

Abstract
CCAAT/enhancer-binding protein β (C/EBPβ) plays a key role in initiation of adipogenesis in adipose tissue and gluconeogenesis in liver; however, the role of C/EBPβ in hepatic lipogenesis remains undefined. Here we show that C/EBPβ inactivation in Leprdb/db mice attenuates obesity, fatty liver, and diabetes. In addition to impaired adipogenesis, livers from C/EBPβ−/− x Leprdb/db mice had dramatically decreased triglyceride content and reduced lipogenic enzyme activity. C/EBPβ deletion in Leprdb/db mice down-regulated peroxisome proliferator-activated receptor γ2 (PPARγ2) and stearoyl-CoA desaturase-1 and up-regulated PPARα independent of SREBP1c. Conversely, C/EBPβ overexpression in wild-type mice increased PPARγ2 and stearoyl-CoA desaturase-1 mRNA and hepatic triglyceride content. In FAO cells, overexpression of the liver inhibiting form of C/EBPβ or C/EBPβ RNA interference attenuated palmitate-induced triglyceride accumulation and reduced PPARγ2 and triglyceride levels in the liver in vivo. Leptin and the anti-diabetic drug metformin acutely down-regulated C/EBPβ expression in hepatocytes, whereas fatty acids up-regulate C/EBPβ expression. These data provide novel evidence linking C/EBPβ expression to lipogenesis and energy balance with important implications for the treatment of obesity and fatty liver disease.
Obesity is the most common nutritional disorder in Western societies. Today in the United States, more than 60% of people are either overweight (body mass index (BMI) > 25) or obese (BMI > 30) (1). Obesity is frequently associated with type II diabetes, hypertension, and hyperlipidemia, all known risk factors for cardiovascular disease (2). Obesity is also a major risk factor for non-alcoholic fatty liver disease, one of the most common emerging liver diseases in Western countries coinciding with the worldwide obesity epidemic (3, 4). The underlying transcriptional events that contribute to obesity and its associated disorders are not well understood. Some of the genes that regulate body weight have been identified as well as additional neuropeptides, hormones, and nutritional factors that play a role in body weight regulation, particularly through the β-ad-renergic system (5, 6). Discovery of the hormone leptin and its receptors, which suppress appetite and reduce fat mass, has dramatically increased our understanding of the regulation of energy balance (7, 8). More recently, the study of specific transcription factor genes and their metabolism has provided powerful new tools for understanding the integrated mechanisms underlying obesity and diabetes (9–11). This is most elegantly illustrated using tissue-specific gene knockouts and overexpression models to elucidate the mechanism of action of the PPAR5 family of nuclear hormone receptors (12).
The CCAAT/enhancer-binding protein (C/EBP) family includes five nuclear transcription factors, C/EBP α, β, γ, δ, and µ, encoded by separate genes located on different chromosomes (13, 14). Collectively, C/EBPs are expressed across a variety of cell types, and a large body of data exists on their expression patterns, the promoters they regulate, and the signals that modulate expression and/or activity (15,16). The great majority of this information was obtained in vitro. Members of the C/EBP family can form homo- and heterodimers within the promoters of genes, often making it difficult to distinguish between unique and redundant functions of the transcription factors on gene expression. However, in recent years mice have been generated with null mutations for each of the C/EBP genes, allowing the identification of unique and physiologically relevant functions.
Although C/EBPα shares a similar tissue distribution with C/EBPβ, they are differentially regulated during development and in response to changes in nutrition and hormonal status. C/EBPα knock-out mice are born without lipid or glycogen reserves and die of neonatal hypoglycemia several hours after birth due in part to the absence of phosphoenolpyruvate carboxykinase, glucose-6-phosphatase, and glycogen synthase (17). There was no significant increase in either C/EBPβ or -δ mRNA in the livers of these mice, suggesting C/EBPα may be part of a developmental program aimed at preparing the fetus for metabolism during the early prenatal period (18). Adult mice lacking C/EBPβ fail to increase gluconeogenesis during fasting and have reduced fat deposition, resulting in hypoglycemia and reduced non-esterified free fatty acids (NEFA) that act systemically to increase insulin sensitivity (19,20). The absence of C/EBPβ leads to lower blood glucose and reduced phosphoenolpyruvate carboxykinase gene induction in diabetes (21), indicating that deleting C/EBPβ may have anti-diabetic as well as anti-obesity effects. Using a gene replacement strategy where C/EBPβ was expressed from the C/EBPα gene locus, Chen et al. (22) showed that C/EBPβ rescued the role of C/EBPα in liver but not in white adipose tissue (WAT), emphasizing the unique role of C/EBPα in adipogenesis and C/EBPβ in gluconeogenesis.
Although C/EBPs regulate adipogenesis and gluconeogenesis, their ability to regulate hepatic lipid metabolism remains relatively unexplored. Recently, Matsusue et al. (23) demonstrated that liver-specific deletion of C/EBPα prevented accelerated lipogenesis in Leprob/ob mice. However, disruption of hepatic C/EBPα in normal adult mice appears to cause an exacerbation of hyperglycemia (24) and age-dependent increase in hepatosteatosis (24, 25), due in part to a decrease in genes encoding lipoprotein transport and fatty acid oxidation. The reason(s) for these differences in C/EBPα−/− mice is unclear but highlight important differences between C/EBPα and C/EBPβ with regard to their regulation and the metabolic phenotype.
To address the potential role of C/EBPβ in the pathogenesis of obesity and related disorders, we generated C/EBPβ−/− mice on a Leprdb/db null background. Here we show that C/EBPβ inactivation in Leprdb/db mice attenuates adipogenesis, obesity, fatty liver, and diabetes. C/EBPβ deletion affected critical genes that regulate hepatic lipogenesis and triglyceride (TG) metabolism resulting in protection from liver steatosis, independent of sterol response element binding protein 1c (SREBP1c). Moreover, forced overexpression of C/EBPβ induced TG accumulation along with PPARγ and stearoyl-CoA desaturase-1 (SCD-1) in vivo, whereas liver-specific inactivation in vivo and in liver cells in vitro can block TG accumulation, coincident with a reduction in PPARγ. Together these results demonstrate that in addition to its role in regulating adipogenesis and gluconeogenesis, C/EBPβ is necessary and sufficient to regulate hepatic lipogenesis independent of SREBP1c.
EXPERIMENTAL PROCEDURES
Animal Crossing and Genotyping
The generation and genotyping procedures for C/EBPβ−/− and Leprdb/db mice have been described previously (26, 27). Mice deficient in C/EBPβ were backcrossed for up to eight generations with C57Bl/6J mice from The Jackson Laboratories (Bar Harbor, ME) and intercrosses between heterozygous mice derived from these littermates. Double heterozygous offspring were then intercrossed to produce offspring with the following genotypes: wild-type (WT) mice (+/+ at the C/EBPβ and db locus), C/EBPβ−/− x Lepr+/+, C/EBPβ+/+ x Leprdb/db, and double knock-out C/EBPβ−/− x Leprdb/db mice. All mice were kept on a 12-h light/dark cycle and were fed a standard mouse chow ad libitum. Experiments and sample collection took place in the early afternoon after a 6-h daytime food withdrawal for steady state measurements. The University of Colorado at Denver and Health Sciences Center Animal Care and Use Committee approved all procedures.
Determination of Serum Insulin, Adiponectin, Free Fatty Acids, and TGs
Blood was collected from the tail vein at the times indicated. Plasma glucose levels were measured using an automatic glucose monitor (Roche Diagnostics). Serum insulin and adiponectin levels (Linco Research, St. Charles, MO and Alpco Diagnostics, Windham, NH), free fatty acids (Waco Diagnostics, Wako, TX), and TGs (Sigma-Aldrich) were measured using commercial kits, according to the manufacturers’ recommended protocols.
Determination of Body Fat Content
To assess body composition (percentage of fat), whole-body measurements of intact mice were performed using dual-energy x-ray absorptiometry (DEXA, PIXImus; GE-Lunar Corp., Madison, WI) as described previously in mice at 16 weeks of age (28, 29). Total fat, lean, and mineral mass were evaluated, excluding the head and tail. The short term precision error for whole-body measurements was <2%. Results are presented as fat content and total body weight.
Indirect Calorimetry
Metabolic measurements were obtained using an open circuit indirect calorimetry system as described previously (30). The system was calibrated against a standard gas mixture to measure O2 consumed and CO2 produced (ml/kg/h). Oxygen consumption was assessed individually in mice. After a 24-h period for adaptation to the metabolic chamber, VO2 and CO2 produced were assessed at 5-min intervals for a 72-h period. Mice had free access to water and food during the 12-h night period. Total oxygen consumption represents the mean of all samples collected during the experiment. Measurements of energy intake and energy expenditure were corrected for lean body weight.
