

Abstract
We report the asymmetric synthesis of the y-amino acid (1R,2R)-2-aminomethyl-1-cyclopentane carboxylic acid (AMCP) and an evaluation of this residue's potential to promote secondary structure in α/γ-peptides. Simulated annealing calculations using NMR-derived distance restraints obtained for α/γ-peptides in chloroform reveal that AMCP-containing oligomers are conformationally flexible. However, additional evidence that suggests an internally hydrogen-bonded helical conformation is partially populated in solution. From these data, we propose characteristic NOE patterns for formation of the α/γ-peptide 12/10-helix and present discussion of the apparent conformational frustration of AMCP-containing oligomers.
INTRODUCTION
γ-Amino acids are appealing as building blocks for peptidic foldamers because the conformational propensities of individual γ subunits can be modulated by varying substitution at the α, β and/or γ carbons.1,2 Pioneering work by Hanessian et al. showed that the folding behavior of γ-amino acid residues bearing one substituent at the α carbon and another at the γ carbon was critically dependent on the relative configurations at the two stereocenters, with one stereoisomeric form supporting helical secondary structure and the other stereoisomeric form favoring reverse turn secondary structure.1b,d These trends were subsequently confirmed by Seebach et al., who also identified an α,β,γ-trisubstitution pattern that promotes a helical conformation (Figure 1a).1c,f,h Incorporation of cyclic constraints into the γ-amino acid backbone can promote sheet1a,g,j,k or helix secondary structure, depending on the ring size, the configurations of the stereogenic centers, and the location of the ring within the γ residue (i.e., incorporation of the Cα-Cβ bond, or the Cβ-Cγ bond, or both the Cα-Cβ and Cβ-Cγ bonds into the ring).1 Although initial studies focused on the folding of pure γ-peptide backbones, subsequent efforts have expanded to include heterogeneous backbones, in which γ residues are combined with α and/or β residues, to generate α/γ−, β/γ− or α/β/γ-peptides.3
Figure 1.

(a) Helix-forming oligomers of γ-residues studied by Hanessian1b,d and Seebach.1cfh (b) Constrained γ-amino acids that promote formation of helices that contain C=O(i)--H-N(i+3) hydrogen bonds.3 (c) The γ-amino acid AMCP.
We have explored two types of γ-residue in which a cyclohexyl ring provides a backbone constraint, I and II (Figure 1b). These residues differ in the position of the cyclohexyl ring along the backbone (involving the Cβ-Cγ vs. the Cα-Cβ bond) and whether or not the third backbone carbon bears a substituent. Trisubstituted residue I strongly promotes formation of helices that contain C=O(i)--H-N(z+3) hydrogen bonds in both homogeneous and heterogenous backbones.3ac Disubstituted residue II has been examined only in α/γ backbones, where it can be accommodated into two different helices, one containing only C=O(i)--H-N(i+3) H-bonds and the other featuring an alternation between C=O(i)--H-N(i+3) H-bonds and C=O(i)--H-N(i-l) H-bonds.3d The former helix appears to reflect the intrinsic preference of residue II, based on the similarity of its observed θ and ζ torsion angle values with those of residue I. Here we describe the stereo-selective synthesis of (1R,2R)-2-(aminomethyl)cyclopentanecarboxylic acid (III; AMCP, Figure 1c),4 and the conformational behavior of α/γ-peptides containing AMCP residues.
RESULTS AND DISCUSSION
Synthesis
Evaluation of the extent to which a chiral amino acid supports the adoption of discrete conformations when incorporated into peptidic oligomers requires access to a protected derivative of that amino acid in enantiopure form. We prepared the Boc-protected form of AMCP as summarized in Scheme 1. This route follows the strategy previously employed to prepare protected derivatives forms of II; the key step is an asymmetric Michael addition of nitromethane to a cycloalkene-1-carboxaldehyde.3d Our approach to this reaction is based on precedents from Hayashi et al. and Wang et al., who reported highly enantioselective addition of nitromethane to β-substituted propenal derivatives catalyzed by pyrrolidine A and benzoic acid.5 The analogous process with cyclohexene-1-carboxaldehyde gave rise to a ~4.5:1 transi:cis product mixture. In situ reduction provided the δ-nitro alcohols, each of which displayed >95% ee (reduction prevents epimerization during isolation).3d Adaptation of this method to the addition of nitromethane to cyclopentene-1-carboxaldehyde required us to replace benzoic acid as co-catalyst with a 3:1 2,4,6-collidine:benzoic acid mixture (see Supporting Information). This reaction produced exclusively the trans δ-nitro alcohol (1) in 95% yield and >99% ee. A three-step procedure, involving nitro group reduction, Boc-protection and primary alcohol oxidation converted δ-nitro alcohol 1 into protected γ-amino acid 2. This AMCP building block can readily be prepared in multi-gram quantities.
The absolute configuration of AMCP prepared in this way was determined from the crystal structure of α/γ-dipeptide 3, which was synthesized by coupling 2 to D-alanine benzyl ester (Figure 2). This analysis showed that use of A as the chiral catalyst (5 configuration at the stereogenic center) provides the γ-amino aldehyde with R configuration at the two new stereogenic centers. The crystal structure of 3 showed θ and ζ torsion angles of 55° and −113°, respectively. This observation is intriguing because our previous crystallographic analysis of oligomers containing residues derived from γ-amino acids I or II show that both favor gauche+,gauche+ (g+,g+, or g−,g−, depending on absolute configuration) θ,ζ torsion angle sequences, which is consistent with the apparent preference of both residues for formation of helices containing exclusively C=O(i)--H-N(i+3) H-bonds.3ac Hofmann et al. have used computational methods to explore the helical conformations available to γ-, α/γ- and β/γ-peptide backbones (no side chains),6 and their results suggest that the distinctive θ,ζ angles manifested by 3 in the solid state could lead to helical preferences for α/γ-peptides containing AMCP (III) that differ from those of analogous oligomers containing I or II.
Figure 2.
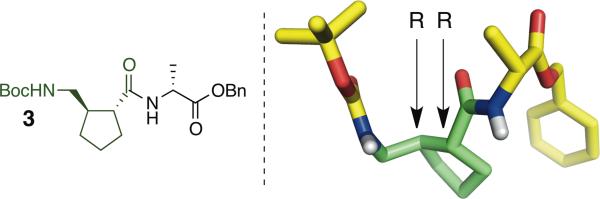
X-ray crystal structure of α/γ-dipeptide 3.
We prepared a series of α/γ-peptides containing γ residue III and D-α-amino acid residues, with 1:1 residue alternation, as summarized in Scheme 2. Tetramer 8, hexamer 9 and octamer 12 all contain D-alanine at the α positions, while decamer 15 contains both D-alanine and D-phenylalanine residues, to promote dispersion of NMR resonances. These oligomers were prepared by condensation of α/γ dimers such as 6 and 7, reactions in which the carboxyl group is provided by a γ-residue. This approach avoids epimerization, which can occur during peptide condensations in which the carboxyl group is provided by an α residue. Because the yields of fragment coupling reactions declined with increasing length of the product oligomer among tetramer 8, hexamer 9 and octamer 10, we replaced HOBt with Oxyma as the nucleophilic additive for the reaction to produce decamer 15.7 In addition, we switched from DMF to a 4:1 NMP:DMSO solvent mixture for this tetramer-plus-hexamer coupling reaction, because the precursors were barely soluble in DMF.
Structural characterization of α/γ-peptides that contain AMCP
α/γPeptides 8, 9 and 12 were analyzed by 1H NMR in CDCl3; 5 % (v/v) CD3OH in CDCl3 was required to ensure complete dissolution of decamer 15.8 We observe invariant chemical shifts for the proton resonances of decamer 15 over a fiftγ-fold range of concentrations (0.04 mM to 2 mM), which suggests that this α/γ-peptide does not self-associate under these conditions. Since 15 is the least soluble of the α/γ-peptides in this study, we suspect that smaller α/γ-peptides 8, 9, and 12 do not aggregate under the conditions used for NMR analysis (2-4 mM in chloroform; see Supporting Information). Well-dispersed and amide proton resonances with chemical shifts > 7.5 ppm were observed for 2-4 mM samples of hexamer 9, octamer 12 and decamer 15 (Figure 3a). Under conditions that do not support self-association, these downfield-shifted NH resonances suggest the formation of internally H-bonded conformations.9 (Non-H-bonded amide NH resonances typically display chemical shifts < 7.0 ppm.)
Figure 3.

(a) Amide proton region of the 1H NMR spectra (CDG3) of α/γ-peptides 8, 9, 12, and 15. (b) HCα and HCγ proton region of the 1H NMR spectra (CDCl3) of α/γ-peptides 8, 9, 12, and 15. Samples were collected at peptide concentrations of 4, 2, 2, and 2 mM, respectively. α/γ-Decamer 15 was dissolved in 5 % (v/v) CD3OH.8
On the other hand, overlap among the D-alanine HCα proton resonances between 4 ppm and 5 ppm and among the γ-residue HCγ proton resonances between 3 ppm and 4 ppm suggest the possibility of conformational averaging on the NMR time scale (Figure 3b). 1H-1H COSY, TOCSY, and ROESY data were obtained for α/γ-peptides 9, 12, and 15 (see Supporting Information for resonance assignment). Several of the observed NOEs suggest at least partial population of folded conformations by these α/γ-peptides, because the protons involved are separated by multiple covalent bonds (Figure 4).
Figure 4.

Selected NOEs observed in ROESY experiments for α/γ-peptides 9, 12, and 15. Data were collected at peptide concentrations 2 mM in CDCl3 solution. α/γ-Decamer 15 was dissolved with addition of 5 % (v/v) CD3OH. See Supporting Information for tabulated sequence assignments and NOEs for each α/γ-peptide.
