji.200737127. [DOI] [PubMed] [Google Scholar]
- 56.Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, Kawakami-Honda N, Goetsch L, Sawamura T, Bonnefoy J, Jeannin P. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. 2002;17:353–362. doi: 10.1016/s1074-7613(02)00388-6. [DOI] [PubMed] [Google Scholar]
- 57.Wang R, Town T, Gokarn V, Flavell RA, Chandawarkar RY. HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J Surg Res. 2006;136:58–69. doi: 10.1016/j.jss.2006.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.