TG Assays
Liver samples were homogenized in microcentrifuge tubes using Kontes disposable pestles, whereas muscle was homogenized in Kontes Duall® homogenizers (Kimble/Kontes, Vineland, NJ). All tissues were initially homogenized in 8 volumes of deionized water to facilitate cell disruption; subsequently, 1 volume of 5 m NaCl was added to enhance partitioning of lipids into the organic phase. Plasma samples were analyzed directly, whereas tissue sample homogenates were extracted using a modification of the method described by Bligh and Dyer (31). Briefly, a 200-µl aliquot of 10% homogenate prepared in 0.5 m NaCl was mixed with 500 µl of methanol, vortexed, then mixed with 250 µl of chloroform and vortexed again for 2 min. The single-phase mixture was broken into two phases by the addition of 250 µl of water followed by 250 µl of chloroform with mixing between each addition. After centrifugation, the lower, organic phase was collected in shell vials. Complete extraction of any residual lipids was ensured by reextracting with 250 µl of chloroform:methanol (9:1). The organic phase was separated by centrifugation and combined with the first extraction. Samples were dried with an N-Evap (Organomation Associates, Berlin, MA) under flowing nitrogen. The lipids were re-dissolved in a solution of 90% isopropanol, 10% Triton X-100 (2%) to disperse the TGs for assay. TGs were measured colorimetrically (TR0100, Sigma-Aldrich). For RNA interference (RNAi) experiments, TGs were extracted from FAO rat liver cells after the tert-butanol extraction method described previously (32).
Histology and Immunofluorescence
Portions of gonadal adipose tissue were removed from anesthetized mice, fixed overnight in 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 µM thickness. Sections were stained with hematoxylin and eosin and photographed at ×50 magnification. For immunohistochemistry, sections were deparafinized in xylenes and rehydrated, and antigen retrieval was performed in heated citrate buffer. Sections were blocked in 1.5% goat serum in phosphate-buffered saline for 1 h at room temperature and then incubated overnight with primary antibody diluted in the PBS, 1.5% goat serum overnight at 4 °C. Primary antibodies to the following targets were: C/EBPα (sc-61, Santa Cruz Biotechnology, Santa Cruz, CA), PPARγ2 (2492, Cell Signaling Technology, Danvers, MA), preadipocyte factor-1 (Pref-1) (PREF-12A, Alpha Diagnostic, San Antonio, TX), and proliferating cell nuclear antigen (M0879, DakoCytomation, Glostrup, Denmark), Akt1 (Upstate, Chicago, IL). Sections were then rinsed 3× in PBS and incubated with Texas Red-conjugated anti-rabbit IgG (Vector Laboratories, Burlingame, CA) for 1 h in PBS. Sections were rinsed three times in PBS, and coverslips were mounted with Vectamount with 4′,6-dia-midino-2-phenylindole (fluorescent nuclear stain, Vector Laboratories). Images of fluorescent-labeled sections were taken with a SPOT RT camera connected to a Nikon Eclipse TE2000U microscope and captured to a PowerMac G4 computer. Phase contrast images were taken with a SPOT Insight camera attached to the same microscope. Images were processed using Adobe Photoshop CS software. Fluorescent terminal dUTP nick-end labeling staining for apoptotic cells was performed using the ApoAlert DNA fragmentation assay kit from Clontech (Mountain View, CA) according to the manufacturer’s protocol.
Western Blot Analysis
To measure nuclear C/EBPβ and SREBP1c protein levels, liver nuclear extracts were prepared from frozen liver samples. Liver tissue (50–70 mg) was homogenized in 300–500 µl of hypotonic buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, 2 µg/ml each of aprotinin and leupeptin, and 0.5 mg/ml benzamidine). The supernatants (cytoplasmic extracts) were saved. The pellets were re-suspended in 40 µl of high salt buffer (20 mM HEPES, 400 mM phenylmethylsulfonyl fluoride, 2 µg/ml aprotinin, 2 µg/ml leupeptin, and 0.5 mg/ml benzamidine) for 30 min on ice with occasional vortexing. After centrifugation at 15,000 rpm for 30 min, the supernatants (nuclear extracts) were saved. The protein concentration was measured with the Bradford assay (Bio-Rad) using bovine serum albumin as standard. Lysates were subjected to SDS/PAGE on 10% gradient gels. Proteins were transferred and immobilized on Immobilon-P membrane. The membranes were immuno-blotted with respective primary and secondary antibodies, and bands were visualized by using enhanced chemiluminescence and quantified by densitometry. Primary antibodies used were SCD-1 (sc-1475, Santa Cruz), PPARα (sc-9000), PPARγ2 (sc-22020), C/EBPβ (sc-150), CD36 (ABM5526, Cascade Biosciences), phospho-AMP-activated protein kinase (AMPK; Thr-172, 2531, Cell Signaling), SREBP1c (sc-366), adipophilin (ADPH; RDI-PROGP40, Fitzgerald Industries, Concord, MA), and GAPDH (sc-20358). The antibody to the LDL receptor was supplied in kind from Dr. Jay Horton (UT-Southwestern). Primary antibodies were diluted 1:300–1:500 in 5% nonfat dry milk. Secondary antibodies were goat anti-mouse and goat anti-rabbit IgG-horseradish peroxidase conjugates (Bio-Rad) and were diluted 1:10000 in 5% nonfat dry milk.
Hepatic Enzyme Activity
Livers were from 16-week-old mice fasted for 6 h. Animals were anesthetized with 150 µl of avertin (2.5%) followed by rapid removal and freezing on dry ice within 1 min. The samples were stored at −80 °C until analysis. Tissue (100 mg) was homogenized for 30 s in 2.0 ml of 0.25 m sucrose, 10 mM Tris acetate, 1 mM EDTA, 1 mM dithiothreitol, pH 7.4, using a Tissumizer (Polytron) homogenizer. The homogenate was centrifuged at 4 °C for 130 min at 68,000 × g. From the supernatant 0.5 ml was gel-filtered using NAP 5 columns (GE Healthcare). For citrate lyase derivation the columns were equilibrated and eluted with 20 mM Tris/HCl, 1 mM EDTA, 1 mM dithiothreitol, pH 7.5, whereas for fatty acid synthase assays the columns were equilibrated and eluted with 0.5 m potassium phosphate buffer, 5 mM dithiothreitol, pH 7.0. Activity of ATP citrate lyase was determined spectrophotometrically as described previously (33). All assays were performed in duplicate and were linear for at least 15 min after the addition of CoA. Fatty acid synthase (FAS) was activated before analysis (34) by incubating the gelfiltered high speed supernatant at 37 °C for 15 min before assay. Activity of FAS was then determined spectrophotometrically (34). All assays were performed in duplicate and were linear for at least 5 min after the addition of malonyl CoA and acetyl CoA.
Adenovirus Purification
Adenovirus was propagated in HEK293 cells. Cells were harvested when cytopathic effects were visible in more than 90% of the cells. Adenovirus was released from cells through rapid freeze/thawing. Adenovirus was purified via cesium chloride gradients and dialyzed into virion dialysis buffer (10 mm Tris-HCl, pH 8.0, 135 mm NaCl, 1 mm MgCl2, and 50% glycerol). Titer was measured using A260.
RNAi Adenovirus Construction
Short hairpin RNA (shRNA) designed for C/EBPβ targeted the 3′-untranslated region (5′-CCGGGCCCTGAGTAAT-CAC-3′). Sense and antisense oligonucleotides were designed and cloned into pSUPER (OligoEngine, Seattle, WA) as described previously (35). Cloning into the adenoviral shuttle vector EH006, and virus propagation was described previously (36).
Adenovirus Transduction
Animals
Adenovirus (1 × 1010–1 × 1011 pfu/ml adenovirus in 150 µl of PBS) expressing the C/EBPβ isoforms, LAP (Adv-LAP) or LIP (Adv-LIP), or GFP (Adv-GFP) alone (control) were injected via tail vein into mice. Animals were sacrificed 4 days post-injection. Liver was removed, and nuclear homogenates were prepared for Western blotting or preserved in RNAlater (Qiagen, Valencia, CA).
Cells
FAO cells were transduced 24 h after plating with Adv-GFP, Adv-LAP, or Adv-LIP and treated with or without 200 µM palmitate (molar ratio of 1 palmitate:3 albumin) for 24 h before harvesting. For RNAi experiments, FAO cells were transduced with or without adenovirus-delivered shRNA targeting C/EBPβ (Adv-C/EBPβ-shRNA) 24 h and treated with or without 200 µM palmitate for 48 h before harvesting.
Treatment of FAO Cells with Metformin
FAO cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and antibiotics (penicillin 100 IU/ml, streptomycin 100 µg/ml) in 5% CO2. Cells were subsequently serum-starved overnight then treated for 4 h with metformin (20,50,200, 500 µM). Separate cytoplasmic and nuclear extracts were prepared as described previously (21). Protein (50 µg) was electrophoresed, blotted, and probed with specific antibodies.
