

Abstract
The immunosuppressive action of the calcineurin inhibitor cyclosporine A (CsA) stems from the inhibition of nuclear factor of activated T cells (NFAT) signaling in T cells. CsA is also used for the treatment of proteinuric kidney diseases. As it stands, the antiproteinuric effect of CsA is attributed to its immunosuppressive action. Here we show that the beneficial effect of CsA on proteinuria is not dependent on NFAT inhibition in T cells, but rather results from the stabilization of the actin cytoskeleton in kidney podocytes. CsA blocks the calcineurin-mediated dephosphorylation of synaptopodin, a regulator of Rho GTPases in podocytes, thereby preserving the phosphorylation-dependent synaptopodin–14-3-3β interaction. Preservation of this interaction, in turn, protects synaptopodin from cathepsin L–mediated degradation. These results represent a new view of calcineurin signaling and shed further light on the treatment of proteinuric kidney diseases. Novel calcineurin substrates such as synaptopodin may provide promising starting points for antiproteinuric drugs that avoid the serious side effects of long-term CsA treatment.
The kidney glomerulus is a highly specialized structure that ensures the selective ultrafiltration of plasma so that essential proteins are retained in the blood. The common denominator in a variety of kidney diseases, including minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS), is podocyte dysfunction involving a massive loss of protein in the urine (proteinuria)1,2. Dynamic regulation of the podocyte actin cytoskeleton is vital to normal kidney filter function, and mutations affecting several podocyte proteins lead to the rearrangement of the actin cytoskeleton and subsequent proteinuria3. Proteins regulating podocyte actin dynamics are therefore crucial for sustained glomerular filter function4.
The actin-binding protein synaptopodin, which is highly expressed in podocytes5, exists in three isoforms, neuronal Synpo-short, renal Synpo-long and Synpo-T6. Synaptopodin-mutant mice lacking Synpo-short and Synpo-long upregulate Synpo-T protein expression in podocytes, thereby rescuing kidney filter function during development6. They do, however, suffer from prolonged proteinuria when challenged with lipopolysaccharide (LPS)6. Synaptopodin is a key regulator of podocyte function because bigenic heterozygosity for synaptopodin and CD2-associated protein results in proteinuria and FSGS-like glomerular damage7. Mechanistically, synaptopodin induces stress fibers by stabilizing the GTPase RhoA8 and suppresses filopodia by disrupting cell division cycle-42–insulin receptor substrate p53–Mena signaling complexes9.
The serine/threonine phosphatase calcineurin is ubiquitously expressed in all mammalian tissues10. The best characterized function of calcineurin is the regulation of NFAT signaling. The immunosuppressive action of the calcineurin inhibitor CsA stems from the inhibition of NFAT signaling in T cells11. In other cell types, calcineurin-NFAT signaling can also cause cardiac hypertrophy, skeletal muscle differentiation or changes in synaptic plasticity11. In addition, calcineurin-NFAT signaling is crucially involved in angiogenesis10, pancreatic beta cell function12 and hair growth13. Likewise, inhibition of calcineurin-NFAT signaling in bone explains the bone loss often experienced during immunosuppressant therapy that targets calcineurin14.
Notably, CsA can induce a remission of proteinuria caused by such diseases as MCD and FSGS15. T cell dysfunction is associated with some forms of proteinuria, including a subset of MCD in children. Therefore, it has been assumed that T cells act on podocytes to cause proteinuria and that the antiproteinuric effect of CsA results from the inhibition of NFAT signaling in T cells15. As it stands, experimental support of this hypothesis is missing. CsA can also reduce proteinuria in human16 and experimental17 Alport’s syndrome, a nonimmunological disease, raising doubts about the above hypothesis. Moreover, LPS-induced proteinuria can develop independently of T cells18, and mice lacking synaptopodin show impaired recovery from LPS-induced proteinuria6. Collectively, these data argue for a direct effect of calcineurin and its inhibitor CsA on podocytes. Here we show that the antiproteinuric effect of CsA is independent of its immunosuppressive function in T cells and results directly from the stabilization of the podocyte actin cytoskeleton. In particular, we show that calcineurin regulates podocyte actin dynamics by dephosphorylating synaptopodin, thereby abrogating the 14-3-3–mediated protection of synaptopodin from cathepsin L (CatL)-mediated proteolysis.
