

Abstract
Infection by Bacillus anthracis in animals and humans results from accidental or intentional exposure, by oral, cutaneous or pulmonary routes, to spores, which are normally present in the soil. Treatment includes administration of antibiotics, vaccination or treatment with antibody to the toxin. A better understanding of the molecular basis of the processes involved in the pathogenesis of anthrax namely, spore germination in macrophages and biological effects of the secreted toxins on heart and blood vessels will lead to improved management of infected animals and patients. Controlling germination will be feasible by inhibiting macrophage paralysis and cell death. On the other hand, the control of terminal hypotension might be achieved by inhibition of cardiomyocyte mitogen-activated protein kinase and stimulation of vessel cAMP.
Keywords: Anthrax, Heart Failure, Rats, Review
2. INTRODUCTION
Because of its antiquity and central role in modern microbiology and immunology, extensive knowledge has been achieved on the history, biology, natural history of the illness and biochemistry. In contrast, little is known about the events in the late stages of anthrax.
2.1. History
Bacillus anthracis chromosomal DNA is closely related to Bacillus cereus DNA. Both organisms may have evolved a symbiotic relationship with plants to avoid mammal and insect consumption, respectively (1). The earliest recorded cases of anthrax may be the “boils” on Pharaoh’s cattle in a 1500BC papyrus (2). Virgil’s Georgics written in 30BC described cattle anthrax as well as its transmission to people. Outbreaks of anthrax were described in records from 14th century Germany and 17th century Russia and central Europe. In 1769, Jean Fournier reported the black anthrax lesions originated from human exposure to animal skins and wool. In 1850, Rayer, Davain and Pollander showed filliform bodies in the blood of anthrax victims and suggested these “bodies” transmitted the disease. In 1876, Robert Koch isolated the Bacillus anthracis organism. Between 1880 and 1881, Greenfield and Pasteur perfected heat-killed vaccines for the disease. In World War I and II, Germany and Japan used anthrax spores as bioweapons (3). In 1979, airborne spores were accidentally released at Sverdlosk, Russia with 97 victims. In 2001, letters contaminated with spores in the U.S. led to 22 cases and 5 deaths. The World Health Organization reported in 1970 that aerosol release of spores by an aircraft over a 5 million population urban area would cause 100,000 casualties and cost $26 billion in medical costs.
2.2. Biology
The spore is synthesized inside the mother cell in response to environmental signals and consists of a central protoplast with dipicolonic acid, a cortex with peptidoglycan and cortex-hydrolyzing enzymes, and the exosporium containing at least 21 proteins including an arginase and small amounts of protective antigen (4). Spore germination is triggered in part by inosine through membrane receptors. Vegetative organisms produce a polyglutamic acid capsule and the tripartite toxin comprised of protective antigen, lethal factor and edema factor. Macrophages in the skin, gastrointestinal tract or lungs phagocytose spores. In a contest between the spore and the macrophage, the macrophage employs toll-like receptors, myeloid differentiation factor 88 (MyD88), NFkappaB transcription factor, pro-interleukin-1beta protein, nucleotide-binding oligomerization domain (NOD)-like receptors, IRAC3, beta-interferon, interferon receptor, signal transducers and activator of transcription (STAT)1, caspase 1, and processed interleukin-1beta to react to the organism and produce local nitric oxide (5). The spore exosporium contains small amounts of the tripartite toxins and arginase to block these macrophage steps and prevent macrophage defenses and block nitric oxide damage, respectively. Once the spore has escaped from the macrophage and produced vegetative offspring and toxins, symptoms and signs of anthrax develop.
2.3. Natural history of infection
Patients exhibit non-specific symptoms at presentation (6). Cutaneous anthrax patients will have an ulcerated small erythematous lesion resembling a spider bite. Gastrointestinal anthrax patients have abdominal discomfort, nausea, vomiting, diarrhea. Pulmonary anthrax patients have fever, cough, dyspnea, chest pain, malaise, diaphoresis, headache and may also have nausea and vomiting. Males are more commonly affected than females (3:1). Treatment with antibiotics and antisera and pleurocentesis reduces mortality. The fulminant phase occurs after 3-5 days and is short with irreversible shock. Unfortunately, detailed hemodynamic monitoring of patients during the “honeymoon” period has not been reported.