We carried out NOE-restrained simulated annealing calculations with the CNS program for 9, 12, and 15.10 In each case, the 10 structures with lowest energy were selected for comparison with one another without any further minimization (see Supporting Information). We did not observe any violation of our NOE-derived distance restraints in the lowest energy structures. The resulting structural ensembles are diverse, lacking a contiguous network of H-bonds or consistent set of backbone torsion angles. As α/γ-peptide length increases from hexamer to octamer to decamer, the RMSD calculated for backbone atoms in the ensembles increases from 0.8 ±0.1 Å to 2.0 ±1.0 Å to 2.4 ±0.4 Å (see Supporting Information). The disordered ensembles suggest that AMCP-containing α/γ-peptides are dynamic in solution, and do not adopt a single highly-populated secondary structure.
The lack of a significant conformational preference implied by the NOE-restrained dynamics analysis of α/γ- peptides 9, 12 and 15 seems somewhat inconsistent with the excellent dispersion of NH resonances observed for these oligomers and a regular pattern of i,i+2 NOEs observed in all three cases (NOE type (i) in Figure 5). This type of i,i+2 NOE has been attributed to 12/10-helix formation for α/γ-peptides of comparable lengths in comparable solvents;11 in these previous α/γ-peptide studies, the γ-residues lacked a cyclic constraint. Our α/γ-peptides display a regular pattern of NOEs between the HN of a γ residue (i) and the HN of an adjacent α residue (NOE type (ii) in Figure 5). This type of NOE was not reported in the previous α/γ-peptide study.11 Based on examination of theoretical models from Hofmann et al.,6 we conclude that this type of i,i+1 NOE is consistent with 12/10-helix formation. Notably absent from our data are NOEs of type (iii) in Figure 5, between the HCγ of a γ-resiude (i) and the HN of an adjacent α residue (i+1); this type of NOE is reported to be characteristic of 12/10 helix formation.11 The lack of type (iii) NOEs in the NMR data for α/γ-peptides 9, 12, and 15 may partially explain why our simulated annealing calculations failed to produce well-defined conformational ensembles comparable to those previously reported.11 Other possible explanations for this difference are mentioned below. To test for the possibility of partial 12/10-helix formation in α/γ-peptides containing AMCP, we conducted a DMSO titration experiment with α/γ-peptide decamer 15 (Figure 6). Amide proton chemical shifts of 15 were monitored as small aliquots of DMSO were added to a solution of the α/γ-peptide in CDCl3 containing 5% (v/v) CD3OH. Since DMSO is a strong H-bond acceptor, added DMSO should form H-bonds with amide protons that are oriented toward the solvent, causing their NMR signals to move downfield. If an amide proton is engaged in intramolecular H-bonds, however, added DMSO should have little or no effect on chemical shift.
Figure 5.

(a) NOEs characteristic of 12/10 helix formation. (i) NOEs observed in α/γ-peptides 9, 12, and 15 consistent with those reported for the α/γ-peptide 12/10-helix by Sharma and Kunwar.11 (ii) NOE observed in our study of α/γ-peptides 9, 12, and 15. (iii) NOE observed in 12/10 helical peptides of Sharma and Kunwar - not observed in NMR study of 9, 12, and 15. (b) Diagram of hydrogen bonding in the α/γ-peptide 12/10-helix. γ-residues shown in green.
Figure 6.

(a) . DMSO titration of 2 mM α/γ-peptide decamer 15 5 % (v/v) CD3OH in CDCl3. (b) Schematic of hydrogen bonding and solvent exposed amide protons in the α/γ-peptide 12/10-helix. Red circles indicate amide protons expected to exhibit the largest chemical shift change upon DMSO addition.
The data in Figure 6 suggest that decamer 15 experiences at least partial 12/10-helix formation in CDCl3 containing 5% (v/v) CD3OH. The NH resonances most strongly affected by DMSO addition are those at the N- and C-termini; the strong downfield shifting in these cases suggests that these NH groups do not form intramolecular H-bonds. In contrast, the resonances from the four internal NHs are only moderately influenced by DMSO addition, which suggests that these NH groups are, on average, at least partially engaged in intramolecular H-bonds. This pattern is consistent with partial population of the 12/10- helical conformation, which features both C=O(i)--H-N(i+3) H-bonds and C=O(i)--H-N(i-l) H-bonds.6d, 11 Because the H-bonds run in both directions relative to the helix axis, non-H-bonded NH groups are found at both termini (Figures 5b, 6b). In contrast, for a helix in which all H-bonds are oriented in the same direction (e.g., an a-helix), non-H-bonded NH groups would be found at only one terminus. The conclusion we draw from the DMSO titration experiment, that the terminal NH groups of 15 do not form intramolecular H-bonds in CDCl3 containing 5% (v/v) CD3OH while the internal NH groups are at least partially intramolecularly H-bonded, is consistent with the pattern in chemical shifts observed in the absence of DMSO: the terminal NH resonances are 2-3 ppm upfield relative to the internal NH resonances.
Overall, our data suggest that α/γ-peptides containing AMCP, such as hexamer 9, octamer 12 and decamer 15, display a modest tendency to adopt 12/10-helical secondary structure. However, this folding propensity is not strong enough to prevent these oligomers from exploring other conformations, even in a nonpolar solvent such as CDCl3.
CONCLUSIONS
We have presented the enantioselective synthesis of a new type of cyclic γ-amino acid, AMCP, and an evaluation of the conformational behavior of α/γ-peptides that contain AMCP residues. Despite the presence of a conformational constraint within these new residues, arising from incorporation of the Cα-Cβ bond into a cyclopentane ring, α/γ-peptides containing AMCP do not display a strong propensity to adopt a specific secondary structure. NMR data suggest that 12/10-helical conformations are moderately populated by these α/γ-peptides in non-polar solvents, but that other conformations are significantly populated as well. We suspect that the lack of a substituent at Cγ of the AMCP backbone causes this new γ residue to be quite flexible in spite of the Cα-Cβ ring constraint. Consistent with this proposal is our previous report on α/γ-peptides in which the y residues are derived from II, which lacks a substituent at Cγ. Crystallographic data showed that this class of α/γ-peptides can accommodate two different helical conformations.3d Further support for the proposed correlation between unsubstituted backbone carbons within a γ-amino acid and flexibility in oligomers that contain the corresponding γ residue is found in the broad conformational diversity evident in crystal structures of oligomers generated from gabapentin, which is disubstituted at Cβ but lacks substituents at Cα and Cγ.12 It is well-known among conventional peptides that glycine residues, which lack a side chain, are inherently more flexible than other a-amino acid residues.
The NMR evidence for partial folding of α/γ-peptides 9, 12 and 15, including dispersion of amide and HCα resonances, patterns of inter-residue NOEs and H-bond pattern implicated by NH chemical shifts and DMSO titration results, are comparable to observations reported for a different set of α/γ-peptides by Sharma and Kunwar.11 NOE-restrained dynamics analysis of these alternative α/γ-peptides, however, seems to suggest a more robust propensity for 12/10-helical secondary structure than we detect for AMCP-containing α/γ-peptides. The difference between the previous conformational conclusions11 and those we draw here may arise because of differences in γ residue substitution (the previous series contained γ residues bearing a single, bulky substituent, at Cγ), differences in the observed NOE patterns (discussed above), differences in treatment of the NMR data (e.g., variations in the distance ranges assigned to varying NOE intensities for NOE-restrained dynamics calculations), or some combination of these factors.
Fülöp and coworkers have proposed that controlling both φ and ψ dihedral angles is critical for manipulating helical secondary structure among α/β-peptides.13 Our study of AMCP-containing α/γ-peptides suggests an analogous importance for φ and ψ dihedral angles in oligomers that contain γ-amino acid residues. Thus, using a ring to constrain one backbone bond within a γ residue may not be sufficient to encode a strong and specific folding propensity.1
EXPERIMENTAL METHODS
General
Solvents and reagents were used as-is unless otherwise indicated. CH2Cl2 and tetrahydrofuran were distilled from CaH2 and sodium-benzophenone ketyl respectively. Flash column chromatography was performed on silica gel (40-63 mm mesh). Thin layer chromatography (TLC) was performed on glass-backed plates with fluorescent indicator. Visualization was carried out with KMnO4 as a general stain and phosphomolybic acid for α/γ-peptides 8, 9, 12, 13, and 15 which stain weakly in other conditions (KMnO4, p-anisaldehyde, ceric ammonium molybdate, ceric ammonium sulfate).