Liver Perfusion Studies
Isolated perfused rat liver preparation has been described previously (37). Livers were perfused with 400 ml of re-circulating oxygenated Krebs-Henseleit buffer with or without leptin (9-fold above basal) for 90 min in the presence or absence of phosphatidylinositol 3-kinase inhibitors wortmannin (100 nm) or LY294002 (10 µM). Livers were removed and kept frozen at −80 °C until analysis of C/EBPβ protein by Western blot analysis.
Quantitative Real-time PCR
Total RNA was extracted from homogenized mouse liver or FAO cells using the RNeasy kit (Qiagen). cDNA was prepared by reverse transcription of 250–1000 ng of total RNA using the Superscript III enzyme and random hexamers (Invitrogen). cDNAs were amplified using Platinum quantitative PCR SuperMix-UDG (Invitrogen) and TaqMan Gene Expression assays (ABI, Foster City, CA), or custom assays were designed using Primer Express (ABI) software on an Opticon 2 (Bio-Rad) or ABI 7700 real-time PCR system. RNA expression data were normalized to levels of 18 S RNA, which was unaffected by adenoviral transduction or animal genotype. The TaqMan ID number for genes analyzed are as follows: acyl-coenzyme A oxidase 1, Mm00443579_m1; FAS, Mm00662319_m1; PPARα, Mm00440939_m1; PPARγ, Mm00440945_m1; SCD-1, Mm00772290_m1; GAPDH, 4308313; 18 S RNA, 4308329. For C/EBPβ, a custom designed assay from ABI was used (forward primer, 5′-AAGA-GCCGCGACAAGGC-3′; reverse primer, 5′-GTCAGCTC-CAGCACCTTGTG-3′, probe 5′-AAGATGCGCAAC-CTGGAGACGCA-3′).
Statistical Analysis
Statistical comparisons between groups were made using Student’s t test or analysis of variance where appropriate. All values are reported as the mean plus or minus S.E., and differences were considered to be statistically significant at p values less than 0.05.
RESULTS
C/EBPβ Deletion Decreases Weight Gain and Fat Mass in Leprdb/db Mice
To determine whether C/EBPβ deficiency can protect against a genetic form of obesity and its related syndromes, we intercrossed C/EBPβ+/− and Leprdb/+ mice and obtained mice deficient in both C/EBPβ and leptin receptors (C/EBPβ−/− x Leprdb/db). The frequency for obtaining C/EBPβ−/− x Leprdb/db progeny was lower than expected due to partial embryonic lethality of C/EBPβ−/− mice (38). At weaning (4 weeks of age) the body weight differences between Leprdb/db and C/EBPβ−/− x Leprdb/db mice were indistinguishable (Fig. 1 A). However, by 12 weeks of age Leprdb/db mice were overtly obese, whereas C/EBPβ−/− x Leprdb/db mice weighed significantly less (p < 0.05). By 16 weeks of age C/EBPβ−/− × Leprdb/db mice weighed 13.21 ± 0.76 g less, a difference of 25% (p < 0.01). The lower body weight in C/EBPβ−/− x Leprdb/db mice was accompanied by a reduction in total fat mass of 10.7 ± 0.56 g as determined by whole-body DEXA scanning (Fig. 1 B). However, C/EBPβ−/− x Leprdb/db mice also had significantly less lean body mass by 3.1 ± 0.15 g, suggesting the decrease in body weight could be associated with increased energy expenditure or perhaps reduced energy intake. To investigate this further, we conducted energy-balance studies in mice housed in metabolic chambers for 72 h. As expected, even when adjusted for lean body mass, Leprdb/db mice had significantly greater energy intake by 24% and significantly lower energy expenditure by 47% compared with WT mice (Fig. 1, C and D). C/EBPβ inactivation in Leprdb/db mice had no effect on this reduced energy expenditure. However, energy intake in CEBPβ−/− x Leprdb/db mice was significantly lower by 33% compared with Leprdb/db mice and was similar to WT or C/EBPβ−/− mice.
FIGURE 1. C/EBPβ deletion reduces obesity in Leprdb/db mice.
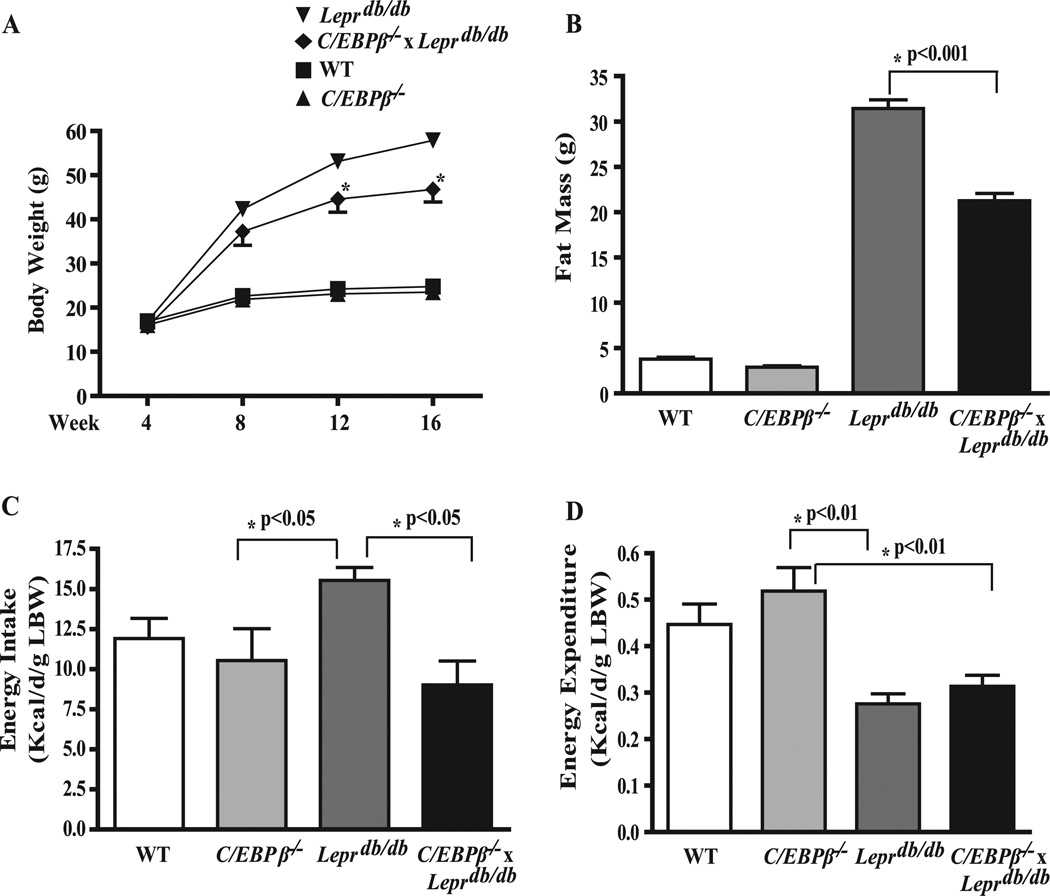
A, growth curve. B, fat mass. C, energy intake. D, energy expenditure. Total fat mass was measured by DEXA at 16 weeks of age. Energy intake and energy expenditure were measured using an open circuit indirect calorimetry system as described previously (30) over a 72-h period and corrected for lean body weight (LBW). Data represent the mean ± S.E. of 8–10 mice/group.
C/EBPβ Deletion Leads to Lower Glucose but Higher Serum-free Fatty Acid in Leprdb/db Mice
To examine the effects of C/EBPβ deficiency on metabolic parameters, blood was collected under fasting conditions for each genotype (Table 1). As observed previously (19), fasting glucose levels were lower in C/EBPβ−/− mice compared with WT controls and were dramatically reduced in C/EBPβ−/− x Leprdb/db mice compared with Leprdb/db mice, consistent with reduced gluconeogenesis and hepatic glucose production in diabetic C/EBPβ−/− mice (19, 21). Interestingly, despite lower glucose and reduced adiposity, fasting insulin levels were no different between Leprdb/db and C/EBPβ−/− x Leprdb/db mice. The levels of plasma TG were significantly elevated in Leprdb/db mice compared with WT mice. However, plasma TGs were also elevated in C/EBPβ−/− mice and showed a higher tendency in C/EBPβ−/− x Leprdb/db mice compared with Leprdb/db, although the latter was not statistically significant. Plasma protein NEFA levels were significantly elevated in C/EBPβ−/− x Leprdb/db mice compared with Leprdb/db mice. Adiponectin levels were unchanged.
TABLE 1. Body weight and clinical characteristics in WT, C/EBPβ−/−, Leprdb/db and C/EBPβ−/− x Leprdb/db mice.