2.4. Biochemistry
Protective antigen is an 83 kDa protein with four domains (Figure 1a) (7). Domain 1 has a beta-sandwich jelly roll topology with four small helices and two calcium ions. Furin cleavage of RKKR at residues 164-167 removes this domain. Domain 2 has a beta-barrel with excursions and a modified Greek-key topology and forms the transmembrane pore when the 2beta2 and 2beta3 loops are unfolded in acidic conditions. Domain 3 has a ferridoxin-like fold and mediates oligomerization by inserting into a cleft in domain 1’ of the adjacent PA63 subunit. Domain 4 is a beta-sandwich with an immunoglobulin-like fold and binds the receptor. Lethal factor is a 90 kDa protein with four domains (Figure 1b) (8). Domain I binds protective antigen. Domains II-IV create a long deep groove which holds the N-terminal tail of mitogen-activated protein kinase (MAPKK, MEK or MKK). Domain IV has the zinc metalloprotease catalytic center. Edema factor is an 89 kDa protein with the protective antigen binding domain (PABD) and a calmodulin-activated adenylyl cyclase domain (Figure 1c) (9). Protective antigen binds receptors present on most cells (10). The receptors are tumor endothelium marker 8 (TEM8) and capillary morphogenesis gene 2 (CMG2). Both receptors contain a von Willebrand factor type A domain with calcium at the binding site-metal ion-dependent adhesion site (MIDAS). Protective antigen domain IV has an aspartate which forms a salt bridge with the receptor calcium ion. Protective antigen domain II has a short alpha-helix which interacts with a hydrophobic cleft on the receptor. After cell binding, protective antigen is cleaved by cell-surface furin and the 63 kDa protective antigen fragments heptamerize and bind 3 molecules of lethal factor or edema factor. The complex moves to lipid rafts and undergoes receptor-mediated endocytosis. In acid endosomes, the protective antigen separates from the receptor with the assistance of protonated His-121 of the receptor. The protective antigen 2beta2-2beta3 loops unfold in acid with protonation of four His in the loops and form a 14-strand beta-barrel pore. Lethal factor and edema factor are unfolded in the low pH endosomes. The N-termini of unfolded lethal factor and edema factor enter the pore first. Translocation is effected by the pH gradient between the endosome and the cytosol. Since the pore is cation-selective, peptides with acidic residues can only enter the low pH side. Phe-427 of protective antigen forms a clamp at the pore entrance to help chaperone the unfolded lethal factor and edema factor to the pore lumen and block ion passage. Once the lethal factor and edema factor travel through the pore to the cytosol, they refold and proteolyze MEKs and generate cAMP, respectively.
Figure 1.
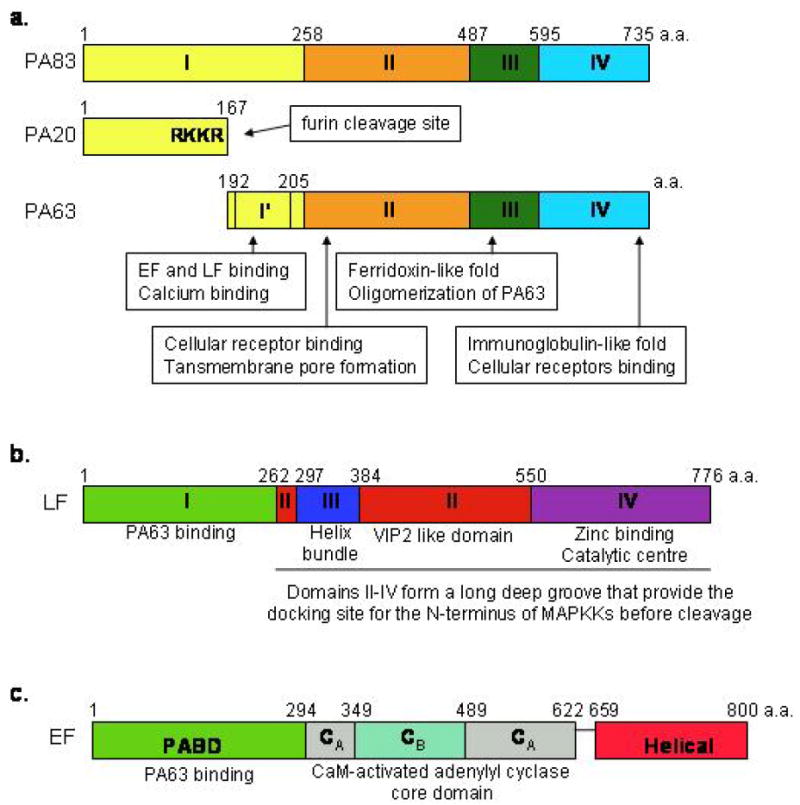
Schematic diagram of domain organization of anthrax toxin molecules. The amino acid (a.a.) boundary is indicated above each domain. (a) protective antigen (PA). (b) lethal factor (LF) (c) edema factor (EF).