HPLC analysis of chiral compounds was carried out on an analytical HPLC system using columns with chiral stationary phases (columns and conditions are indicated in Supporting Information). One-dimensional 1H NMR spectra were recorded on 300 MHz spectrometers; results are reported in parts per million (ppm, d) relative to tetramethylsilane. First-order 1H NMR splitting patterns were assigned on the basis of their appearance as singlet (s), doublet (d), triplet (t), or quartet (q). Resonances that could not be interpreted were designated multiplet (m) or broad (b). One-dimensional 13C NMR spectra were recorded on a 300 MHz spectrometer (@ 75 MHz). For some compounds, higher field 500 MHz spectrometers were utilized to obtain 13C spectra (indicated on spectra). 13C NMR chemical shifts are reported in the text to ± 0.1 ppm, rounded from values to two decimal places obtained from spectra included in the Supporting Information. Two-dimensional 1H-1H NMR COSY, TOCSY, and ROESY experiments were carried out on a 600 MHz spectrometer. High-resolution mass spectra (HRMS) were measured using a an electrospray ionization spectrometer (TOF detector). Optical rotations were measured using a 1 mL cell with a 0.5 dm path length on a digital polarimeter (Na lamp) and are reported as follows: [a]rtD (c in g per 100 mL). Images of crystal and NMR structures shown throughout the manuscript and the Supporting Information were prepared in MacPymol.14
Synthetic procedures
1-cyclopentene-1-carboxaldehyde
1-cyclopentene-1-carboxaldehyde is a known compound characterized previously in the literature.15 We prepared it for use according to the following two step procedure. We note that due to its propensity for co-evaporation even in low-boiling solvents our yields were reduced following the oxidation step. We isolated 1-cyclopentene-1-carboxaldehyde as a solution of known weight percent in ether/pentane, quantified by 1H NMR using 1,4-dioxane as an internal standard. To a stirring 0.5 M solution of methyl-1-cyclopentane-1-carboxylate (4.84 mL, 39.6 mmol, 1.0 equiv.) in anhydrous hexanes at −78°C (80 mL) was added DIBAL-H via syringe (1.0 M solution in hexanes; 80 mL, 80.0 mmol, 2.0 equiv.) in four 20 mL aliquots over a period of 15 minutes under N2 atmosphere. The reaction was warmed to 0°C (ice bath) at stirred for 1.5 hours. The reaction was quenched with 50 mL ice cold Rochelle's salt (saturated Na/K tartrate) and allowed to stir at 0°C open to air. The reaction became a graγ-white gelatinous suspension that solubilized over the course of 30 minutes to 1 hour stirring at room temperature open to air. The mixture was poured into a 1 L separatory funnel containing 200 mL diethyl ether and 200 mL distilled H2O. The layers were separated and the aqueous layer was extracted twice more with 200 mL diethyl ether. The organic layers were combined, dried over MgSO4, and concentrated to afford a pale clear oil which was purified via silica gel column chromatography, eluting with 3:1 Hexanes:EtOAc. 1-cyclopentene-1-methanol product was isolated as a clear oil (3.50 g, 90 % yield). For the purposes of scaled up amino acid synthesis this reaction was often done in multiple passes. 1H 1D NMR in CDCl3 agree with literature values.14a 1H NMR literature (300 MHz, CDCl3):15a d 5.62-5.59 (m, 1H), d 4.18 (m, 2H), d 2.39-2.27 (m, 4H), 1.96-1.86 (m, 2H), 1.46 (s, 1H). 1H NMR observed (300 MHz, CDCl3): d 5.62 (app. quintet, 1H, J = 1.8 Hz), d 4.18 (m, 2H), d 2.42-2.26 (m, 4H), ), d 1.92 (quintet, 2H, J = 7.5 Hz), d 1.08 (broad s, 1H).
To a stirring suspension of pyridinium chlorochromate (PCC) (13.2 g, 61.4 mmol, 1.2 equiv.) in 50 mL distilled CH2Cl2 with ~3 g 4 Å molecular sieves at 0°C (ice bath) was added 1-hydroxymethyl-1-cyclopentene (5.02 g, 51.2 mmol, 1.0 equiv.) as a solution in 25 mL distilled CH2Cl2 drop-wise over 20 mintues via addition funnel under N2 atmosphere. The reaction was allowed to stir for ~6 hours as it warmed to r.t. and was monitored via TLC (3:1 Hexanes:EtOAc, KMnO4 stain). The reaction was stirred 4 hours at r.t. at which point additional PCC (2.6 g, 12.1 mmol, 0.25 eqiuv.) was added. The reaction was monitored by TLC as described and complete (loss of alcohol starting material spot) 1 hour following addition of more PCC. The reaction was diluted with 50 mL diethyl ether and filtered through a plug of silica. 50 mL additional diethyl ether was used to wash the silica plug. The filtrate was filtered a second time as described above (plug washed as above). The filtrate was concentrated carefully to yield a malodorous, pale yellow/green oil. The crude material was purified via silica gel column chromatography eluting with 13 % (v/v) diethyl ether in pentane. The product was isolated as a colorless solution of approximately 88 wt. % 1-cyclopenete-1-carboxaldehyde in ether/pentane (solution quantified by 1H NMR with 1,4-dioxane as internal standard; 1.19g in solution, 24 % yield). Significant yield was lost despite extra care in concentrating product (ice in rotovap bath). For the purposes of scaled up amino acid synthesis this reaction was often done in multiple passes. 1H 1D NMR of 1-cyclopenete-1-carboxaldehyde in CDCl3 agree with literature values.15b 1H NMR literature (300 MHz, CDCl3):14b d 9.80 (s, 1H), d 6.88-6.86 (m, 1H), d 2.63-2.59 (m, 2H), d 2.55-2.51 (m, 2H), d 2.01 (app. t, 1H, J=7.5 Hz), d 1.98 (app. t, 1H, J=7.5 Hz) 1H NMR observed (300 MHz, CDCl3): d 9.80 (s, 1H), 6.89-6.86 (m, 1H), d 2.65-2.58 (m, 2H), d 2.55-2.50 (m, 2H), d 2.00 (app. quintet, 2H, J=7.6 Hz) note: this last resonance would occur from the superposition of the two triplets (d 2.01, d 1.98) reported in the cited reference, given their identical three-bond J coupling constants.
(lR,2R)-1-(hydroxymethyl)-2-(nitromethyl) cyclopentane (1)
S-(-)-α,α-diphenylpyrrolidnemethanol trimethylsilyl ether (2.0 g, 6.0 mmol, 0.2 equiv.) was dissolved in EtOH. 2,4,6-collidine (617 μL, 4.7 mmol, 0.15 equiv.) was added, followed by benzoic (189 mg, 1.6 mmol, 0.05 equiv.) and the solution was stirred about 5-10 min until all species dissolved. 1-cyclopentene-1-carboxaldehyde was added (2.98 g, 31 mmol, 1.0 equiv.) and the solution was stirred at room temperature as it slowly took on a yellow-orange color. Nitromethane (3.0 equiv.) was added in a single portion and the solution immediately turned a deep green color. The total reaction volume was ~62 mL (0.5 M in 1-cyclopentene-1-carboxaldehyde). The reaction was sealed with a septum and stirred at room temperature for 2 hours.
The solution was diluted with an equivalent volume of MeOH and cooled to 0°C in an ice bath. NaBH4 (1.76 g, 46.5 mmol, 1.5 equiv.) was added slowly over a period of 15 minutes causing the reaction to bubble vigorously. The solution was stirred at 0°C for an additional 15-30 minutes or until all bubbling subsided. The reaction was quenched at 0°C via slow addition of an equal volume of saturated (aq) NH4O and stirred for ~30 minutes. The mixture was stirred until all precipitates dissolved and then was allowed to warm to room temperature. The reaction mixture was then diluted in a separatory funnel with brine and extracted five times with Et2O. The organic layers were combined, dried over MgSO4, filtered, and concentrated to afford a yellow-orange oil. The crude material was purified via flash column chromatography on silica gel, eluting with a gradient of EtOAC in hexanes (Hexanes:EtOAC (v/v) 20:1 to 3:1) to afford pure nitroalcohol 1 as a clear, pale yellow oil (4.93 g, > 95 % yield). Rf (3:1 (v/v) Hexanes:EtOAc) = 0.07. 1H NMR (300 MHz, CDCl3):
δ 4.45 (ABX, Jax = 6.0 Hz, Jbx = 9.0 Hz, Jab = 12.0 Hz, vab = 83.4 Hz, 2H), δ 3.61 (ABX, Jax = 5.4 Hz, Jbx = 6 .9 Hz, Jab = 10.3 Hz, vab = 831.2 Hz, 2H), δ 2.47 (sextet, J = 6.0 Hz, 1H), δ 1.93 (sextet, J = 6.6 Hz, 1H), δ 1.85 (m, 2H), δ 1.66 (quintet, J = 7.5 Hz, 2H), δ 1.43 (quintet, J = 6.3 Hz, 2H). 13C NMR (75.4 MHz, CDCl3): δ 80.3, 66.2, 45.4, 41.8, 31.1, 29.1, 24.3. Calculated [M+Na+]/observed for C7H13NO3Na+: 182.08/182.00. [α]Dr.t. = −15.8° (c=0.6).
(lR,2R)-2-((tert-butoxycarbonyl)amino)methyl)-1-cylopentanecarboxylic acid (2)
Nitroalcohol 1 (3.36 g, 21.1 mmol, 1.0 equiv.) was taken up in MeOH that had been pre-saturated with N2 for 30 minutes, and the solution was transferred to a high-pressure flask. A catalytic amount (spatula tip, ~50 mg) of Raney-Ni was added and the flask was sealed with a pressure gauge and three-way valve. The flask was evacuated with an aspirator and filled with H2 at 20 psi three times. The flask was brought up to 40 psi H2 and stirred vigorously at room temperature for 18 hours. The pressure was vented into a fume hood, and the reaction mixture was filtered through a plug of celite topped with a small layer of silica. The filtrate was carefully concentrated via rotovap to remove MeOH with minimal heating (significant product is lost due to co-evaporation with MeOH, even in a cooled rotovap bath) to yield an oily residue ((1R,2R)-1-(hydroxymethyl)-2-(aminomethyl)-cyclopentane; 1.68 g, 62 % yield), which was carried forward without any additional purification. The oily residue was taken up in distilled dichloromethane (100 mL) and the solution was transferred to a roundbottom flask. iPr2EtN (4.53 mL, 26 mmol, 2.0 equiv.) was added, followed by Boc2O (4.26 g, 19.5 mmol, 1.5 equiv.). The flask was flushed with N2, sealed with a septum and stirred at room temperature for 3-5 hours. Reaction completion was determined via TLC using ninhydrin stain. The solution was diluted threefold with EtOAc and washed once with saturated (aq) NaHSO4, washed once with brine (50 mL each wash), dried over anhydrous Na2SO4, filtered, and concentrated to afford a pale orange-yellow oil. The N-Boc amino alcohol product ((1R,2R)-1-(hydroxymethyl)-2-((tert-butoxycarbonyl)amino)methyl)-cyclopentane) was isolated as a clear, colorless oil following column chromatography of the crude reaction product, eluting with 1:1 EtOAc : Hexanes (2.11 g, 71 % yield). Rf (1:1 (v/v) Hexanes:EtOAc) = 0.44. 1H NMR (300 MHz, CDCl3): δ 5.17 (broad, 1H), δ 3.57 (ABX, Jax = 4.9 Hz, Jbx = 6.8 Hz, Jab = 10.0 Hz, vab = 51.1 Hz, 2H), δ 3.11 (ABX, unresolved, 2H), δ 2.40 (broad, 1H), δ 1.80 (m, 4H), δ 1.59 (m, 2H), δ 1.44 (s, 9H), δ 1.30 (m, 2H). 13C NMR (75.4 MHz, CDCl3): δ 156.7, 79.3, 66.7, 45.8, 45.4, 43.6, 31.4, 29.7, 28.6, 24.6. Calculated [M+Na+]/observed for C12H23NO3Na+: 252.1571/252.1578. [α]Dr.t. = −2.8° (c=4.8).