RESULTS
Synaptopodin is a 14-3-3 and calcineurin-binding protein
14-3-3 proteins are chaperone-like phospho–serine/threonine–binding proteins that can alter the structure or function of target proteins19. Human Synpo-short and Synpo-long contain two consensus 14-3-3–binding motifs (amino acids 212–218: RAAATTP, and amino acids 615–621: RPSRSSP; Fig. 1a). Using a yeast two-hybrid screen6, we found that 14-3-3β and 14-3-3η bound synaptopodin. In the kidney, 14-3-3 colocalized with synaptopodin in podocytes (Fig. 1b). In differentiated cultured podocytes, 14-3-3 colocalized with synaptopodin along stress fibers, and both proteins remained associated after disruption of stress fibers by latrunculin A (Fig. 1c). Biochemically, GST–14-3-3β specifically bound synaptopodin from isolated glomeruli (Fig. 1d), and glomerular 14-3-3β precipitated with endogenous synaptopodin (Fig. 1e). To map 14-3-3 interaction sites in synaptopodin, we focused on the two 14-3-3–binding sites (Fig. 1a) that fit the mode 2 consensus 14-3-3–binding motif (RXXXpSXP or RXXXpTXP)20. We hypothesized that Thr216 and Ser619 are phosphoacceptor sites within the first (RAAApTTP) and the second (RPSRpSSP) motif, respectively, whose phosphorylation is required for 14-3-3 binding. To test this hypothesis, we expressed Flag-synaptopodin proteins with GFP–14-3-3β in HEK293 cells and conducted coimmunoprecipitation studies. Wild-type synaptopodin and phosphomimetic Synpo-ED (containing T216E and S619D mutations) interacted with 14-3-3β (Fig. 1f). In contrast, the destruction of the phosphoacceptor sites by alanine substitution (T216A and S619A; Synpo-AA) abrogated 14-3-3β binding (Fig. 1f).
Figure 1.

Synaptopodin specifically interacts with 14-3-3. (a) Schematic of synaptopodin isoforms. The white box shows the first 676 amino acids that are shared between Synpo-long and Synpo-short. This fragment contains two 14-3-3 binding motifs (black ovals) and two CatL cleavage sites (arrows). (b) In the adult mouse kidney, 14-3-3β colocalizes with synaptopodin in podocytes. Scale bar, 30 μm. (c) In differentiated cultured podocytes, 14-3-3β colocalizes with synaptopodin along stress fibers (top), and both proteins remain associated after disruption of actin filaments with latrunculin A (bottom). Scale, 25 μm. (d) Synaptopodin from isolated mouse glomerular extracts (input) specifically binds GST–14-3-3β but not GST alone. (e) Coimmunoprecipitation experiments show that endogenous synaptopodin interacts with endogenous 14-3-3β in isolated mouse glomeruli. An antibody to GFP serves as negative control. IP, immunoprecipitation. (f) GFP–14-3-3β precipitates with wild-type (WT) Flag-synaptopodin and phosphomimetic Flag–Synpo-ED but not with phosphoresistant Flag–Synpo-AA or Flag-raver (control).
Myopodin, another member of the synaptopodin gene family, binds the catalytic calcineurin subunits CnAα and CnAβ21. Similarly, GFP–Synpo-alt, the C-terminal fragment of Synpo-long not present in Synpo-short (Fig. 1a), precipitated with CnAα from co-transfected HEK293 cells, whereas both GFP–Synpo-short and GFP–Synpo-alt interacted with constitutively active calcineurin (aCnA)22 (Fig. 2a). Furthermore, synaptopodin could be immunoprecipitated with endogenous CnAα from isolated glomeruli (Fig. 2b), and calcineurin colocalized with synaptopodin in podocytes in the kidney and in culture (Fig. 2c).