3. GERMINATION VERSUS SPORE DESTRUCTION
The interplay of the spore and macrophage determines the fate of the host organism. The spore needs time to germinate. The host needs to rapidly respond by either triggering superoxide-mediated destruction of the spore in the phagosome or exuberant cytokine and chemokine signaling to bring fresh innate and adaptive immune cells to the site. Recent data suggests toxins secreted by both the germinating spore in the phagolysosome and toxins from surrounding vegetative organisms can impact the outcome. Immune paralysis clearly favors bacterial survival. Rapid macrophage lysis may adversely impact the organisms by not allowing sufficient time and proper environment for germination. Most studies have examined the effects of toxins on macrophages and spore-macrophage combinations in tissue culture.
3.1. Macrophage response
The host macrophage engulfs the spores and reacts by recognizing pathogen associated molecular patterns with its toll-like receptors and NOD-like receptors as described above. The defense by the organism to macrophage signaling is complex. Germinating spores may release toxins intracellularly which modify macrophage functions (11). Vegetative bacilli also release lethal toxin; the toxin enters macrophages and inhibits further spore phagocytosis (12). The lethal toxin also inhibits macrophage release of cytokines (13). Lethal toxin prevents up-regulation of co-stimulatory molecules (14). Lethal factor cleaves MKK3 and MKK6; p38 stress-activated protein kinase fails to be activated; interferon-regulatory factor 3 is not phosphorylated and activated; chemokine and nitric oxide synthetase proteins are not expressed (15). Edema toxin triggered increased toxin receptor expression and inhibited some cytokine secretion (16). Both toxins inhibit cell migration (17).
3.2. Macrophage cell death
Anthrax lethal toxin causes rapid macrophage cell death in some rodent species (18). Slower and less potent cytotoxicity is observed with human monocytes and endothelial cells (19). Several experiments show cell death depends upon the lethal factor enzyme activity (20), Nalp1b protein (18), potassium ion fluxes (21), proteasome activity (21), and caspase 1 (21). How these different events are tied together is currently unknown. Conversely, non-human primate cells are less sensitive to lethal toxin (12).
4. HEMODYNAMIC RESPONSE TO SECRETED TOXINS
Systemically administered anthrax toxins reproduce many of the symptoms, signs, laboratory abnormalities, hemodynamic changes and pathology of the late stages of anthrax (22, 23). In addition, infection of animals with mutant Bacillus anthracis which lacked toxin genes (pOX1-) yielded a thousand-fold reduction in toxicity (24). Animals were protected from death due to anthrax bacilli infections by prophylactic treatments with anti-toxin antibodies (25, 26). Further, immunization with protective antigen protected animals from anthrax (27), and vaccine protection correlated with titers of anti-protective antigen antibodies (28). Thus, secreted toxins may be responsible for the late lethal stage of anthrax, particularly in cases where antibiotics reduced growth of vegetative organisms.
4.1. Lethal toxin-induced shock
Anthrax lethal toxin has been administered to mice and rats. Equal amounts of protective antigen and lethal factor given at 1-4 mg/kg each to Balb/c and C57BL/6J mice produced malaise and death by 60-100 hours (23). Similar doses produced death within 24-72 hours in CAST mice (29). Blood tests showed hypoalbuminemia, hypofibrinogenemia, thrombocytopenia, elevated prothrombin time, and elevated partial thromboplastin time. Circulating inflammatory cytokines were not present. Pathology showed disseminated fibrin deposition, hemorrhage, tissue hypoxic damage, and evidence of vascular leak consistent with shock. Corticosteroids and aldosterone did not prevent the shock or lethality suggesting endocrine dysfunction was not responsible (30). Rats received twice as much protective antigen as lethal factor in a series of studies. Doses of 0.04mg/kg to 10mg/kg protective antigen and half as much lethal factor given intravenously to Sprague-Dawley and Fischer rats produced death in 2-21 hours with refractory hypotension, lactic acidosis, bradycardia, pleural effusions and secondary tissue hypoxic injury (26, 31). Fisher rats treated with a mutant form of protective antigen which only binds the CMG2 anthrax receptor combined with lethal factor developed hypotension, bradycardia and death similar to rats treated with wild-type protective antigen and lethal factor (32). Saline infusions failed to reverse lethal toxin-induced shock in rats (33). Thus, dehydration or hypovolemia alone does not account for the shock. Animals infected with Bacillus anthracis spores produce blood levels of protective antigen and lethal factor prior to death similar to those observed with toxin infusions (34, 35). Interestingly, anti-protective antigen antibodies could be given to rats even after the onset of lethal toxin-mediated shock and both blood pressure and survival improved (26). Thus, the observations with purified toxin should have relevance to clinical anthrax disease—particularly in the late stage. Importantly, patients with late stages of pulmonary anthrax often have refractory shock unresponsive to antibiotics, pressors or volume replacement—a scenario similar to the toxin-treated rodents described above (6, 36).