The N-Boc amino alcohol product ((1R,2R)-1-(hydroxymethyl)-2-((tert-butoxycarbonyl)amino)methyl)-cyclopentane) (2.11 g, 9.2 mmol, 1.0 equiv.) was dissolved in acetone (~50 mL) and cooled to 0°C in an ice bath. Jones’ reagent (27.6 mL, 13.8 mmol, 1.5 equiv.; see below) was added drop-wise via addition funnel with vigorous stirring. The reaction mixture was stirred at 0°C for 30 minutes and then allowed to gradually warm up to room temperature as it stirred for ~18 hours. The reaction was quenched with an equal volume of iPrOH and the resulting mixture stirred for about 1 hour until the mixture became a deep green solution. The solution was acidified with 1 N HCl and extracted exhaustively with dichloromethane (10 times), until product was not observed via TLC (bromocresol green stain). The organic layers were combined, dried over MgSO4, filtered, and concentrated to afford a yellow-green crude oil. Crude carboxylic acid was purified via column chromatography eluting with EtOAc in hexanes (1:1 v/v) to afford Boc-amino acid 2 as an amorphous solid (1.39 g, 66 % yield). TLC was visualized with bromocresol green – carboxylic acid product appears as yellow spot on blue/green background. Rf (1:1 (v/v) Hexanes:EtOAc) = 0.32. 1H NMR (300 MHz, CDCl3): δ 11.14 (broad, 1H), δ 6.47 & 4.92 (NH peak split due to N-Boc rotamers; broad, 1H total), δ 3.20 (m, 2H), δ 2.43 (m, 1H), δ 2.32 (m, 1H), δ 1.90 (broad, 3H), δ 1.69 (sextet, J = 6.3 Hz, 2H), δ 1.44 (shoulder at 1.46 due to N-Boc rotamers; s, 9H total), δ 1.34 (m, 1H). 13C NMR (75.4 MHz, CDCl3): δ 181.1, (158.1, 156.8 – split due to N-Boc rotamers), 100.7, (80.8, 79.7 – split due to N-Boc rotamers),(48.8, 48.0 – split due to N-Boc rotamers), (46.3, 44.5 – split due to N-Boc rotamers), (31.0, 30.1 – split due to N-Boc rotamers), 30.6, 28.6, 24.9. Calculated [M-H−]/observed for C12H20NO4: 242.1397/242.1389. [α]Dr.t = −21.8° (c=3.8).
Jones’ reagent preparation
Sodium dichromate dihydrate (29.8 g, 100 mmol, 1.0 eq) was weighed into a flask cooled to 0°C. H2SO4 (21.6 mL, 400 mmol, 4.0 eq) was added slowly to the flask at 0°C. The solution was diluted to a volume of 200 mL with distilled H2O to afford a 0.5 M solution of H2Cr2O7.
Dipeptide (3)
N-Boc-amino acid 2 (180 mg, 0.74 mmol, 1.0 equiv.) was dissolved in distilled dichloromethane (5 mL). iPr2NEt (387 μL, 2.2 mmol, 3.0 equiv.) was added followed by HOBt (120 mg, 0.89 mmol, 1.2 equiv.) and then EDCI (170 mg, 0.89 mmol, 1.2 equiv.). The reaction was stirred for 15 minutes at room temperature at which point D-alanine benzyl ester tosylate salt was added (260 mg, 0.74 mmol, 1.0 equiv). The coupling reaction was stirred for 24 hours at room temperature. The reaction was diluted with EtOAc (50 mL) and washed with 1 N (aq) NaHSO4, saturated (aq) NaHCO3, and brine (25 mL each). The acidic aqueous washes were extracted twice with dichloromethane (25 mL each) and the same was done for the combined basic/brine washes. All organic layers were combined, dried over MgSO4, filtered, and concentrated to afford crude semisolid material. Dipeptide 3 was isolated via flash column chromatography on silica gel, eluting with a gradient of EtOAc in hexanes to afford a white solid (300 mg isolated, 51 % yield). Small diastereomeric impurities (inseparable) appeared to be present in 1H NMR spectrum. We were able to crystallize a small quantity of 3 and obtain diffraction quality crystals. Crystallographic characterization of dipeptide 3 is detailed in the Supporting Information. The 1H spectrum shown for 3 in the SI is from a crystal and a small amount of retained mother liquor that was dried on high vacuum and re-dissolved in CDCl3. Rf (2.5 % (v/v) MeOH in CH2Cl3) = 0.16. 1H NMR (300 MHz, CDCl3): δ 7.35 (m, 5H), δ 5.16 (ABq, Jab = 13.8 Hz, vab = 25.1 Hz, 2H), δ 4.88 (broad, 1H), δ 4.59 (quintet, J = 8.7 Hz, 1H), δ 3.54 (m, 1H), δ 2.90 (m, 1H), δ 2.29 (quartet, J = 8.7 Hz, 1H), δ 2.05 (broad, 2H), δ 1.89-1.52 (broad, 6H), δ 1.45 (d, J = 7.5 Hz, 3H), δ 1.44 (s, 9H), δ 1.33 (broad, 1H). Calculated [M+Na+]/observed for C22H32N2O5Na+: 427.2204/427.2206.
The x-ray crystal structure of 3 revealed the configuration at C1 and C2 of cyclopentyl ring of AMCP to be (R,R). These stereocenters would have been set in the asymmetric conjugate addition of nitromethane to 1-cyclopentene-1-carboxaldehyde. Thus, we propose that the crystal structure of dipeptide 3 reveals the configuration of the sole observed product of the reaction sequence to produce nitroalcohol 1 to also be (R,R).
Boc-NH-AMCP-OBn (4)
Boc-NH-AMCP-OH (2) (1g 4.1 mmol, 1.0 eq) was dissolved in 20 mL DMF (N,N-dimethylformamide). Cs2CO3 (1.34 g, 4.1 mmol, 1.0 eq) was added to the reaction mixture was was stirred rapidly at room temperature. Benzyl bromide (487 μL, 4.9 mmol, 1.2 eq) was added, the reaction flask was sealed with a septum and the reaction mixture was stirred vigorously for 18 h at room temperature. The reaction mixture was then diluted with EtOAc (~100 mL) and transferred to a separatory funnel. DMF was removed by washing five times with 50 mL H2O. Each H2O wash was back-extracted four times with 50 mL Et2O. All organic layers were combined, dried over MgSO4, filtered, and concentrated to a pale, yellow oil. The crude material was purified to a clear colorless oil via column chromatography on silica, eluting with 3:1 Hexanes:EtOAC. Rf (3:1 hexanes : EtOAC) = 0.27. Product was isolated in > 95 % yield (colorless oil, 1.37 g). 1H NMR (300 MHz, CDCl3): δ 7.53 (m, 5H), δ 5.13 (ABq, Jab = 12.4 Hz, vab = 13.5 Hz, 2H), δ 4.78 (broad, 1H), δ 3.151 (m, 2H), δ 2.50 (q, J = 7.5 Hz, 1H), δ 2.33 (sextet, J = 7.2 Hz, 1H), δ 1.89 (m, 3H), δ 1.68 (m, 2H), δ 1.43 (s, 9H), δ 1.31 (m, 1H). 13C NMR (75.4 MHz, CDCl3): δ 176.1, 156.2, 136.3, 128.8, 128.4, 128.3, 79.3, 66.6, 48.4, 45.1, 44.3, 30.7, 30.5, 28.6, 25.0. Calculated [M+Na+]/observed for C19H27NO4Na+: 356.1833/356.1830. [α]Dr.t. = −15.3°.
General Procedures for α/γ-peptide synthesis. General procedure A - Benzyl deprotection
Peptide was dissolved in MeOH (~20 mL/mmol peptide) that had been saturated with bubbling N2 for thirty minutes prior to reaction. A catalytic quantity (spatula tip) of 10 wt %
(dry) Pd/C (wet) was added to the solution and the reaction flask was flushed with N2 and sealed with a septum. The flask was evacuated on an aspirator and back-filled from an H2 balloon three times. The flask was equipped with a H2 balloon and the reaction was stirred for 5 hours at room temperature. Following observation of the loss of a benzyl signal from the crude 1H NMR spectrum of the reaction mixture (~5 hours; samples were filtered to remove catalyst), the mixture was filtered at once through a pad of celite topped with a thin layer of silica. The filtrate was evaporated to yield peptide acid fragments as white foams, typically in > 95 % yield based on the mass of isolated crude material. This material was taken forward into coupling reactions without further purification in all cases.