Data are from equal numbers of male and female mice fasted for 6 h at 16 weeks of age. Values are the mean ± S.E. of 7–12 mice per genotype.
| WT | C/EBPβ−/− | Leprdb/db | C/EBPβ−/− x Leprdb/db | |
|---|---|---|---|---|
| Weight (male) (g) | 26.4 ± 0.4 | 24.4 ± 0.8 | 58.5 ± 1.3 | 43.1 ± 1.1a |
| Weight (female) (g) | 24.5 ± 0.3 | 22.6 ± 0.5 | 49.7 ± 0.9 | 35.3 ± 1.2a |
| Plasma glucose (mg/dl) | 148 ±6 | 109 ±3b | 448 ±26 | 192 ± 12c |
| Plasma insulin (ng/ml) | 2.8 ± 0.3 | 3.2 ± 0.2 | 19.4 ± 1.6 | 19.0 ± 2.7 |
| Plasma triglyceride (mg/dl) | 22 ±3 | 33 ± 3b | 31 ±5 | 35 ±8 |
| Plasma NEFA (µmol/l) | 0.65 ± 0.04 | 0.62 ± 0.06 | 0.74 ± 0.08 | 1.00 ± 0.05a |
| Plasma adiponectin (ng/ml) | 8.86 ± 0.90 | 6.78 ± 1.50 | 7.08 ± 0.93 | 7.68 ± 0.62 |
p < 0.05, C/EBPβ−/− versus WT.
p < 0.05, Leprdb/db versus C/EBPβ−/− x Leprdb/db.
p < 0.01, Leprdb/db versus C/EBPβ−/− x Leprdb/db.
Impaired Adipogenesis in C/EBPβ−/− x Leprdb/db Mice
C/EBPβ is a well established transcription factor necessary for adipogenesis in vitro. We examined WAT morphology in all four genotypes at 16 weeks of age. The majority of adipocytes from WT mice were between 75 and 125 µm in diameter (Fig. 2 A). Relatively few non-adipocytes were observed. However, adipocytes from C/EBPβ−/− mice were smaller with an average diameter of approximately half that of the adipocytes from WT mice (Fig. 2 B). Furthermore, there appeared to be a slight increase in both the extracellular matrix and the number of cells between the adipocytes suggesting an increase in mesenchy-mal-derived connective tissue cells, consistent with a reduction in adipocyte-differentiation potential (39). As might be predicted, adipocytes from Leprdb/db mice were extremely hypertrophic with an average diameter 1.5–2.0-fold higher than that of the WT adipocytes and with a correspondingly larger amount of lipid (Fig. 2 C). In striking contrast, adipocytes from C/EBPβ−/− x Leprdb/db mice showed a highly variable but marked reduction in both cell size and lipid content (Fig. 2 D). Although some cells appear to have a near normal content of lipid, they lacked the typical appearance of unilocular white adipose cells. In addition, there was a remarkable increase in extracellular matrix and clusters of small, undifferentiated cells in the inter-adipocyte spaces. Inflammatory cells (neutrophils and lymphocytes) were searched for, but no increased number of either cell type was found in Fig. 2, A–D (data not shown). Also, apoptotic cells were searched for but were not found in Fig. 2, A–D (data not shown).
FIGURE 2. Impaired adipogenesis in C/EBPβ−/− x Leprdb/db mice.

A–D, portions of gonadal adipose tissue were removed from anesthetized mice, fixed overnight in 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 µM thickness. Sections were stained with hematoxylin and eosin and photographed at 50× magnification. The bar in the lower right corner of each photo equals 100 µm.E, for immunohistochemistry, sections were deparafinized in xylenes and rehydrated, and antigen retrieval was performed in heated citrate buffer. Sections were immunoblotted with Pref-1 antibody.
To determine the differentiation state of these cells, we performed immunohistochemistry for Pref-1 (Fig. 2 E), which is used as a marker of preadipocytes (40). Pref-1 was absent in adipocytes from WT mice but strongly positive in the adipocytes from C/EBPβ−/− and C/EBPβ−/− x Leprdb/db mice, indicating that the majority of cells were likely preadipocytes. Propidium iodide staining was used to identify nuclei (not shown). The number of positive cells was slightly higher in the interadipocyte spaces from C/EBPβ−/− and C/EBPβ−/− x Leprdb/db mice. To rule out the possibility that cells were undergoing apoptosis, terminal dUTP nick-end labeling staining was performed and was not present in any of the cells examined. Sections were also stained for proliferating cell nuclear antigen as a marker for cell proliferation, and proliferating cell nuclear antigen was not present in any of the sections from the four genotypes (results not shown). These data suggest that C/EBPβ deletion in Leprdb/db mice reduced fat mass due to arrested differentiation of cells along the adipogenic lineage and that a higher percentage of adipocytes that are present are immature and have a reduced capacity for lipid storage.
C/EBPβ Deletion Prevents Hepatic Steatosis in Leprdb/db Mice
The lack of fully differentiated WAT and reduced adiposity in the double knock-out mice prompted us to examine whether C/EBPβ deletion had an effect on reducing lipid deposition in tissues other than adipose tissue. At 16 weeks of age the livers from Leprdb/db mice were enlarged compared with WT (not shown), and the hepatic TG content was 7–8-fold greater than WT (Fig. 3 A). No phenotypic differences were noted between WT and C/EBPβ−/− mice. However the TG content in livers from C/EBPβ−/− x Leprdb/db mice was significantly reduced by 75% to levels similar to WT mice. For comparative purposes, we also measured TG levels in skeletal muscle, a tissue that does not express C/EBPβ (16). Leprdb/db mice demonstrated a 10-fold increase in TG content compared with WT and C/EBPβ−/− mice and remained remarkably elevated in C/EBPβ−/− x Leprdb/db mice (Fig. 3 A). These data suggest that C/EBPβ deletion has a tissue-specific effect on preventing fatty liver and adipogenesis in Leprdb/db mice.
FIGURE 3. C/EBPβ deletion prevents fatty liver and reduces lipogenic enzyme activity in Leprdb/db mice.

A, hepatic and muscle TGs. Data represent the mean ± S.E. of 5–8 mice/group. Enzyme activities for FAS (B) and ATP-citrate lyase (C). Enzyme assays were carried out on fresh or frozen livers as outlined under “Experimental Procedures.” Data represent the mean ± S.E. of 5–8 mice/group.
C/EBPβ Deletion Decreases Hepatic Lipogenic Enzyme Activity and Expression of Proteins That Control TG Metabolism
To determine the potential regulatory pathways responsible for the decrease in hepatic TG in the C/EBPβ−/− x Leprdb/db mice, the activity of two key regulatory enzymes in the lipogenic pathway, ATP-citrate lyase and FAS, were measured (Fig. 3, B and C). The enzymatic activity for both hepatic ATP-citrate lyase and FAS were unaffected in C/EBPβ−/− mice but were clearly elevated by 3–4-fold in Leprdb/db mice compared with WT mice and dramatically lowered in CEBPβ−/− x Leprdb/db mice. We next turned our attention to the possible transcription factors and genes that control hepatic lipogenesis. A wealth of literature supports an important role for the transcriptional regulator SREBP1c in control of genes in the lipogenic pathway in the liver (11, 41). We examined the level of the mature nuclear form of SREBP1c by Western blot analysis and found it was 2.1-fold higher in the livers from Leprdb/db mice compared with WT or C/EBPβ−/− mice (Fig. 4 A). However, there was no difference between WT and C/EBPβ−/− livers or between Leprdb/db and C/EBPβ−/− x Leprdb/db mice (Fig. 4 A). These results indicate the reduced TG content and lipogenic enzyme activity in C/EBPβ−/− x Leprdb/db mice occurred without a reduction in SREBP1c.
FIGURE 4. Effect of C/EBPβ deletion on proteins that regulate hepatic lipid balance in Leprdb/db mice.

A and B, representative immunoblots and densitometric values for proteins involved in lipid metabolism. Hepatic nuclear or cytosolic extracts were isolated from liver of each group, and equivalent amounts of protein were electrophoresed and blotted with respective antibodies. The representative immunoblots were from the same gel and exposure for each protein. Results were corrected for loading differences using GAPDH and expressed relative to WT control mice. Representative blots are shown. Data represent the mean ± S.E. of 4–6 mice/group. LDLR, LDL receptor.
Both PPARγ and PPARα are critical regulators of hepatic lipid metabolism and steatosis (9,42). Excess TG accumulation in the liver of Leprob/ob mice is associated with a dramatic increase in PPARγ expression, primarily PPARγ2 (43, 44). Studies in 3T3L1 adipocytes in culture (45) and our data in C/EBPβ−/− mice suggests that C/EBPβ plays a direct role in controlling PPARγ expression in adipose tissue. To examine whether C/EBPβ deletion affected the levels of PPARγ or PPARα in the liver, Western blots were performed on liver extracts (Fig. 4 A). There was no difference in PPARγ in C/EBPβ−/− compared with WT mice; however, the levels of PPARγ2 were increased 1.5-fold in Leprdb/db mice compared with WT mice and normalized in C/EBPβ−/− x Leprdb/db mice, suggesting that C/EBPβ controls PPARγ2 induction in the liver. For PPARα, there was no difference between WT and C/EBPβ−/− mice; however, it was significantly lower in Leprdb/db mice and was sharply increased by 3.4-fold in livers of C/EBPβ−/− x Leprdb/db mice compared with Leprdb/db mice. PPARα activation increases transcription of numerous genes involved in fatty acid uptake and metabolism including LDL receptor (46) and CD36 (47), a scavenger receptor believed to facilitate fatty acid transport and function as a receptor for oxidized LDL (48). CD36 was up-regulated significantly in the livers from C/EBPβ−/− compared with WT mice and was increased in C/EBPβ−/− x Leprdb/db compared with Leprdb/db mice (Fig. 4 B). Induction of CD36 promotes the uptake of long chain fatty acids (49), potentially promoting lipid storage or oxidation. However, the level of LDL receptor showed the opposite pattern and was lower in C/EBPβ and in C/EBPβ−/− x Leprdb/db mice (Fig. 4 B) despite elevated circulating TGs and increased SREBP1c. ADPH is a lipid drop-let-associated protein that plays a key role in ectopic accumulation of triacylglycerols in non-adipose tissues (50, 51). ADPH was significantly elevated by 4.5-fold in liver extracts from Leprdb/db compared with WT mice and was restored to normal in C/EBPβ−/− x Leprdb/db mice, suggesting that ADPH may play an important role in promoting excess lipid storage found in steatotic livers.