4.2. Edema toxin-induced shock
Edema toxin was administered to Balb/c mice with equal doses of protective antigen and edema factor ranging from 1-4mg/kg of each protein (37). Death occurred within 60 h and profound hypotension occurred. Pathology showed fluid accumulation in the intestines, adrenal hemorrhage, lymphocytolysis, osteoblast necrosis, cardiomyocyte necrosis, elevated transaminases, lactate dehydrogenase, urea nitrogen, phosphorus and amylase. The results were consistent with irreversible shock and hypoxic tissue injury. Rats treated similarly with 0.2-0.8mg/kg edema toxin developed hypotension and tachycardia (38). The combination of anthrax lethal toxin and edema toxin in rats gave additive effects.
5. HYPOTHESES FOR MOLECULAR PATHOGENESIS OF SPORE AND BLOOD PRESSURE CONTROL
The clinical course of patients exposed to Bacillus anthracis spores depends upon several critical events. First, spores are phagocytosed by macrophages in the skin, gastrointestinal tract or lungs and must germinate. Second, vegetative organisms release toxins which incapacitate the host. The first event is a contest between the macrophage and spore. The traditional view is that immune paralysis and cell death are equivalent and favor the pathogen. We hypothesize that rapid macrophage lysis does not provide sufficient time for spore germination and will favor the host. If valid, then hosts with sensitive macrophages will tolerate higher spore numbers and have better survival than hosts with resistant macrophages. Monocytes were isolated from the blood of human, baboon and rats by density gradient centrifugation and plastic adherence and then cultured with human or rat granulocyte-macrophage colony-stimulating factor for one week. Then aliquots of cells were incubated for 48 h with protective antigen (different concentrations) and lethal factor (1nM). 3H-leucine was added for an additional 18 h and then cells harvested and counted. The IC50 is the concentration of protective antigen yielding a 50% reduction in protein synthesis. Rat macrophages had a median IC50 of 60pM; baboon macrophages had a median IC50 of 180pM human macrophages had a median IC50 of 200pM. Published data on spore sensitivity estimates of the human, monkey and rat aerosol spore number leading to death were 10, 50 and 1500, respectively (39). A plot of macrophage sensitivity vs. spore tolerance shows an inverse relationship with an r2=0.99 and a p value of 0.07 (Figure 2).
Figure 2.
Plot of macrophage IC50 (pM) vs. spore tolerance based on experiments above and spore data from Young (39).
Shock associated with the late stages of many bacterial pathogens is associated with an inflammatory cytokine storm and responsiveness to volume replacement and pressors. In contrast, anthrax shock lacks these properties. We hypothesize that anthrax lethal toxin induces cardiogenic shock, and anthrax edema toxin worsens this state by vascular dysfunction leading to preload reduction. Cohorts of Sprague Dawley rats were treated with 0.1mg/kg protective antigen plus 0.045mg/kg lethal factor or 0.45mg/kg protective antigen plus 0.225mg/kg edema factor and monitored by telemetry (40). These doses yielded 50% mortality within 4 days. Serum protective antigen levels reached 1.5μg/mL. Mean arterial pressure, systolic blood pressure and diastolic blood pressure were significantly lowered by lethal toxin and edema toxin (Figure 3). These results confirm the incidence of refractory hypotension with anthrax toxins.
Figure 3.
Effect of bolus anthrax toxins on mean arterial pressure (MAP). MAP in rats was recorded continuously via telemetry before and after toxin injection.
We then administered similar doses of anthrax toxins to rats and performed echocardiography at various times post-toxin infusion (41). Lethal toxin reduced ejection fraction by 30% in 11/14 surviving animals (Figure 4A). Edema toxin did not reduce ejection fraction in any animal. There were associated increased left ventricular end-systolic areas but not increased end-diastolic areas in treated rats. Heart-rate-corrected-velocity of circumferential fiber shortening (vcfc) was decreased indicating decreased left ventricular contractility. Edema toxin did not reduce ejection fraction but did decrease left ventricular end-systolic and diastolic areas consistent with decreased preload (Figure 4B). These results are consistent with lethal toxin effects on the heart and edema toxin effects on blood vessels.