General procedure B - N-Boc deprotection
Peptide was dissolved in minimal 1,4-dioxane. 5-10 mL of a solution of 4 N HCl in 1,4-dioxane (or approx. 15 equiv. HCl relative to peptide) was added to the solution and the reaction flask was sealed with a septum. The reaction was stirred for 3-4 hours at room temperature. Following loss of the N-Boc signal from the crude 1H NMR spectrum of an aliquot of the reaction mixture, the reaction flask was evacuated on an aspirator for 30 minutes to remove dissolved HCl. 1,4-Dioxane was removed via rotovap to yield dipeptide HCl salts typically in > 95 % yield based on the mass of crude isolated material. This material was taken forward into coupling reactions without further purification in all cases.
Boc-HN-DAla-AMCP-OBn (5)
Benzyl ester 4 (Boc-HN-AMCP-OBn; 1.37 g, 0.41 mmol, 1.0 equiv.) was Boc-deprotected according to General Procedure B in > 95 % yield to give H2N-AMCP-OBn•HCl, which was carried forward without further purification. Boc-HN-DAla-OH (776 mg, 4.1 mmol, 1.0 equiv.) was dissolved in 10 mL distilled CH2Cl2. iPr2EtN (4.29 mL, 24.6 mmol, 6.0 equiv.) was added followed by HOBt (665 mg, 4.92 mmol, 1.2 equiv.) and EDCI (942 mg, 4.92 mmol, 1.2 equiv.). The solution was stirred for 5 minutes. H2N-AMCP-OBn•HCl was then added and the solution was stirred overnight at room temperature. The solution was diluted four-fold with EtOAc and washed twice each (25 mL each wash) with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2O2. All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with 1:1 (v/v) EtOAc:Hexanes to give an amorphous solid. Rf (1:1 EtOAc:Hexanes) = 0.18. 1.275 g isolated, 77 % yield. 1H NMR (300 MHz, CDCl3): δ 7.36 (m, 5H), δ 6.50 (broad, 1H), δ 5.14 (s, 2H), δ 4.93 (broad, 1H), δ 4.05 (m, 1H), δ 3.32 (m, 2H), 62.51 (q, J = 8.9 Hz, 1H), δ 2.34 (sextet, J = 7.7 Hz, 1H), δ 1.99 (m, 1H), δ 1.89 (m, 2H), δ 1.64 (quintet, J = 6.9 Hz), δ 1.44 (s, 9H), δ 1.34 (m, 1H), δ 1.28 (6, J = 6.9 Hz, 3H). 13C NMR (75.4 MHz, CDCl3): δ 176.3, 172.9, 169.6, 136.2, 128.9, 128.5, 128.4, 66.7, 48.7, 44.0, 43.5, 30.8, 30.5, 28.6, 25.0, 18.8. Calculated [M+Na+]/observed for C22H32N2O5Na+: 427.2204/427.2210.
Boc-HN-(DAla-AMCP)2-OBn (8)
Two 405 mg (1.0 mmol, 1.0 equiv.) portions of α/γ-peptide dimer 5 were deprotected orthogonally according to General Procedure A and General Procedure B to yield an α/γ-dipeptide acid 6 and α/γ-dipeptide amine HCl salt 7 both in > 95 % yield. α/γ-Dipeptide acid 6 (1.0 mmol, 1.0 equiv.) was dissolved in DMF (2.5 mL). iPr2EtN (1.045 mL, 6.0 mmol, 6.0 equiv.) was added followed by HOBt (162 mg, 1.2 mmol, 1.2 equiv.) and EDCI (230 mg, 1.2 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 15 minutes at which point a solution of α/γ-dipeptide amine HCl salt 7 (1.0 mmol, 1.0 equiv.) in DMF was added (2.5 mL). The reaction flask was sealed with a septum and the reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was diluted ten-fold with EtOAc and washed (25 mL each wash) twice each with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2O2. All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient of 3:1 Hexanes:EtOAc to 1:3 Hexanes:EtOAc. Product eluted when the column was flushed with neat EtOAc. Rf (2.5 % (v/v) MeOH in CH2Cl2) = 0.20. Tetramer 8 was isolated as a white solid (530 mg, 88 % yield). 1H NMR (300 MHz, CDCl3): δ 7.28 (m, 5H), δ 7.24 (broad, 1H), δ 7.17 (broad, 1H), δ 6.79 (broad, 1H), δ 5.13 (δ, J = 7.2 Hz, 1H), δ 5.07 (ABq, Jab = 13.7 Hz, vab = 9.9 Hz, 2H), δ 4.29 (quintet, J = 7.5 Hz, 1H), δ 4.06 (quintet, J = 6.9 Hz, 1H), δ 3.28 (broad, 3H), δ 3.11 (broad, 1H), δ 2.48 (q, J = 7.8 Hz, 1H), δ 2.30 (sextet, J = 8.1 Hz, 1H), δ 2.19 (broad, 2H), δ 1.96-1.52 (broad, 11H), δ 1.36 (s, 9H), d 1.31 (m, 2H), δ 1.27 (d, J = 7.2 Hz, 3H), δ 1.23 (d, J = 7.5 Hz, 3H). 13C NMR (75.4 MHz, CDCl3): δ 177.0, 176.3, 174.1, 173.9, 156.0, 136.2, 128.8, 128.4, 128.2, 80.3, 66.7, 50.7, 49.6, 49.0, 48. 6, 44.7, 44.1, 43.4, 43.0, 31.1, 30.8, 30.5, 28.5, 25.8, 25.0, 18.3, 18.1. Calculated [M+H+]/observed for C32H48N4O7H+: 601.3596/601.3591.
Boc-HN-(DAla-AMCP)3-OBn (9)
A 0.33 mmol portion of α/γ-peptide dimer 5 was deprotected according to General Procedure A (to yield 6) and a 0.33 mmol portion of α/γ-peptide tetramer 8 was deprotected according General Procedure B. α/γ-Dipeptide acid 6 (0.33 mmol, 1.0 equiv.) was dissolved in 2.5 mL DMF. iPr2EtN (348 μL, 2 mmol, 6.0 equiv.) was added followed by HOBt (54 mg, 0.4 mmol, 1.2 equiv.) and EDCI (77 mg, 0.4 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 15 minutes at which point a solution of N-deprotected 8 in 2.5 mL DMF was added. The reaction flask was sealed with a septum and the reaction mixture was stirred at room temperature for 48 hours. The reaction mixture was diluted ten-fold with EtOAc and washed (25 mL each wash) twice each with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2O2. All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient of 1:1 Hexanes:EtOAc to neat EtOAc. Rf (neat EtOAc) = 0.32. Hexamer 9 was isolated as a white solid (200 mg, 76 % yield). 1H NMR (300 MHz, CDCl3): 6 8.09 (d, J = 6.9 Hz, 1H), δ 7.79 (d, J = 7.2 Hz, 1H), δ 7.54 (d, J = 6.6 Hz, 1H), δ 7.34 (broad, 1H), δ 7.29 (m, 5H), δ 6.56 (broad, 1H), δ 5.07 (ABq, Jab = 13.1 Hz, vab = 7.7 Hz, 2H), δ 5.02 (d, J = 3Hz, 1H), δ 4.26 (quintet, J = 7.5 Hz, 1H), δ 4.21 (quintet, J = 7.5 Hz, 1H), δ 4.13 (quintet, J = 5.0 Hz, 1H), δ 3.55-3.29 (broad, 3H), δ 3.22-2.98 (broad, 3H), δ 2.48 (q, J = 8.1 Hz), δ 2.22 (broad, 5H), 1.80 (broad, 11H), δ 1.59 (broad 7H), δ 1.36 (s, 9H), δ 1.32 (m, 2H), δ 1.28 (d, J = 9.9 Hz, 3H), δ 1.25 (d, J = 9.9 Hz, 3H), δ 1.16 (d, J = 9.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 178.0, 177.9, 176.2, 174.5, 174.5, 173.8, 155.8, 135.8, 128.7, 128.3, 128.1, 80.1, 66.7, 50.4, 50.3, 49.6, 49.2, 49.1, 48.7, 44.4, 44.3, 44.1, 44.0, 43.0, 43.03, 31.6, 31.4, 31.2, 31.1, 30.7, 30.3, 28.3, 26.6, 26.1, 24.8, 18.1, 17.3, 16.9. Calculated [M+Na+]/observed for C42H64N6O9Na+: 819.4627/819.4606.
Boc-HN-(DAla-AMCP)4-OBn (12)
Two 0.33 mmol portions of α/γ-peptide tetramer 8 were deprotected orthogonally according to General Procedure A and General Procedure B to yield α/γ-tetrapeptide acid 10 and α/γ-tetrapeptide amine HCl salt 11, respectively, with both reactions proceeding in > 95 % yield. Tetrapeptide acid 10 (0.33 mmol, 1.0 equiv.) was dissolved in 2.5 mL DMF. iPr2EtN (348 μL, 2 mmol, 6.0 equiv.) was added followed by HOBt (54 mg, 0.4 mmol, 1.2 equiv.) and EDCI (77 mg, 0.4 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 15 minutes at which point a solution of α/γ-tetrapeptide amine HCl salt 11 in DMF (2.5 mL) was added. The reaction flask was sealed with a septum and the reaction mixture was stirred at room temperature for 72 hours. The reaction mixture was diluted tenfold with EtOAc and washed twice each with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2O2. All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient of 1% (v/v) to 5 % MeOH in CH2Cl2. Rf (5 % MeOH) = 0.14. Octamer 12 was isolated as a white solid (190 mg, 58 % yield). 1H NMR (300 MHz, CDCl3): δ 8.74 (d, J = 6.3 Hz, 1H), δ 8.37 (d, J = 6.3 Hz, 1H), δ 7.96 (d, J = 4.8 Hz, 1H), δ 7.60 (broad, 2H), δ 7.45 (d, J = 6.0 Hz, 1H), δ 7.36 (m, 5H), δ 6.60 (broad, 1H), δ 5.13 (ABq, Jab = 11.0 Hz, vab = 9.1 Hz, 2H), δ 5.10 (d, J = 9.9 Hz, 1H), δ 4.24 (m, 1H), δ 4.27 (broad, 3H), δ 3.54 (broad, 4 H), δ 3.13 (broad, 4H), δ 2.54 (q, J = 8.4 Hz, 1H), δ 2.32 (broad, 7H), δ 1.87 (broad, 15 H), δ 1.64 (broad, 13H), δ 1.43 (s, 9H), δ 1.33 (broad, 10H), δ 1.26 (d, J = 7.8 Hz, 3H), δ 1.21 (d, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 178.4, 178.2, 178.1, 176.2, 174.9, 174.7, 174.6, 173.8, 171.2, 155.8, 135.8, 128.7, 128.3, 128.1, 80.1, 66.7, 60.4, 50.4, 50.4, 49.5, 49.3, 48.8, 44.5, 44.4, 44.2, 44.1, 44.0, 43.7, 43.0, 31.7, 31.6, 31.6, 31.5, 31.5, 31.4, 31.2, 30.7, 30.2, 29.7, 28.3, 26.8, 26.7, 26.3, 24.8, 22.7, 21.1, 18.1, 17.2, 17.1, 16.6, 14.2, 14.1. Calculated [M+H+]/ observed for C52H80N8O11H+:993.6020/993.6027.