Recent studies have demonstrated that the ability of leptin to reduce hepatic steatosis is mediated in large part by repression of SCD-1 activity and expression (52). The levels of SCD-1 protein were measured in microsomal liver fractions and were lower by 3-fold in C/EBPβ−/− mice compared with WT controls. As expected, SCD-1 was elevated in Leprdb/db mice, whereas C/EBPβ deletion in Leprdb/db mice significantly reduced SCD-1 by 3-fold (Fig. 4 B). Thus, C/EBPβ deletion in Leprdb/db mice dramatically reduced hepatic steatosis along with activity and expression of enzymes associated with lipogenesis, including FAS, ATP-citrate lyase, SCD-1, LDL receptor, and ADPH, while increasing PPARα and CD36. Together, these data suggest that a significant portion of the effect of C/EBPβ deletion on reducing hepatic steatosis in C/EBPβ−/− x Leprdb/db may be mediated by inhibiting induction of lipogenic genes.
Increased C/EBPβ Expression Increases Hepatic TG and Lipogenic Genes in Vivo
To substantiate the direct importance of liver C/EBPβ in regulating hepatic lipogenic gene expression and TG deposition in vivo, we treated WT mice with an adenovirus carrying the full-length C/EBPβ gene or GFP control. A single C/EBPβ mRNA produces three isoforms through alternative translation from three in-frame AUG codons. The primary isoforms in the liver are the full-length p34-LAP liver-enriched activator protein and p20-LIP, a truncated liver-enriched inhibitory protein. Western blot analysis of C/EBPβ expression shown in three representative animals 5 days after adenovirus administration demonstrated that the full-length p34-LAP protein was increased up to 6-fold in the liver of mice receiving Adv-C/EBPβ (Fig. 5 A). Expression of the p20-LIP isoform is also increased by 2 – 4-fold in these livers, yielding an average 2-fold change in the LAP/LIP ratio. No changes in C/EBPβ expression were found in brain, kidney, muscle, or adipose tissue in Adv-C/EBPβ treated mice (not shown). Importantly, mice with increased C/EBPβ in their livers showed a 2.5-fold increase in liver TG content (Fig. 5 B) and an increase in glucose levels at 30,60, and 90 min upon glucose tolerance testing (Fig. 5 C), suggesting glucose intolerance accompanied increased hepatic TG levels. Analysis of hepatic gene expression showed that the mRNA level for PPARγ2 and SCD-1 was increased up to 3-fold in the liver from mice receiving Adv-C/EBPβ compared with the Adv-GFP mice (Fig. 5 D), whereas the levels of PPARα were reduced by 50% in the liver (Fig. 5 D). The PPARα target gene acyl-coenzyme A oxidase 1, which encodes peroxisomal fatty acid oxidation, tended to be lower in the Adv-C/EBPβ-treated mice.
FIGURE 5. Effect of C/EBPβ overexpression by adenovirus on the fatty liver phenotype.

A, representative immunoblot analysis showing LAP and LIP expression 5 days after adenovirus delivery. B, hepatic TG levels in C57Bl/6J mice transduced with increasing levels of Adv-C/EBPβ (0,1 × 1010,and 1 × 1011 pfu/mouse).*, p < 0.05 versus Adv-GFP transduced mice. C, intraperitoneal glucose tolerance test in Adv-GFP and Adv-LAP transduced mice (4 days post-injection) using 2 g glucose/kg body weight. *, p < 0.05 versus Adv-GFP-transduced mice. D, relative mRNA levels for PPARγ, SCD-1, PPARα, and acyl-coenzyme A oxidase 1 (ACO) quantified by real-time quantitative PCR. RNA expression data were normalized to the levels of 18 S RNA and expressed relative to control mice receiving Adv-GFP. *, p < 0.05 versus Adv-GFP transduced mice.
The truncated form of C/EBPβ, termed LIP, is synthesized from an internal translation initiation site (53) and contains the carboxyl-terminal dimerization and DNA binding domains but lacks the amino-terminal activation domain of LAP and is thought to function as a natural negative regulator of transcription (54). Previous studies by Duong et al. (55) and our own results (56) indicate the LIP to LAP ratio is critical for control of phosphoenolpyruvate carboxykinase gene expression and gluconeogenesis in liver cells. To test whether increasing the LIP to LAP ratio affects lipid metabolism in the liver in vivo, we treated WT mice with an adenoviral vector carrying a truncated form of C/EBPβ lacking the transactivation domain (Adv-LIP). Western blot analysis performed after 4 days revealed a 3-fold increase in the LIP/LAP ratio in Adv-LIP-treated mice (Fig. 6 A). Liver lipids were extracted, and TG was measured as above. The data show that increasing the LIP to LAP ratio reduced hepatic TG by 50% (Fig. 6 B). Furthermore, there was a 4-fold decrease in the levels of PPARγ2 (Fig. 6 C) and a significant increase in PPARα protein as measured by Western blot analysis (Fig. 6 D). These data are consistent with results obtained in the global C/EBPβ−/− x Leprdb/db mice and suggest that C/EBPβ may play a role in regulation of TG content by affecting expression of PPARγ and PPARα.
FIGURE 6. Increasing the LIP:LAP ratio in liver lowers hepatic TG and alters PPAR expression.

A, LAP and LIP expression in Adv-LIP-infected mice. C57Bl/6J mice were treated with increasing levels of Adv-LIP (0,1 × 109, 3 × 1010, and 1 × 1011 pfu/mouse) or Ad-GFP (1 × 1010 pfu/mouse). After 4 days livers were harvested after a 12-h fast. Representative immunoblots are shown. B, hepatic TG levels. *,p < 0.05 versus Adv-GFP transduced mice. C–D, immunoblots and densitometric values of PPARγ and PPARα. Data represent the mean ± S.E. of two independent experiments. n = 5 mice/group. *, p < 0.05 versus Adv-GFP transduced mice.
Palmitate Induces C/EBPβ mRNA, and Inhibiting C/EBPβ Has a Direct Effect on PPAR Expression and TG Accumulation in FAO Cells
The reduction in hepatic TG in Adv-LIP-treated mice and reciprocal changes in PPARγ and PPARα prompted us to examine whether this effect could be demonstrated directly in liver cells. To test whether increasing LIP could inhibit lipid accumulation under lipogenic conditions, we transduced FAO cells with adenovirus for GFP (control) or Adv-LIP, and 24 h later treated the cells with 200 µM palmitate for 24 h. More than 80% transduction efficiency was obtained using 100 pfu/cell of adenovirus, as assessed by fluorescence microscopy (not shown). Cells were harvested, TGs were extracted, total mRNA was isolated, and PPARγ, PPARα, and C/EBPβ mRNA levels were measured by real-time quantitative PCR. As shown in Fig. 7 A, treatment of FAO cells with palmitate for 24 h significantly increased TG accumulation 1.8-fold (p < 0.05), whereas adenovirus encoding LIP suppressed the accumulation of TGs to levels similar to control cells. Adv-LIP prevented the induction of PPARγ mRNA by palmitate (Fig. 7 B) and increased PPARα expression 3.5-fold in the presence of palmitate. Interestingly, we also observed that incubation with palmitate alone increased C/EBPβ mRNA expression significantly by 2-fold.
FIGURE 7. Dominant negative C/EBPβ (LIP) and C/EBPβ shRNA prevent TG accumulation in FAO cells.

FAO cells were transduced 24 h after plating with Adv-GFP or Adv-LIP (30 pfu/cell) and treated with or without 200 µM palmitate for 24–48 h before harvesting.A, cellular TG levels. *, p < 0.05 versus control. B, relative mRNA levels for PPARγ, PPARα, and C/EBPβ quantified by real-time quantitative PCR. RNA expression data were normalized to the levels of 18S RNA and expressed as percent change over control. Data represent the mean ± S.E. of two independent experiments performed in triplicate. *, p < 0.05 versus control. C, immunoblot showing knockdown of C/EBPβ LAP and LIP isoforms with Adv-C/EBPβ-shRNA. FAO cells were treated with adenovirus-delivered shRNA targeting C/EBPβ (Adv-C/EBPβ-shRNA) for 24 h and treated with or without 200 µM palmitate for 48 h before harvesting. D, cellular TG levels. *, p < 0.05 versus control.