Figure 4.
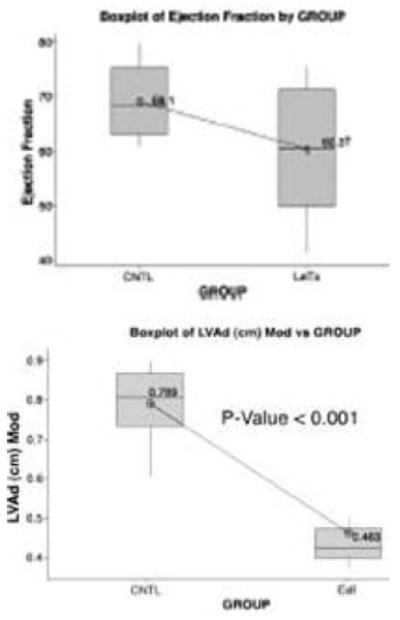
Rat echocardiography studies performed as described (41). A. Ejection fraction of lethal toxin treated rats; B. Left ventricular end-diastolic area of edema toxin treated rats.
We followed up this study with a examination of treated rat chemistries and tissue pathology (Figure 5) (42). Lethal toxin produced transaminasemia, elevated lactate dehydrogenase and elevated hematocrit. Edema toxin produced transaminasemia, elevated lactate dehydrogenase and lymphopenia. The only observed pathology was in the lungs. Lethal toxin produced pulmonary edema and edema toxin produced pulmonary vascular hemorrhage. Protective antigen, edema factor and lethal factor alone had no effect on chemistries or pathology.
Figure 5.
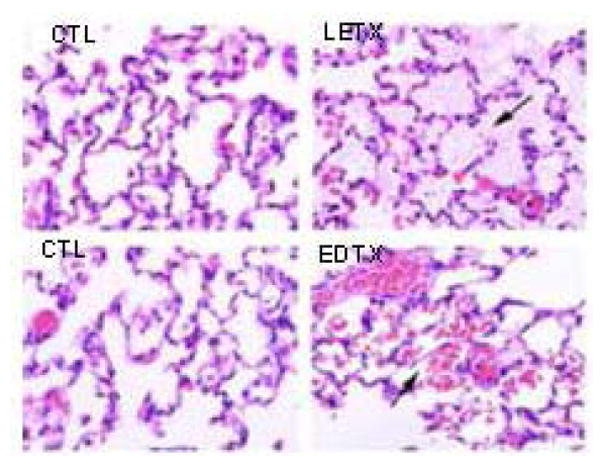
Lung pathology of control, anthrax lethal toxin and anthrax edema toxin treated rats. Arrows point to interstitial protein/fluid with lethal toxin and vascular disruption/hemorrhage with edema toxin.
These results are all consistent with a model of anthrax lethal toxin-induced cardiogenic shock and anthrax edema toxin-induced vascular damage and preload reduction. However, our results were limited to rodents. To extend these findings, we treated dogs with 0.27mg/kg protective antigen and 0.14mg/kg lethal factor and monitored hemodynamics periodically (43). After 72 h, heart rate was 129bpm vs. 95bpm control. Left ventricular end-diastolic pressure of treated vs. control was 21mmHg vs. 9mmHg, time constant of relaxation was 41ms vs. 30ms, stroke volume was 8.4mL vs. 15.8mL, end-systolic pressure was 92mmHg vs. 118mmHg, left ventricular contractility was 4.1mmHg/mL vs. 8.1mmHg/mL, and MSW was 42.mmHg vs. 88.9 mmHg. There was a rightward shift in the left ventricular pressure-volume loops (Figure 6). At 96 h, MSW, systolic volume and ejection fraction were further decreased accompanied by severe heart failure. Histology showed left ventricular dilatation. Toxin treated dogs died by day 4. Thus, the cardiovascular effects of anthrax lethal toxin were reproducible in a non-rodent species.
Figure 6.