Compound (Si): Boc-HN-(DPhe-AMCP)-OBn
See Supporting Information for scheme. Benzyl ester 4 (Boc-HN-AMCP-OBn; 1.09 g, 3.3 mmol, 1.0 equiv.) was Boc-deprotected according to General Procedure B in > 95 % yield to give H2N-AMCP-OBn•HCl, which was carried forward without further purification. Boc-HN-DPhe-OH (1.041 g, 3.9 mmol, 1.2 equiv.) was dissolved in 10 mL distilled CH2Cl2. iPr2EtN (3.5 mL, 19.6 mmol, 6.0 equiv.) was added followed by HOBt (530 mg, 3.9 mmol, 1.2 equiv.) and EDCI (751 mg, 3.9 mmol, 1.2 equiv.). The solution was stirred for 10 minutes. H2N-AMCP-OBn•HCl was then added and the reaction mixture was stirred 24 hrs at room temperature. The reaction mixture was diluted four-fold with EtOAc and washed twice each (25 mL each wash) with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2Cl2. All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient from 1:5 (v/v) to 1:1 EtOAc:Hexanes to give dipeptide S1 as an amorphous solid. 1.027 g isolated, 65 % yield. Rf (20:1 CH2Cl2:MeOH) = 0.49. 1H NMR (300 MHz, CDCl3): δ 7.32 (broad m, 10H), δ 6.13 (broad, 1H), δ 5.09 (ABq, Jab = 12.0 Hz, vab = 16.8 Hz, 2H), δ 4.96, (broad, 1H), δ 4.27 (q, J = 6.9 Hz, 1H), δ 3.11 (superimposed ABX patterns, 4H), δ 2.38 (q, J = 8.4 Hz, 1H), δ 2.19 (sextet, J = 7.8 Hz, 1H), δ 1.85 (m, 3H), δ 1.62 (broad, 2H), δ 1.40 (s, 9H), δ 1.22 (m 1H). 13C NMR (75.4 MHz, CDCl3): δ 176.0, 171.4, 137.1, 136.2, 129.5, 128.8, 128.5, 128.3, 127.1, 66.6, 56.2, 48.4, 43.9, 43.1, 38.9, 30.7, 30.3, 28.5, 24.9, 19.0. Calculated [M+Na+]/observed for C28H36N2O5Na+: 503.2517/503.2528.
Boc-HN-(DPhe-AMCP)2-OBn (13)
See Supporting Information for scheme. Two 199 mg (0.42 mmol, 1.0 equiv.) portions of α/γ-peptide dimer S1 (See Supporting Information for preparation of this dipeptide) were deprotected orthogonally according to General Procedure A and General Procedure B to yield a α/γ-dipeptide acid S2 and a α/γ-dipeptide amine HCl salt S3, respectively, both in > 95 % yield. The α/γ-dipeptide acid S2 (0.42 mmol, 1.0 equiv.) was dissolved in 5 mL CH2Cl2. iPr2EtN (444 μL, 2.5 mmol, 6.0 equiv.) was added followed by HOBt (67 mg, 0.5 mmol, 1.2 equiv.) and EDCI (95.4 mg, 0.5 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 15 minutes at which point a solution of dipeptide amine HCl salt S3 in 5 mL CH2Cl2 was added. The reaction flask was sealed with a septum and the reaction mixture was stirred at room temperature for 48 hours. The reaction mixture was diluted four-fold with EtOAc and washed twice each (25 mL each wash) with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with 25 mL CH2Cl2 All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient of 1 % (v/v) to 5 % MeOH in CH2Cl2. Rf (5 % (v/v) MeOH in CH2Cl2) = 0.30. Tetramer 13 was isolated as an amorphous (183.9 mg, 58 % yield). note: small additional peaks in 1H NMR spectrum attributed to rotamers, which were not observed in 13C NMR spectrum. 1H NMR (300 MHz, CDCl3): δ 7.34 (m, 15H), δ 6.81 (broad, 3H), δ 5.31 (d, J = 8.7 Hz, 1 H), δ 5.10 (ABq, Jab = 12.3 Hz, vab = 14.0 Hz, 2H), δ 4.64 (q, J = 7.8 Hz, 1H), δ 4.43 (q, J = 7.2 Hz, 1H), δ 3.16 (broad, 8H), δ 2.46 (q, J = 9 Hz, 1H), δ 2.29 (sextet, J = 6.3 Hz, 1H), δ 2.13 (broad, 1H), 1.85 (m, 4H), δ 1.64 (broad, 6H), δ 1.38 (s, 9H), δ 1.24 (m, 3H). 13C NMR (75.4 MHz, CDCl3): δ 176.3, 176.2, 172.6, 172.1, 156.0, 137.5, 137.2, 136.3, 129.5, 128.8, 128.8, 128.8, 128.6, 128.4, 128.2, 127.1, 127.0, 80.5, 56.3, 55.1, 48.6, 48.5, 44.3, 44.1, 44.0, 43.3, 42.1, 38.4, 38.1, 30.7, 30.5, 30.3, 28.5, 25.1, 25.0. Calculated [M+Na+]/observed for C44H56N4O7Na+: 775.4042/775.4057.
Boc-HN-(DAla-AMCP)3-(DPhe-AMCP)2-OBn (15)
A 0.11 mmol portion of α/γ-peptide hexamer 9 was deprotected according to General Procedure A and a 0.11 mmol portion of α/γ-peptide tetramer 13 was deprotected according General Procedure B to yield an α/γ-hexapeptide acid and α/γ-tetrapeptide HCl salt 14, respectively, with both reactions proceeding in > 95 % yield. The α/γ-hexapeptide acid (0.11 mmol, 1.0 equiv.) was dissolved in 2.5 mL 4:1 NMP:DMSO. iPr2EtN (0.118 mL, 0.66 mmol, 6.0 equiv.) was added followed by Oxyma (20.3 mg, 0.14 mmol, 1.3 equiv.) and EDCI (25.3 mg, 0.13 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 15 minutes at which point a solution of α/γ-tetrapeptide amine HCl salt 14 in 2.5 mL 4:1 NMP:DMSO was added. The reaction flask was sealed with a septum and the reaction mixture was stirred at room temperature for 5 days. The reaction was diluted with EtOAc and transferred to a separatory funnel. NMP/DMSO were removed by washing the diluted reaction five times with 50 mL H2O. Each H2O wash was back-extracted four times with 50 mL Et2O. The combined organic layers were washed twice each with 1 M aqueous NaHSO4, then saturated (aq) NaHCO3, then brine. Each aqueous layer was extracted twice with CH2Cl2 All organic layers were combined, dried over MgSO4, filtered, and evaporated to afford a crude solid. The product was purified via column chromatography eluting with a gradient of 1 % (v/v) to 10 % MeOH in CH2O2.. Rf (5 % MeOH) = 0.15. Product was isolated as an amorphous solid (116 mg, 77 % yield). 1H NMR (300 MHz, CDCl3 with 5 % CD3OH): δ 8.63 (d, J = 6.3 Hz, 1H), δ 8.51 (d, J = 7.5 Hz, 1H), δ 8.19 ppm (d, J = 9.3 Hz, 1H), δ 8.12 (broad, 2H), δ 8.03 (d, J = 3.9 Hz, 1H), δ 7.88 (d, J = 3.9 Hz, 1H), δ 7.79 (d, J = 6.3 Hz, 1H), δ 7.25 (m, 15H), δ 6.97 (broad, 1H), δ 5.61 (d, J = 9.3 Hz, 1H), δ 5.09 (ABq, Jab = 11.7 Hz, vab = 15.2 Hz, 2H), δ 4.60 (q, J = 7.5 Hz, 1H), δ 4.45 (broad, 2H), δ 4.33 (broad, 1H), δ 4.18 (broad, 1H), δ 3.13 (broad, 15H), δ 2.36 (broad, 8H), δ 1.77 (broad, 25H), δ 1.44 (s, 9H), δ 1.33 (broad, 14H). 13C NMR (125 MHz, CDCl3 with 5 % CD3OH): Carbonyl (11 observed, 11 in molecule) and aromatic peaks are listed below as these were diagnostic of decamer 15. Full 13C Spectrum is provided in the Supporting Information.: 179.2, 178.7, 178.3, 178.1, 176.2, 175.4, 174.8, 174.6, 174.6, 173.8, 173.0, 156.2, 151.6, 137.8, 137.5, 136.1, 136.0, 129.6, 129.4, 129.5, 129.3, 128.7, 128.6, 128.4, 128.3, 128.1, 126.9, 126.8, 125.7. Calculated [M+H+]/observed for C74H104N410O13H+: 1365.1/ 1365.1.