Whereas an increase in LIP was sufficient to inhibit lipogenesis in FAO cells, we wanted to further investigate whether a reduction in both C/EBPβ isoforms using RNAi was capable of suppressing lipogenesis, as it did in C/EBPβ−/− x Leprdb/db mice. We constructed a recombinant adenovirus to express a shRNA sequence against the 3′-untranslated region for C/EBPβ. Treatment of FAO cells with a non-targeting shRNA control virus had no effect on C/EBPβ, whereas C/EBPβ shRNA-treated cells resulted in a 75–85% reduction in both LAP and LIP protein levels (Fig. 7 C). Importantly, TG levels were unchanged in C/EBPβ shRNA-treated cells in the absence of palmitate (Fig. 7 D); however, in the presence of 200 µM palmitate TG levels were reduced by 43 ± 3% in C/EBPβ shRNA-treated cells compared with control virus-treated cells. These results indicate that suppression of C/EBPβ either by dominant negative or RNAi reduces hepatic TG levels, consistent with our in vivo data.
Leptin and the AMPK Activator Metformin Reduce Hepatic C/EBPβ Expression
Because C/EBPβ deletion had a major effect on reducing the expression of lipogenic genes in Leprdb/db mice lacking the leptin receptor and in cells in response to lipid loading, we considered the possibility that C/EBPβ may be a target of leptin signaling. To explore this further, we examined the acute effects of leptin on hepatic C/EBPβ expression using a perfused rat liver preparation (37). Perfusion of normal livers with leptin at physiological levels (9-fold above basal) had a powerful effect on reducing C/EBPβ expression by more than 50% relative to control livers perfused without leptin (Fig. 8 A). This effect was not blocked by inhibiting phosphatidylinositol 3-kinase either with LY294002 or wortmannin. We did not explore whether blocking AMPK, another mediator of leptin signaling, was involved in down-regulating C/EBPβ. However, we did explore the effects of the anti-diabetic drug metformin, which has been shown to decrease hepatic glucose production and increase fatty acid oxidation in part through activation of AMPK (57, 58). Treatment of FAO cells with increasing doses of metformin progressively increased AMPK phosphorylation at Thr-172 (Fig. 8 B). At the higher metformin concentrations C/EBPβ protein was reduced ~50–70% relative to basal levels (Fig. 8 B). These results suggest that exposure to metformin increases AMPK activation and suppresses C/EBPβ expression and that acute exposure to leptin, also an AMPK activator (59), dramatically down-regulates C/EBPβ expression in the liver.
FIGURE 8. Leptin and metformin acutely down-regulate C/EBPβ expression in perfused rat livers.

A, immunoblot and densitometric values for C/EBPβ (LAP) in leptin-perfused rat liver. Isolated livers were perfused with or without leptin (9-fold above basal) for 90 min in the presence or absence of phosphatidylinositol 3-kinase inhibitors wortmannin (100 nM) or LY294002 (10 µM) as described by us previously (37). Data represent the mean ± S.E. of 5–8 rats livers/group normalized to GAPDH. B, immunoblots and densitometric values for phospho-AMPK and C/EBPβ in metformin-treated FAO cells. FAO cells were serum-starved overnight and treated with increasing concentrations of metformin for 4 h. Data represent the mean ± S.E. of two independent experiments expressed as percent change over control normalized to AMPK and GAPDH for phospho-AMPK and C/EBPβ, respectively.
DISCUSSION
Members of the C/EBP family control the transcription of genes involved in a broad range of integrated metabolic processes ranging from the acute phase response to the control of hepatic glucose homeostasis to adipose tissue differentiation. In the present study C/EBPβ−/− x Leprdb/db mice revealed several unexpected findings. Despite reduced adipose tissue differentiation, C/EBPβ−/− x Leprdb/db mice were protected from fatty liver infiltration. The absence of liver steatosis and diabetes in C/EBPβ−/− x Leprdb/db mice is remarkable given the compromised adipogenesis, elevated NEFA, and hyperinsu-linemia. This is particularly striking given that mouse models, where adipogenesis is disrupted (i.e. lipodystrophy), hepatic steatosis is increased, and animals have severe insulin resistance and diabetes (60, 61).
Reduced hepatic TG accumulation was accompanied by decreased activity and expression of key genes for lipogenesis including decreased PPARγ2 expression. Similarly, overexpression of the dominant negative C/EBPβ isoform LIP in FAO cells and in the liver of WT animals reduced hepatic TG levels and PPARγ expression. Although PPARγ2 is normally only minimally expressed in hepatocytes, hepatic TG accumulation is associated with a dramatic increase in PPARγ expression (44). Experiments using a liver-specific PPARγ deletion in Leprob/ob mice prevented steatosis and caused a pronounced delay of hepatic TG uptake (62). Conversely, overexpression of PPARγ in normal mouse liver or PPARγ2 in liver cell lines results in a striking increase in steatosis and expression of genes implicated in de novo lipogenesis and lipid storage (63, 64). Our finding that C/EBPβ expression is sufficient to induce PPARγ2 and TG in mice treated with C/EBPβ adenovirus argues for an important role for C/EBPβ in directing hepatic lipogenesis in part through increased PPARγ2 expression, independent of changes in SREBP1c.
PPARα is a key transcription factor regulating several genes important for β-oxidation of fatty acids in the liver. In FAO cells and in mice transduced with adenovirus for C/EBPβ, we show that PPARα was suppressed, whereas in C/EBPβ−/− x Leprdb/db mice, PPARα was up-regulated. These animals also demonstrated an increase in CD36 expression, a PPARα target gene involved in fatty acid uptake and oxidation (65). Mice lacking PPARα develop hypoglycemia, hyperlipidemia, and fatty liver (66), whereas PPARα activation increases fatty acid oxidation, reduces adiposity, and improves hepatic steatosis (67), suggesting a mechanistic link between PPARα and liver steatosis. In addition to PPARα and PPARγ, a host of other transcription factors including PPARδ (68)), SREBP1c (69), pregnane X receptor (70), and carbohydrate response element-binding protein (ChREBP) (71), and the nuclear receptor LXRβ (72) and co-activator PGC-1α/β (73) have been implicated in pathways for fatty liver development. The lipogenic activity of LXRβ and PGC1α has been attributed in large part to interaction with SREBP1c (74, 75). Although we did not measure the levels of LXRβ or PGC-1 in this study, if C/EBPβ deletion did affect LXRβ or PGC1β, we would have expected to observe a decrease in SREBP1c. The level of mature nuclear SREBP1c protein was increased in the liver of Leprdb/db mice but remained elevated in C/EBPβ−/− x Leprdb/db mice. This may be related to the significant hyperinsulinemia remaining in these animals that is known to regulate SREBP1c expression (76). Thus, although other co-activators or transcription factors may interact with C/EBPβ in reducing lipogenic gene expression, our data suggest that C/EBPβ may control hepatic TG levels by altering the levels of PPARγ and PPARα expression. It is important to note, however, that C/EBPs may affect other aspects of lipid metabolism, including apolipoprotein gene expression (23) and lipid uptake by the liver (24, 77) that could synergistically reduce lipid accumulation.
SCD-1 catalyzes the rate-limiting reaction of monounsaturated fatty acid synthesis and plays an important role in the development of obesity (78). We found that C/EBPβ deletion suppressed SCD-1 levels in C/EBPβ−/− x Leprdb/db mice despite hyperinsulinemia, whereas C/EBPβ overexpression increased SCD-1 expression in the liver. Although other transcription factors cannot be ruled out, our results suggest that SCD-1 may be regulated by C/EBPβ to control lipogenesis. We also observed an increase in the lipid droplet protein ADPH in Leprdb/db mice that was normalized in C/EBPβ−/− x Leprdb/db mice. ADPH is a member of the lipid droplet-associated PAT protein family and plays a role in TG accumulation in non-adipose tissues (51). Arecent report has suggested a role for PPARγ in inducing ADPH and steatosis in liver cell lines (64).
A large number of in vitro studies have established that C/EBPβ is a major regulator of adipose tissue development. The effect of C/EBPβ deficiency on mitotic clonal expansion and subsequent differentiation was investigated by Tang et al. (79) using mouse embryonic fibroblasts isolated from C/EBPβ−/− mouse embryos. Unlike WT mouse embryonic fibroblasts, C/EBPβ−/− mouse embryonic fibroblasts neither formed mitotic foci nor gave rise to cells with adipocyte characteristics, e.g. the accumulation of cytoplasmic TG. Our results in WAT of mature C/EBPβ−/− mice show they are smaller in size and have reduced expression of adipocyte markers (C/EBPα, PPARγ, not shown), consistent with C/EBPβ’s role in mitotic clonal expansion. In addition, C/EBPβ deletion arrested adipocyte differentiation in knock-out mice and in Leprdb/db mice, as evidenced by increased Pref-1 expression and morphology, indicating that C/EBPβ is important for differentiation to the mature adipocyte phenotype in vivo. These data suggest that the markedly reduced WAT mass in C/EBPβ−/− mice was not due to a reduced adipocyte number but instead to the inhibition of TG accumulation in WAT.