Figure 2.

Identification of synaptopodin as calcineurin binding protein. (a) GFP–Synpo-short and GFP–Synpo-alt precipitate with Flag-tagged aCnA from co-transfected HEK293 cells. GFP–Synpo-alt can also interact with WT calcineurin. No binding is found with Flag-raver (control). (b) Endogenous coimmunoprecipitation experiments show that synaptopodin interacts with calcineurin in isolated mouse glomeruli. An antibody specific for GFP serves as negative control. (c) In the adult mouse kidney (top) and differentiated cultured podocytes (bottom), calcineurin partially colocalizes with synaptopodin. Scale bars, 30 μm (top panels) and 25 μm (bottom panels).
Phosphorylation of synaptopodin and binding to 14-3-3
Similar to the vast majority of 14-3-3–interacting proteins23, myopodin is a phosphoprotein that binds to 14-3-3β in a serine/threonine phosphorylation–dependent fashion24. To test whether synaptopodin is also a phosphoprotein, we incubated purified Flag–Synpo-short from transfected HEK293 cells with lambda protein phosphatase (λ-PPase). As shown by SDS-PAGE and western blot analysis, λ-PPase reduced the molecular size of synaptopodin (Fig. 3a), thereby showing that synaptopodin is phosphorylated in vivo. Consensus 14-3-3–binding motifs serve as substrates for serine/threonine protein kinases25 including protein kinase A (PKA) and calcium-dependent protein kinase II (CaMKII)26, two kinases that can phosphorylate the 14-3-3–binding motifs of myopodin21. To test whether PKA and CaMKII can phosphorylate synaptopodin, we dephosphorylated purified wild-type Flag–Synpo-short and Flag–Synpo-AA with λ-PPase and incubated it with PKA or CaMKII in the presence of [γ32P]ATP. 32P labeling of wild-type synaptopodin by PKA was first detected after 2 min and increased over time (Fig. 3b). In contrast, 32P labeling of Synpo-AA was strongly reduced (Fig. 3b). 32P labeling of synaptopodin by CaMKII was first detected after 15 min and increased thereafter (Fig. 3b). In contrast, CaMKII caused virtually no 32P labeling of Synpo-AA (Fig. 3b). To further explore the phosphorylation of synaptopodin by PKA and CaMKII, we transfected HEK293 cells with Flag–Synpo-short and incubated them in 32P-containing medium for 2 h. After immunoprecipitation, Flag–Synpo-short showed strong 32P labeling, which was markedly reduced in the presence of the PKA inhibitor H89 or the CaMKII inhibitor KN62 (Fig. 3c). Combined inhibition of both kinases further decreased the 32P labeling of synaptopodin (Fig. 3c).
Figure 3.