2D NMR Procedures
Sample Preparation
NMR samples were prepared by dissolving the α/γ-peptide in CDCl3 (0.03 %) TMS. CD3OH was added to a concentration of 5 % (v/v) to the solution of decamer 15 to fully solubilize the α/γ-peptide; the total concentration of this sample was 2 mM. Samples were prepared with 500-600 μL solvent for use in 5 mm NMR tubes. Spectra were referenced to tetramethylsilane. Samples showed the same spectral features after several weeks in solution, without showing visible signs of precipitation. All 2D NMR experiments were preformed on 600 MHz spectrometer. Data were collected at 10 °C, 20 °C, and 24°C, respectively, for α/γ-peptides 9, 12, and 15. Sample concentrations for all three α/γ-peptides were 2 mM.
gCOSY, TOCSY,16 ROESY:17
Standard pulse sequences were used and data were processed using the spectrometer's software. Assignments and analyses of spectra were performed in the SPARKY program.18 Spectral windows, f2 resolutions, and mixing times are as follows:
Hexamer 9 – 6492.5 Hz ; points in f2 = 1664 ; TOCSY mixing time = 120 ms; ROESY mixing time = 300 ms
Octamer 12 – 6999.7 Hz ; points in f2 = 1792 ; TOCSY mixing time = 80 ms; ROESY mixing time = 200 ms
Decamer 15 – 8000 Hz ; points in f2 = 2048 ; TOCSY mixing time = 80 ms; ROESY mixing time = 200 ms. Note: the CD3OH solvent peak was suppressed for this sample, which was essential in allowing for the assignnment of AMCP Hg protons.
Shifted sinebell window functions were applied before Fourier transformation of 2D data. For all samples, gCOSY spectra were obtained in absolute mode with gradient echo coherence selection. TOCSY and ROESY spectra were acquired in the sensitive mode with hypercomplex phase cycling. Data were collected with 1600-2048 points in f2 (above) and 200-600 points in f1. TOCSY experiments employed a standard MLEV-17 spin lock sequence with a spin lock field of 7-8 KHz. ROESY experiments utlilized spin-locking fieds of ~3 KHz.
A note on mixing times
Our choice of mixing times of 200-300 ms in ROESY experiments is due to the exploratory nature of our study. Medium-range ROESY crosspeaks give the most compelling evidence for helical folding (e.g., between residues i and i+4 in an α-helix, or between residues i and i+2 in our α/γ-peptides). These ROEs arise from through-space interactions between H atoms that are many bonds apart. Such ROEs are fundamentally slower in their rate of buildup than short-range ROEs that arise from H atoms separated by only a few bonds. We can use extended mixing times confidently because the ROESY experiment allows one to identify (and disregard) crosspeaks that arise from spin diffusion or TOCSY artifacts since these crosspeaks would be of the same sign as the diagonal of the 2D spectrum; the true ROESY crosspeaks that we seek are opposite in sign relative to the diagonal.
Sequence assignment
Chemical shift assignment was accomplished via sequential procedures.9 Briefly, the N-terminal Boc NH amide peak was identified for each peptide as the most upfield of the amide chemical shifts (this is due to the increased shielding of a urethane carbonyl versus an amide carbonyl). The resonances of the N-terminal residue were assigned using TOCSY and ROESY crosspeaks (with COSY to assist in cases of ambiguous chemical shifts). The next residue in the sequence was assigned on the basis of a strong NOE (1.8-3.0 Å) crosspeak between the HCα of the N-terminal residue and the NH amide resonance of the next residue. Once the NH peak of the following residue was identified, the above procedure was repeated proceeding from the N-terminus to the C-terminus of each peptide sequence. 1H chemical shift assignments and structural NOEs for α/γ-peptides 9, 12, and 15 are tabulated in the Supporting Information.
A note on NOEs in α/γ-peptides
Mid-range and long-range NOEs between non-adjacent amino acids (between residue i and i+2 or greater) are typically viewed in α-peptides as crosspeaks that do not arise simply because of proximity in a peptide sequence and are therefore indicative of some type of compact conformation. γ-amino acids, however, span a significant distance in space in a given α/γ-peptide sequence. For this reason, we define sequential NOEs specifically as the signals between the HCα of residue i and the NH amide proton resonance of residue Furthermore, we view signals between γ-residue i and α-residue i+1 other than the characteristic sequential NOE as indicative of some type of structure, even though the residues are adjacent.
A note on the selection of ROESY experiments in this study
We routinely select ROESY in preference to NOESY for 2D NMR studies of peptidic oligomers between 500 and 1500 Daltons. In general, molecules in this mass range display attenuated or very weak NOESY spectra, presumably because their rotational correlation times fall into a range such that their product with the Larmor frequency approaches unity; the net effect of this is an NOE value of low or zero magnitude. The buildup of crosspeaks in ROESY, while fundamentally lower in signal to noise than for NOESY, always leads to positive signals and is independent of the correlation time/Larmor frequency mathematical relationship. In addition, ROESY allows for the unique assignment of crosspeaks involving one proton from a diastereotopic methylene unit (spin diffusion effects in NOESY experiments lead to signals to both protons, when only one may in fact be interacting), which is useful given our desire to calculate NMR structures.
Derivation of distance restraints from integration of ROESY spectra crosspeaks
Crosspeaks were integrated in the program SPARKY18 using the Gaussian mode. Integrations were converted into distances using equation (1):
Where rij is the distance between two protons calculated from measured integrations, rref is the reference distance, Iref is the corrected integration corresponding to the reference distance, and Iij is the integration corrected according to equation (2):
Where m and n are the multiplicities of each of the interacting protons i and j. For example, if two methylene protons yield a single crosspeak with three methyl protons, the measured integration is divided by 6, the product of the multiplicities, prior to being entered into equation (1).
Selection of the reference distance
The most obvious choice for reference distance was to use the crosspeak between the diastereotopic benzyl ester protons in each peptide. However, the proximity of this crosspeak to the diagonal in the ROESY spectra of α/γ-peptide 9 precluded accurate integration. Because the diagonal is opposite in sign from ROESY crosspeaks by definition, this proximity reduces the value of the benzyl methylene crosspeak. This would give an artificially low integration, resulting in weak NOEs appearing stronger than they actually are. Therefore, to avoid this possible error, we selected the crosspeaks between DAlanine Hα and methyl Hβ signals for hexamer 9. The reference distance was calculated to 2.4 Å, according to the method of Wuthrich.9a Iref was then calculated as the average integration of the three crosspeaks resulting from the DAlanine Hα and methyl Hβ signals in hexamer 9, corrected according to equation (2). The diastereotopic methylene crosspeaks of the benzyl groups of octamer 12 and decamer 15 were sufficiently resolved from the diagonal of the ROESY spectrum and were used as distance references with a value of 1.6 Å.
NMR Structure Calculations for α/γ-peptides 9, 12, and 15
The nonsequential NOEs tabulated above were used as distance restraints in simulated annealing/torsional dynamics calculations in the CNS software package (v. 1.3) for α/γ-peptides 9, 12, and 15.10 We manually created CNS-formatted topology and parameter files for AMCP. We increased the scale of the dihedral energy term by 20% during both torsional and cartesian slow-cooling stages that follow the high temperature annealing stage in the “anneal.inp” protocol downloaded with CNS. All other parameters were used in the default setting from the “anneal.inp” protocol downloaded with the CNS software package. For each peptide we generated 1000 initial structures in the annealing stage and then selected for the ten lowest energy structures using the “accept.inp” protocol, again with increased dihedral energy terms, but no other specified selection criteria. The raised energy terms were necessary to prevent the cyclopentyl rings of AMCP residues from being forced into planar conformations. It was necessary to, additionally, place dihedral constraints, measured from the crystal structure of dipeptide 3, on the ring dihedrals to maintain cyclopentane envelope conformations. This was done while leaving the backbone dihedral about the cyclopentyl ring, ζ, free to float with the rest of the backbone torsion angles during the annealing runs. We note that i to i+1 NOEs are typically thought of as a consequence of peptide sequence, often occurring independently of conformation. However, we consider NOEs across the full distance of a γ-residue to the adjacent amino acid to be mid-range as these distances would correspond to those found from residue i to residue i + 2 in α-peptides. We did not include the strong sequential NOEs, defined earlier, in our calculations. In all cases, the lowest energy structures selected for analysis following calculations did not display any violations of our distance constraints derived from NMR study.
Supplementary Material
Scheme 1.

Synthesis of N-Boc-(1R,2R)-2-aminomethyl-1-cyclopentanecarboxylic acid 2 (AMCP).
Scheme 2.

Solution-phase synthesis of α/γ-peptides that contain AMCP.
ACKNOWLEDGEMENTS
The authors would like to thank: the NSF for financial support (Grant CHE-0848847 to S.H.G.), Dr. Dr. Charles G. Fry for assistance in collection of 13C 1D NMR spectra (@ 125 MHz) on a 500 MHz spectrometer at NMRFAM equipped with a cryoprobe for compounds 9, 12, and 15, Dr. Tomohisa Sawada for helpful discussions regarding simulated annealing calculations in CNS, and Prof. A.C. Kunwar (Indian Institute of Chemical Technology) for helpful email correspondence regarding molecular dynamics calculations of α/γ-peptides. NMR spectrometers were purchased with partial support from the NSF and NIH.
NMRFAM acknowledgement statement: This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41RR02301 (BRTP/ NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Footnotes
In memory of Professor Howard E. Zimmerman, 1926-2012
The authors declare no competing financial interest.