The present study also demonstrates that C/EBPβ deletion in Leprdb/db mice limited the development of obesity in part by reducing hyperphagia. While this manuscript was in preparation, another report appeared suggesting that C/EBPβ−/− mice are protected from dietary obesity due to an increase in fatty acid oxidation primarily associated with a shift in gene expression in brown adipose tissue (BAT) despite reduced BAT mass (80). Interestingly, C/EBPβ−/− mice have less brown adipose tissue, and C/EBPβ deletion by itself has very little effect on the expression of lipogenic genes. However, fatty acids induced C/EBPβ mRNA in liver cells, and increasing C/EBPβ expression in vivo increased liver TG and lipogenic genes. Because C/EBPβ deletion mainly affected lipogenic genes in Leprdb/db mice rather than in C/EBPβ−/− mice, it suggests that C/EBPβ likely controls the inducible expression of these genes under lipogenic conditions in different tissues. C/EBPβ is expressed at relatively high levels in the hippocampus where it regulates stress behavior in response to adrenal-glucocorticoids (81, 82). In pituitary cells, C/EBPβ overexpression stimulates expression of both pro-opiomelanocortin-α (an anorectic peptide) and suppressor of cytokine signaling-3 (SOC-3) (83), a feedback inhibitor of leptin receptor signaling (84). Our data showing that leptin down-regulates C/EBPβ in the liver suggests that leptin could be involved in reducing nuclear C/EBPβ expression, which might be a molecular pathway for the long-term regulation of genes that control both lipogenesis and energy balance. Whether C/EBPβ functions as a nuclear fuel sensor in the central nervous system, as it does in liver in response to glucose (56), amino acids (85), and leptin (shown herein), warrants further investigation.
Non-alcoholic fatty liver disease is a common finding in patients and various animal models with obesity, insulin resistance, diabetes, and dyslipidemia (86). Although C/EBPβ deletion prevented hepatic steatosis and hyperglycemia, it did not suppress circulating NEFA or TG or rescue the hyperinsulinemia in Leprdb/db mice. This is likely due to the fact that C/EBPβ deletion reduced adiposity and hepatic TG deposition in C/EBPβ−/− x Leprdb/db mice (Figs. 2 and 3), thereby removing two major storage depots for excess lipids. This resulted in excess skeletal muscle TG deposition in C/EBPβ−/− x Leprdb/db mice (Fig. 3). It has been proposed that accumulation of excess lipids within tissues may be a mechanism of activation of inflammatory pathways and insulin resistance (87, 88). These results suggest that on the whole body level, inhibition of adipogenesis and hepatic steatosis may cause excess fuels to be diverted to skeletal muscle, resulting in hyperinsulinemia and insulin resistance.
In summary, our findings identify C/EBPβ as a novel target for the regulation of hepatic lipid metabolism and energy balance. The data also reinforce the idea that the C/EBPs are major players in the modulation of lipogenesis and adipogenesis as well as gluconeogenesis shown previously and emphasize the therapeutic potential of reducing the expression of C/EBPβ in controlling pathological responses in the liver.
Acknowledgments
We thank Dr. Hiroshi Sakaue, Kobe University Graduate School of Medicine, Kobe, Japan for the LAP and LIP adenoviruses. We also thank Uma Pugnizthenti, University of Colorado at Denver and Health Sciences Cancer Center for performing the quantitative real-time PCR.
Footnotes
This work was supported by National Institutes of Health Grants DK59767 and P30-DK-48520 (to J. E. F.), DK-058855 and DK-072162 (to R. M.O.), HD-045962 and HD-38129 (to J. L M.), U01-DK-56047 (to T. C. B.), DK-53969 (to D. J. K.), and DK-26356 (to R. H. E.).
The abbreviations used are: PPAR, peroxisome proliferator-activated receptor; ADPH, adipophilin; AMPK, AMP-activated protein kinase; C/EBP, CCAAT/enhancer-binding protein; FAS, fatty acid synthase; LAP, liver-enriched activating protein; Leprdb/db, leptin receptor mutant mouse; LIP, liver-enriched inhibitory protein; LXR, liver X receptor; NEFA, non-esterified free fatty acids; PGC-1, peroxisomal proliferated-activated receptor γ co-activator-1; Pref-1, preadipocyte factor-1; SCD-1, stearoyl-CoA desaturase-1; SREBP, sterol response element-binding protein; TG, triglycerides; WAT, white adipose tissue; WT, wild type; RNAi, RNA interference; PBS, phosphate-buffered saline; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LDL, low density lipoprotein; shRNA, short hairpin RNA; pfu, plaque-forming units; Adv, adenovirus; GFP, green fluorescent protein.
REFERENCES
- 1.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. J. Am. Med. Assoc. 2001;286:1195–2000. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 2.Burton BT, Foster WR, Hirsch J, Van Itallie TB. Int. J. Obes. 1985;9:155–170. [PubMed] [Google Scholar]
- 3.Clark JM. J. Clin. Gastroenterol. 2006;40(Suppl. 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 4.Marchesini G, Babini M. Minerva Cardioangiol. 2006;54:229–239. [PubMed] [Google Scholar]
- 5.Collins S, Surwit RS. Recent Prog. Horm. Res. 2001;56:309–328. doi: 10.1210/rp.56.1.309. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 7.Chen G, Koyama K, Yuan X, Lee Y, Zhou YT, O’Doherty R, New-gard CB, Unger RH. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14795–14799. doi: 10.1073/pnas.93.25.14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman JM. Nutr. Rev. 1998;56:S38–S46. doi: 10.1111/j.1753-4887.1998.tb01685.x. and S54-S75 (discussion) [DOI] [PubMed] [Google Scholar]
- 9.Beaven SW, Tontonoz P. Annu. Rev. Med. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- 10.Finck BN, Kelly DP. J. Clin. Investig. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. J. Clin. Investig. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chinetti-Gbaguidi G, Fruchart JC, Staels B. Curr. Opin. Pharmacol. 2005;5:177–183. doi: 10.1016/j.coph.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Landschulz WH, Johnson PF, Adashi EY, Graves BJ, McKnight SL. Genes Dev. 1988;2:786–800. doi: 10.1101/gad.2.7.786. [DOI] [PubMed] [Google Scholar]
- 14.Williams SC, Cantwell CA, Johnson PF. Genes Dev. 1991;5:1553–1567. doi: 10.1101/gad.5.9.1553. [DOI] [PubMed] [Google Scholar]
- 15.Poli V. J. Biol. Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- 16.Ramji DP, Foka P. Biochem. J. 2002;365:561–575. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 18.Darlington GJ, Wang N, Hanson RW. Curr. Opin. Genet. Dev. 1995;5:565–570. doi: 10.1016/0959-437x(95)80024-7. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Croniger C, Arizmendi C, Harada-Shiba M, Ren J, Poli V, Hanson RW, Friedman JE. J. Clin. Investig. 1999;103:207–213. doi: 10.1172/JCI4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Shao J, Muhlenkamp P, Liu S, Klepcyk P, Ren J, Friedman JE. J. Biol. Chem. 2000;275:14173–14181. doi: 10.1074/jbc.m000764200. [DOI] [PubMed] [Google Scholar]
- 21.Arizmendi C, Liu S, Croniger C, Poli V, Friedman JE. J. Biol. Chem. 1999;274:13033–13040. doi: 10.1074/jbc.274.19.13033. [DOI] [PubMed] [Google Scholar]
- 22.Chen SS, Chen JF, Johnson PF, Muppala V, Lee YH. Mol. Cell. Biol. 2000;20:7292–7299. doi: 10.1128/mcb.20.19.7292-7299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsusue K, Gavrilova O, Lambert G, Brewer HB, Jr, Ward JM, Inoue Y, LeRoith D, Gonzalez FJ. Mol. Endocrinol. 2004;18:2751–2764. doi: 10.1210/me.2004-0213. [DOI] [PubMed] [Google Scholar]
- 24.Inoue Y, Inoue J, Lambert G, Yim SH, Gonzalez FJ. J. Biol. Chem. 2004;279:44740–44748. doi: 10.1074/jbc.M405177200. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Croniger CM, Lekstrom-Himes J, Zhang P, Fenyus M, Tenen DG, Darlington GJ, Hanson RW. J. Biol. Chem. 2005;280:38689–38699. doi: 10.1074/jbc.M503486200. [DOI] [PubMed] [Google Scholar]
- 26.Nizielski SE, Arizmendi C, Shteyngarts AR, Farrell CJ, Friedman JE. Am. J. Physiol. 1996;270:R1005–R1012. doi: 10.1152/ajpregu.1996.270.5.R1005. [DOI] [PubMed] [Google Scholar]