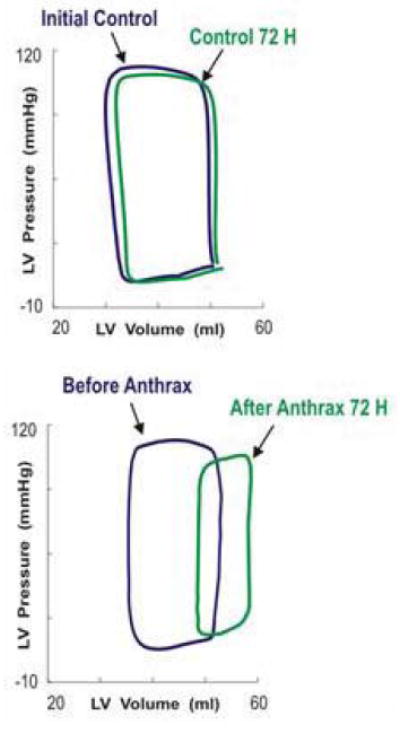
Dog left ventricle pressure-volume loops after anthrax lethal toxin infusion
With confirmation of the physiological effects of lethal toxin in vivo, we next wanted to examine whether the effects were due to direct action on cardiomyocytes and vascular cells or indirect effects. Myocytes were isolated as previously described by collagenase digestion of left ventricular biopsies (44). Lethal toxin-treated myocytes had an increase in length to 201μm vs. 122μm control. Toxin-treated myocytes had a change in cell contraction with DL/dtmax of 87μm/s vs. 148μm/s control. Examples of analog recordings in electrically-stimulated myocytes are shown in Figure 7. Compared to control myocyte, lethal toxin treated myocytes had reductions in SA, dL/dtmax, and dR/dtmax. All these findings suggest a direct effect of lethal toxin on cardiomyocyte contractile function.
Figure 7.
Isolated dog cardiomyocyte in vitro contractility parameters. Cells were from anthrax toxin treated or control dogs.
Proof that anthrax toxins directly alter hemodynamics will require prolonged incubations of toxins with isolated normal cardiovascular tissues. These studies will help provide data for a molecular mechanism of toxin induced shock and also facilitate testing of novel anti-toxin therapeutics which may be employed during late stage anthrax. Evaluation of the proposed molecular pathophysiology of anthrax should generate advances in both microbiology and medicine.
Acknowledgments
This work was supported by the Van Andel Research Foundation and R21CA108438 (NSD).
References
- 1.Sternbach G. The history of anthrax. J Emerg Med. 2003;24:463–467. doi: 10.1016/s0736-4679(03)00079-9. [DOI] [PubMed] [Google Scholar]
- 2.Wenner KA, Kenner JR. Anthrax. Dermatol Clin. 2004;22:247–256. doi: 10.1016/j.det.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Kasten FH. Biological weapons, war crimes, and WWI. Science. 2002;296:1235–1237. [PubMed] [Google Scholar]
- 4.Steichen CT, Kearney JF, Turnbough CL. Non-uniform assembly of the Bacillus anthracis exosporium and a bottle cap model for spore germination and outgrowth. Mol Microbiol. 2007;64:359–367. doi: 10.1111/j.1365-2958.2007.05658.x. [DOI] [PubMed] [Google Scholar]
- 5.Hughes MA, Green CS, Lowchyj L, Lee GM, Grippe VK, Smith MF, Huang LY, Harvill ET, Merkel TJ. MyD88-dependent signaling contributes to protection following Bacillus anthracis spore challenge of mice: implications for Toll-like receptor signaling. Infect Immun. 2005;73:7535–7540. doi: 10.1128/IAI.73.11.7535-7540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holty JE, Bravata DM, Liu H, Olshen RA, McDonald KM, Owens DK. Systematic review: a century of inhalational anthrax cases from 1900 to 2005. Ann Intern Med. 2006;144:270–280. doi: 10.7326/0003-4819-144-4-200602210-00009. [DOI] [PubMed] [Google Scholar]
- 7.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 8.Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, Hanna P, Liddington RC. Crystal structure of the anthrax lethal factor. Nature. 2001;414:229–233. doi: 10.1038/n35101998. [DOI] [PubMed] [Google Scholar]
- 9.Drum CL, Yan SZ, Bard J, Shen YQ, Lu D, Soelaiman S, Grabarek Z, Bohm A, Tang WJ. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 2002;415:396–402. doi: 10.1038/415396a. [DOI] [PubMed] [Google Scholar]
- 10.Young JAT, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 11.Banks DJ, Barnajian M, Maldonado-Arocho FJ, Sanchez AM, Bradley KA. Anthrax toxin receptor 2 mediates Bacillus anthracis killing of macrophages following spore challenge. Cell Microbiol. 2005;7:1173–1185. doi: 10.1111/j.1462-5822.2005.00545.x. [DOI] [PubMed] [Google Scholar]