Supporting Information. The following are provided: Schemes for the preparation of S1, S2, S3, and 13 (procedures and data reported above). HPLC trace and description of conjugate addition optimization for nitroalochol 1. Tabulated 2D NMR data, chemical shift assignments, and 2D NMR spectra for α/γ-peptides 9, 12, and 15. Statistics and ensembles generate from NMR-restrained molecular dynamics calculations. 1H and 13C NMR spectra for all new compounds. Aggregation control and DMSO titration experiments for α/γ-peptide decamer 15. Crystallographic report and CIF file for the structure of dipeptide 3. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a Hagihara M, Anthony NJ, Stout TJ, Clardy J, Schreiber SL. J. Am. Chem. Soc. 1992;114:6568. Studies of secondary structure in oligomers that contain γ-amino acids. [Google Scholar]; b Hanessian S, Luo XH, Schaum R, Michnick S. J. Am. Chem. Soc. 1998;120:8569. [Google Scholar]; c Hintermann T, Gademann K, Jaun B, Seebach D. Helv. Chim. Acta. 1998;81:983. [Google Scholar]; d Hanessian S, McNaughton-Smith G, Lombart HG, Lubell WD. Tetrahedron. 1997;53:12789. [Google Scholar]; e Hanessian S, Luo XH, Schaum R. Tet. Lett. 1999;40:4925. [Google Scholar]; f Seebach D, Brenner M, Rueping M, Schweizer B, Jaun B. Chem. Commun. 2001:207. [Google Scholar]; g Woll MG, Lai JR, Guzei IA, Taylor SJC, Smith MEB, Gellman SH. J. Am. Chem. Soc. 2001;123:11077. doi: 10.1021/ja011719p. [DOI] [PubMed] [Google Scholar]; h Seebach D, Brenner M, Rueping M, Jaun B. Chem. Eur. J. 2002;8:573. doi: 10.1002/1521-3765(20020201)8:3<573::AID-CHEM573>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]; i Seebach D, Hook DF, Glattli A. Biopolymers. 2006;84:23. doi: 10.1002/bip.20391. [DOI] [PubMed] [Google Scholar]; j Khurram M, Qureshi N, Smith MD. Chem. Commun. 2006:5006. doi: 10.1039/b611882h. [DOI] [PubMed] [Google Scholar]; k Jones CR, Qureshi MKN, Truscott FR, Hsu STD, Morrison AJ, Smith MD. Angew. Chem. Int. Ed. 2008;47:7099. doi: 10.1002/anie.200802648. [DOI] [PubMed] [Google Scholar]
- 2.Selected reviews on foldamer structure: Gellman SH. Acc. Chem. Res. 1998;31:173. Hill DJM, M.J., Prince RB, Hughes TS, Moore JS. Chem. Rev. 2001;101:3893. doi: 10.1021/cr990120t. Goodman CM, Choi S, Shandler S, DGrado WF. Nat. Chem. Biol. 2007;3:252. doi: 10.1038/nchembio876. Seebach D, Gardiner J. Acc. Chem. Res. 2008;41:1366. doi: 10.1021/ar700263g. Horne WS, Gellman SH. Acc. Chem. Res. 2008;41:1399. doi: 10.1021/ar800009n. Martinek TA, Fülöp F. Chem. Soc. Rev. 2012;41:687. doi: 10.1039/c1cs15097a.
- 3.a Guo L, Chi YG, Almeida AM, Guzei IA, Parker BK, Gellman SH. J. Am. Chem. Soc. 2009;131:16018. doi: 10.1021/ja907233q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guo L, Almeida AM, Zhang W, Reidenbach AG, Choi SH, Guzei IA, Gellman SH. J. Am. Chem. Soc. 2010;132:7868. doi: 10.1021/ja103233a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Guo L, Zhang WC, Reidenbach AG, Giuliano MW, Guzei IA, Spencer LC, Gellman SH. Angew. Chem. Int. Ed. 2011;50:5843. doi: 10.1002/anie.201101301. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Guo L, Zhang WC, Guzei IA, Spencer LC, Gellman SH. Org. Lett. 2012;14:2582. doi: 10.1021/ol3008815. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sawada T, Gellman SH. J. Am. Chem. Soc. 2011;133:7336. doi: 10.1021/ja202175a. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shin Y-H, Mortenson DE, Satyshur KA, Forest KT, Gellman SH. J. Am. Chem. Soc. 2013;135:8149. doi: 10.1021/ja403319q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Previously reported stereoselective synthesis of the cis isomer of 2-aminomethyl-1-cyclopentane carboxylic acid: Baxendale IR, Ernst M, Krahnert W-R, Ley SV. Synlett. 2002:1641.
- 5.a Hayashi Y, Gotoh H, Hayashi T, Shoji M. Angew. Chem. Int. Ed. 2005;44:4212. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]; b Marigo M, Wabnitz TC, Fielenbach D, Jorgensen KA. Angew. Chem. Int. Ed. 2005;44:794. doi: 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]; c Gotoh H, Ishikawa H, Hayashi Y. Org. Lett. 2007;9:5307. doi: 10.1021/ol702545z. [DOI] [PubMed] [Google Scholar]
- 6.a Baldauf C, Gunther R, Hofmann HJ. Helv. Chim. Acta. 2003;86:2573. [Google Scholar]; b Baldauf C, Gunther R, Hofmann HJ. Angew. Chem. Int. Ed. 2004;43:1594. doi: 10.1002/anie.200353249. [DOI] [PubMed] [Google Scholar]; c Baldauf C, Gunther R, Hofmann HJ. Biopolymers. 2005;80:675. doi: 10.1002/bip.20249. [DOI] [PubMed] [Google Scholar]; d Baldauf C, Gunther R, Hofmann HJ. J. Org. Chem. 2006;71:1200. doi: 10.1021/jo052340e. [DOI] [PubMed] [Google Scholar]
- 7.Subiros-Funosas R, Prohens R, Barbas R, El-Faham A, Albericio F. Chem. Eur. J. 2009;15:9394. doi: 10.1002/chem.200900614. [DOI] [PubMed] [Google Scholar]
- 8.Decamer 15 showed poor solubility, precipitating rapidly from DMSO, CH2Cl2, CH3CN, or acetone. This decamer formed organogels in CDCl3 or in CD3OH within one hour at room temeprature. In 5 % (v/v) CD3OH in CDCl3, however, samples of 15 remained soluble and freely flowing for more than seven days. Additionally, we observed no change in the chemical shifts of the amide protons of 15 upon dilution (see Supporting Information). We therefore believe that 15 and the far more soluble α/γ-peptides 9 and 12, for which we also conducted 2D NMR experiments, do not self-associate under conditions employed for the 2D NMR studies.
- 9.a Billeter M, Braun W, Wuthrich K. J. Mol. Biol. 1982;155:321. doi: 10.1016/0022-2836(82)90008-0. [DOI] [PubMed] [Google Scholar]; b Wuthrich K, Wider G, Wagner G, Braun W. J. Mol. Biol. 1982;155:311. doi: 10.1016/0022-2836(82)90007-9. [DOI] [PubMed] [Google Scholar]; c Wuthrich K, Billeter M, Braun W. J. Mol. Biol. 1984;180:715. doi: 10.1016/0022-2836(84)90034-2. [DOI] [PubMed] [Google Scholar]; d Wüthrich K. NMR of proteins and nucleic acids. Wiley; New York: 1986. [Google Scholar]
- 10.a Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998;54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]; b Brunger AT. Nat. Protoc. 2007;2:2728. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 11.Sharma GVM, Jadhav VB, Ramakrishna KVS, Jayaprakash P, Narsimulu K, Subash V, Kunwar AC. J. Am. Chem. Soc. 2006 doi: 10.1021/ja064875a. [DOI] [PubMed] [Google Scholar]
- 12.Vasudev PG, Chatterjee S, Shamala N, Balaram P. Acc. Chem. Res. 2009;42:1628. doi: 10.1021/ar9001153. [DOI] [PubMed] [Google Scholar]
- 13.a Martinek TA, Mandity IM, Fulop L, Toth GK, Vass E, Hollosi M, Forro E, Fülöp F. J. Am. Chem. Soc. 2006;128:13539. doi: 10.1021/ja063890c. [DOI] [PubMed] [Google Scholar]; b Mandity IM, Weber E, Martinek TA, Olajos G, Toth GK, Vass E, Fülöp F. Angew. Chem. Int. Ed. 2009;48:2171. doi: 10.1002/anie.200805095. [DOI] [PubMed] [Google Scholar]; c Mandity IM, Fulop L, Vass E, Toth GK, Martinek TA, Fülöp F. Org. Lett. 2010;12:5584. doi: 10.1021/ol102494m. [DOI] [PubMed] [Google Scholar]; d Berlicki L, Pilsi L, Weber E, Madity IM, Cabrele C, Martinek TA, Fülöp F, Reiser O. Angew. Chem. Int. Ed. 2012;51:2208. doi: 10.1002/anie.201107702. [DOI] [PubMed] [Google Scholar]
- 14.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 15.a Kapat A, Nyfeler E, Giuffredi GI, Renaud P. J. Am. Chem. Soc. 2009;131:17746. doi: 10.1021/ja908933s. [DOI] [PubMed] [Google Scholar]; b Ryu HD, Lee TW, Corey EJ. J. Am. Chem. Soc. 2002;124:9992. doi: 10.1021/ja027468h. [DOI] [PubMed] [Google Scholar]
- 16.Bax A, Davies DG. J. Mag. Res. 1985;65:355. [Google Scholar]
- 17.Bothner-By AA, Stephens RL, Lee J, Warren CD, Jeanloz RW. J. Am. Chem. Soc. 1984;106:811. [Google Scholar]
- 18.Goddard DT, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.