- 27.Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, Sellitto C, Scarpa S, Bellavia D, Lattanzio G, Bistoni F, Frati L, Cortese R, Gulino A, Ciliberto G, Costantini F, Poli V. EMBO J. 1995;14:1932–1941. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbour LA, Shao J, Qiao L, Pulawa LK, Jensen DR, Bartke A, Garrity M, Draznin B, Friedman JE. Am. J. Obstet. Gynecol. 2002;186:512–517. doi: 10.1067/mob.2002.121256. [DOI] [PubMed] [Google Scholar]
- 29.Haugen BR, Jensen DR, Sharma V, Pulawa LK, Hays WR, Krezel W, Chambon P, Eckel RH. Endocrinology. 2004;145:3679–3685. doi: 10.1210/en.2003-1401. [DOI] [PubMed] [Google Scholar]
- 30.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr Nat. Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 31.Bligh EG, Dyer WJ. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 32.Perdomo G, Commerford SR, Richard AT, Adams SH, Corkey BE, O’Doherty RM, Brown NF. J. Biol. Chem. 2004;279:27177–27186. doi: 10.1074/jbc.M403566200. [DOI] [PubMed] [Google Scholar]
- 33.Corrigan AP, Rider CC. Biochem. J. 1983;214:299–307. doi: 10.1042/bj2140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nepokroeff CM, Lakshmanan MR, Porter JW. Methods Enzymol. 1975;35:37–44. doi: 10.1016/0076-6879(75)35136-7. [DOI] [PubMed] [Google Scholar]
- 35.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 36.Bain JR, Schisler JC, Takeuchi K, Newgard CB, Becker TC. Diabetes. 2004;53:2190–2194. doi: 10.2337/diabetes.53.9.2190. [DOI] [PubMed] [Google Scholar]
- 37.Huang W, Dedousis N, Bhatt BA, O’Doherty RM. J. Biol. Chem. 2004;279:21695–21700. doi: 10.1074/jbc.M401546200. [DOI] [PubMed] [Google Scholar]
- 38.Croniger C, Trus M, Lysek-Stupp K, Cohen H, Liu Y, Darlington GJ, Poli V, Hanson RW, Reshef L. J. Biol. Chem. 1997;272:26306–26312. doi: 10.1074/jbc.272.42.26306. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka T, Yoshida N, Kishimoto T, Akira S. EMBO J. 1997;16:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smas CM, Sul HS. Crit. Rev. Eukaryotic Gene Expression. 1997;7:281–298. doi: 10.1615/critreveukargeneexpr.v7.i4.10. [DOI] [PubMed] [Google Scholar]
- 41.Stoeckman AK, Towle HC. J. Biol. Chem. 2002;277:27029–27035. doi: 10.1074/jbc.M202638200. [DOI] [PubMed] [Google Scholar]
- 42.Rao MS, Reddy JK. Hepatology. 2004;40:783–786. doi: 10.1002/hep.20453. [DOI] [PubMed] [Google Scholar]
- 43.Rahimian R, Masih-Khan E, Lo M, van Breemen C, McManus BM, Dube GP. Mol. Cell. Biochem. 2001;224:29–37. doi: 10.1023/a:1011927113563. [DOI] [PubMed] [Google Scholar]
- 44.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. J. Biol. Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 45.Wu Z, Bucher NL, Farmer SR. Mol. Cell. Biol. 1996;16:4128–4136. doi: 10.1128/mcb.16.8.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M. Diabetes. 2002;51:3486–3491. doi: 10.2337/diabetes.51.12.3486. [DOI] [PubMed] [Google Scholar]
- 47.Benton CR, Koonen DP, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Heikkila JJ, Bonen A. J. Physiol. (Lond) 2006;573:199–210. doi: 10.1113/jphysiol.2006.106013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. J. Biol. Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 49.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. J. Biol. Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- 50.McManaman JL, Zabaronick W, Schaack J, Orlicky DJ. J. Lipid Res. 2003;44:668–673. doi: 10.1194/jlr.C200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Sztalryd C, Bell M, Lu X, Mertz P, Hickenbottom S, Chang BH, Chan L, Kimmel AR, Londos C. J. Biol. Chem. 2006;281:34341–34348. doi: 10.1074/jbc.M602497200. [DOI] [PubMed] [Google Scholar]
- 52.Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, Soukas AA, Kahn CR, Ntambi JM, Socci ND, Friedman JM. J. Clin. Investig. 2004;113:414–424. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calkhoven CF, Muller C, Leutz A. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- 54.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 55.Duong DT, Waltner-Law ME, Sears R, Sealy L, Granner DK. J. Biol. Chem. 2002;277:32234–32242. doi: 10.1074/jbc.M204873200. [DOI] [PubMed] [Google Scholar]
- 56.Shao J, Qiao L, Janssen RC, Pagliassotti M, Friedman JE. Diabetes. 2005;54:976–984. doi: 10.2337/diabetes.54.4.976. [DOI] [PubMed] [Google Scholar]
- 57.Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. J. Biol. Chem. 2004;279:47898–47905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- 58.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. J. Clin. Investig. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 60.Kim JKOG, Chen Y, Reitman ML, Shulman GI. J. Biol. Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 61.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Nat. Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- 62.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B, Jr, Reitman ML, Gonzalez FJ. J. Clin. Investig. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. J. Biol. Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 64.Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. Am. J. Physiol. Endocrinol. Metab. 2005;288:1195–1205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- 65.Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, Koonen DP, Glatz JF, Luiken JJ. Proc. Nutr. Soc. 2004;63:245–249. doi: 10.1079/PNS2004331. [DOI] [PubMed] [Google Scholar]
- 66.Sugden MC, Bulmer K, Gibbons GF, Knight BL, Holness MJ. Biochem. J. 2002;364:361–368. doi: 10.1042/BJ20011699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC, Berge RK, Staels B. J. Biol. Chem. 2000;275:16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- 68.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 69.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, Gotoda T, Ishibashi S, Yamada N. J. Biol. Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, Ren S, Evans RM, Xie W. J. Biol. Chem. 2006;281:15013–15020. doi: 10.1074/jbc.M511116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishii S, Iizuka K, Miller BC, Uyeda K. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lund EG, Peterson LB, Adams AD, Lam MH, Burton CA, Chin J, Guo Q, Huang S, Latham M, Lopez JC, Menke JG, Milot DP, Mitnaul LJ, Rex-Rabe SE, Rosa RL, Tian JY, Wright SD, Sparrow CP. Biochem. Pharmacol. 2006;71:453–463. doi: 10.1016/j.bcp.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 73.Vega RB, Huss JM, Kelly DP. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin J, Handschin C, Spiegelman BM. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 76.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. J. Biol. Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 77.Qiao L, MacLean PS, You H, Schaack J, Shao J. Endocrinology. 2006;147:3060–3069. doi: 10.1210/en.2005-1507. [DOI] [PubMed] [Google Scholar]
- 78.Miyazaki M, Dobrzyn A, Sampath H, Lee SH, Man WC, Chu K, Peters JM, Gonzalez FJ, Ntambi JM. J. Biol. Chem. 2004;279:35017–35024. doi: 10.1074/jbc.M405327200. [DOI] [PubMed] [Google Scholar]
- 79.Tang QQ, Otto TC, Lane MD. Proc. Natl. Acad. Sci. U. S. A. 2003;100:850–855. doi: 10.1073/pnas.0337434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Millward CA, Heaney JD, Sinasac DS, Chu EC, Bederman IR, Gilge DA, Previs SF, Croniger CM. Diabetes. 2007;56:161–167. doi: 10.2337/db06-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen J, Newton SS, Zeng L, Adams DH, Dow AL, Madsen TM, Nestler EJ, Duman RS. Neuropsychopharmacology. 2004;29:23–31. doi: 10.1038/sj.npp.1300289. [DOI] [PubMed] [Google Scholar]
- 82.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. Nat. Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- 83.Abbud RA, Kelleher R, Melmed S. Endocrinology. 2004;145:867–880. doi: 10.1210/en.2003-0897. [DOI] [PubMed] [Google Scholar]
- 84.Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. J. Biol. Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 85.Chen H, Pan YX, Dudenhausen EE, Kilberg MS. J. Biol. Chem. 2004;279:50829–50839. doi: 10.1074/jbc.M409173200. [DOI] [PubMed] [Google Scholar]
- 86.Chitturi S, Farrell GC. Semin. Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 87.Perseghin G, Petersen K, Shulman GI. Int. J. Obes. Relat. Metab. Disord. 2003;27(Suppl. 3):6–11. doi: 10.1038/sj.ijo.0802491. [DOI] [PubMed] [Google Scholar]
- 88.Boden G. Curr. Diab. Rep. 2006;6:177–181. doi: 10.1007/s11892-006-0031-x. [DOI] [PubMed] [Google Scholar]