- 12.Ribot WJ, Panchal RG, Brittingham KC, Ruthel G, Kenny TA, Lane D, Curry B, Hoover TA, Friedlander AM, Bavari S. Anthrax lethal toxin impairs innate immune functions of alveolar macrophages and facilitates Bacillus anthracis survival. Infect Immun. 2006;74:5029–5034. doi: 10.1128/IAI.00275-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L, Frucht DM. Bacillus anthracis: a multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int J Biochem Cell Biol. 2007;39:20–24. doi: 10.1016/j.biocel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Fukao T. Immune system paralysis by anthrax lethal toxin: the roles of innate and adaptive immunity. The Lancet Infectious Diseases. 2004;4:166–170. doi: 10.1016/S1473-3099(04)00940-5. [DOI] [PubMed] [Google Scholar]
- 15.Dang O, Navarro L, Anderson K, David M. Cutting edge: anthrax lethal toxin inhibits activation of IFN-regulatory factor 3 by lipopolysaccharide. J Immunol. 2004;172:747–751. doi: 10.4049/jimmunol.172.2.747. [DOI] [PubMed] [Google Scholar]
- 16.Cleret A, Quesnel-Hellmann A, Mathieu J, Vidal D, Tournier JN. Resident CD11c+ lung cells are impaired by anthrax toxins after spore infection. J Infect Dis. 2006;194:86–94. doi: 10.1086/504686. [DOI] [PubMed] [Google Scholar]
- 17.Baldari CT, Tonello F, Paccani SR, Montecucco C. Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol. 2006;27:434–440. doi: 10.1016/j.it.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 19.Abi-Habib RJ, Urieto JO, Liu S, Leppla SH, Duesbery NS, Frankel AE. BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase ½ activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol Cancer Ther. 2005;4:1303–1310. doi: 10.1158/1535-7163.MCT-05-0145. [DOI] [PubMed] [Google Scholar]
- 20.Shoop WL, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Felcetto T, Michael BF, Bansal A, Cummings RT, Cunningham BR, Friedlander AM, Douglas CM, Patel SB, Wisniewski D, Scapin G, Salowe SP, Zaller DM, Chapman KT, Scholnick EM, Schmatz DM, Bartizal K, MacCoss M, Hermes JD. Anthrax lethal factor inhibition. Proc Natl Acad Sci USA. 2005;102:7958–7963. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol Epub. 2007 doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezzell JW, Irvins BE, Leppla SH. Immunoelectrophoretic analysis, toxicity, and kinetics of in vitro production of the protective antigen and lethal factor components of Bacillus anthracis toxins. Infect Immun. 1984;45:761–767. doi: 10.1128/iai.45.3.761-767.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNFα-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pezard C, Berche P, Mock M. Contributions of individual toxin components to virulence of Bacillus anthracis. Infect Immun. 1991;59:3472–3477. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Little SF, Leppla SH, Cora E. Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infect Immun. 1990;58:1606–1613. doi: 10.21236/ada216203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui X, Li Y, Moayeri M, Choi GH, Subramanian GM, Li X, Haley M, Fitz Y, Feng J, Banks SM, Leppla SH, Eichacker PQ. Late treatment with a protective antigen-directed monoclonal antibody improves hemodynamic function and survival in a lethal toxin-infused rat model of anthrax sepsis. J Infect Dis. 2005;191:422–434. doi: 10.1086/427189. [DOI] [PubMed] [Google Scholar]
- 27.Marcus H, Danieli R, Epstein E, Velan B, Shafferman A, Reuveny S. Contribution of immunological memory to protective immunity conferred by a Bacillus anthracis protective antigen-based vaccine. Infect Immun. 2004;72:3471–3477. doi: 10.1128/IAI.72.6.3471-3477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuveny S, White MD, Adar YY, Kafri Y, Altboum Z, Gozes Y, Kobiler D, Shafferman A, Velan B. Search for correlates of protective immunity conferred by anthrax vaccine. Infect Immun. 2001;69:2888–2893. doi: 10.1128/IAI.69.5.2888-2893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culley NC, Pinson DM, Chakrabarty A, Mayo MS, Levine SM. Pathophysiological manifestations in mice exposed to anthrax lethal toxin. Infect Immun. 2005;73:7006–7010. doi: 10.1128/IAI.73.10.7006-7010.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moayeri M, Webster JI, Wiggins JF, Leppla SH, Sternberg EM. Endocrine perturbation increases susceptibility of mice to anthrax lethal toxin. Infect Immun. 2005;73:4238–4244. doi: 10.1128/IAI.73.7.4238-4244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui X, Moayeri M, Li Y, Li X, Haley M, Fitz Y, Correa-Araujo R, Banks SM, Leppla SH, Eichacker PQ. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R699–R709. doi: 10.1152/ajpregu.00593.2003. [DOI] [PubMed] [Google Scholar]
- 32.Scobie HM, Wigelsworth DJ, Marlett JM, Thomas D, Rainey JA, Lacy DB, Manchester M, Collier RJ, Young JAT. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLos Pathogens. 2006;2:949–955. doi: 10.1371/journal.ppat.0020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherer K, Li Y, Cui X, Li X, Subramanian M, Laird MW, Moayeri M, Leppla SH, Fitz Y, Su J, Eichacker PQ. Fluid support worsens outcome and negates the benefit of protective antigen-directed monoclonal antibody in a lethal toxin-infused rat Bacillus anthracis shock model. Crit Care Med. 2007;35:1560–1567. doi: 10.1097/01.CCM.0000266535.95770.A2. [DOI] [PubMed] [Google Scholar]
- 34.Boyer AE, Quinn CP, Woolfitt AR, Pirkle JL, McWilliams LG, Stamey KL, Bagarozzi DA, Hart JC, Barr JR. Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal Chem. 2007 doi: 10.1021/ac701741s. Epub Oct 12. [DOI] [PubMed] [Google Scholar]
- 35.Mabry R, Brasky K, Geiger R, Carrion R, Hubbard GB, Leppla SH, Patterson JL, Georgiou G, Iverson BL. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin Vaccine Immunol. 2006;13:671–677. doi: 10.1128/CVI.00023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mina B, Dym JP, Kuepper F, Tso R, Arrastia C, Kaplounova I, Faraj H, Kwapniewski A, Krol CM, Grosser M, Glick J, Fochios S, Remolina A, Vasovic L, Moses J, Robin T, DeVita M, Tapper ML. Fatal inhalational anthrax with unknown source of exposure in a 61-year-old woman in New York city. JAMA. 2002;287:858–862. doi: 10.1001/jama.287.7.858. [DOI] [PubMed] [Google Scholar]
- 37.Fivored AM, Miller GF, Moayeri M, Kakkar R, Shen Y, Wiggins JF, McNally EM, Tang WJ, Leppla SH. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167:1309–1320. doi: 10.1016/S0002-9440(10)61218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui X, Li Y, Li X, Laird MW, Subramanian M, Moayeri M, Leppla SH, Fitz Y, Su J, Sherer K, Eichacker PQ. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J Infect Dis. 2007;195:572–580. doi: 10.1086/510856. [DOI] [PubMed] [Google Scholar]
- 39.Young GA, Zelle MR, Lincoln RE. Respiratory pathogenicity of Bacillus anthracis spores. J Infect Dis. 1946;79:233–246. doi: 10.1093/infdis/79.3.233. [DOI] [PubMed] [Google Scholar]
- 40.Watson LE, Kuo S, Katki K, Dang T, Park SK, Dostal DE, Tang WJ, Leppla SH, Frankel AE. Anthrax toxins induce shock in rats by depressed cardiac ventricular function and reduced preload. PLosONE. 2007;2:e466. doi: 10.1371/journal.pone.0000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson LE, Mock J, Lal H, Lu G, Bourdeau RW, Tang WJ, Leppla SH, Dostal DE, Frankel AE. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Frontiers in Biosciences. 2007;12:4670–4675. doi: 10.2741/2416. [DOI] [PubMed] [Google Scholar]
- 42.Kuo SR, Willingham MC, Bour SH, Andreas EA, Park SK, Jackson C, Duesbery NS, Leppla SH, Tang WJ, Frankel AE. Anthrax toxin-induced shock in rats is associated with pulmonary edema and hemorrhage. Microb Pathog. doi: 10.1016/j.micpath.2007.12.001. in press. [DOI] [PubMed] [Google Scholar]
- 43.Cheng CP, Masutani S, Cheng HJ, Cross M, Zhang CX, Zhou P, Cann J, Cline JM, Little WC, Kuo SR, Frankel AE. Progressive left ventricle, myocyte dysfunction, and heart failure in the lethality of anthrax toxin in conscious dogs. Circulation. 2007;116(Suppl II):758. [Google Scholar]
- 44.Cheng CP, Suzuki M, Ohte N, Ohno M, Wang ZM, Little WC. Altered ventricular and myocyte response to angiotensin II in pacing-induced heart failure. Circ Res. 1996;78:880–892. doi: 10.1161/01.res.78.5.880. [DOI] [PubMed] [Google Scholar]