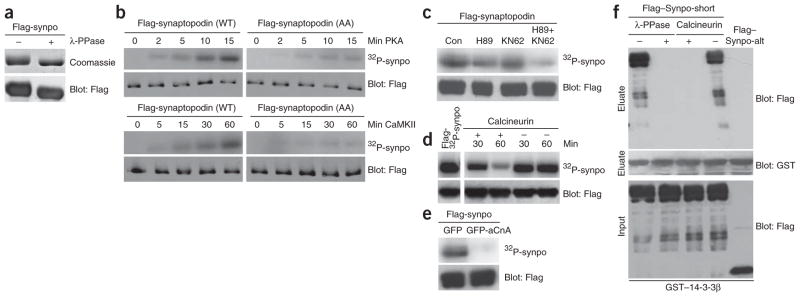
The synaptopodin–14-3-3 interaction is antagonistically regulated by PKA, CaMKII and calcineurin. (a) SDS-PAGE (top) and western blot (bottom) analyses show a reduced molecular weight of purified Flag-synaptopodin (Flag-Synpo) after dephosphorylation with λ-PPase. (b) Time-dependent phosphorylation of purified Flag–Synpo-short by PKA (top panels) and CaMKII (bottom panels). Alanine substitution of Thr216 and Ser619 (AA) strongly reduces 32P labeling when compared to WT synaptopodin. Flag blots show equal protein loading. (c) Phosphorylation of synaptopodin by PKA and CaMKII in HEK293 cells cultured in the absence of inhibitors (Con), KN62 and H89. (d) Time-dependent dephosphorylation of purified 32P-labeled Flag–Synpo-short by calcineurin (left) or in its absence (right). (e) Dephosphorylation of 32P–Flag–Synpo-short in HEK293 cells by calcineurin. (f) Dephosphorylation of purified Flag–Synpo-short by λ-PPase or calcineurin abrogates the binding of synaptopodin to immobilized GST–14-3-3β. Flag–Synpo-alt does not bind 14-3-3 and serves as negative control.
Consensus 14-3-3–binding motifs can be dephosphorylated by serine/threonine protein phosphatases, including calcineurin26. Calcineurin dephosphorylates myopodin, thereby abrogating the myopodin–14-3-3 interaction21. To test whether synaptopodin is also a substrate of calcineurin, we used PKA to label purified Flag–Synpo-short with 32P before incubation for 30 or 60 min in the presence or absence of calcineurin. 32P-synaptopodin abundance decreased over time only in the presence of calcineurin (Fig. 3d). To further test whether calcineurin can dephosphorylate synaptopodin, we expressed Flag–Synpo-short in HEK293 cells with dominant active GFP-aCnA20 in the presence of 32P followed by immunoprecipitation with an antibody to Flag. 32P labeling of Flag–Synpo-short immunoprecipitated from cells expressing GFP-aCnA was almost completely abolished when compared to cells expressing GFP (Fig. 3e). Next, we examined the ability of purified Flag–Synpo-short to bind immobilized GST–14-3-3β before and after dephosphorylation. Flag–Synpo-alt, which cannot bind 14-3-3β (data not shown), served as a negative control. In the absence of phosphatase treatment, synaptopodin strongly and specifically bound to immobilized GST–14-3-3β (Fig. 3f). In contrast, the dephosphorylation of Flag–Synpo-short with λ-PPase or calcineurin abrogated the binding of synaptopodin to 14-3-3β (Fig. 3f).
Synaptopodin-14-3-3 binding maintains stress fibers
Synaptopodin, which is required for stress fiber formation in podocytes6,8, can be phosphorylated by PKA and CaMKII (Fig. 3b,c) and dephosphorylated by calcineurin (Fig. 3d,e). Therefore, we tested whether the pharmacological inhibition of calcineurin, PKA or CaMKII modulates podocyte actin dynamics. Inhibition of calcineurin with CsA increased stress fiber content and synaptopodin expression (Supplementary Fig. 1a online) without inducing podocyte apoptosis (Supplementary Table 1 online). The effect on stress fiber content was mediated by synaptopodin, because it did not occur in synaptopodin-knockdown podocytes6. The PKA inhibitor H89 and, more strongly, the combined application of H89 and the CaMKII inhibitor KN62 caused the loss of stress fibers and synaptopodin expression, which could be rescued by CsA (Supplementary Fig. 1a). Similarly, the transfection of differentiated podocytes with yellow fluorescence protein–tagged difopein, a specific inhibitor of 14-3-3–ligand interactions27, caused the loss of stress fibers and synaptopodin expression in transfected podocytes but not in neighboring nontransfected cells (Supplementary Fig. 1b).
Synaptopodin proteolysis by CatL and protection by 14-3-3
Upregulation of CatL activity in podocytes is associated with the development of proteinuria28,29. Glomerular synaptopodin is highly unstable, leading to the generation of a 44-kDa fragment5. Synaptopodin contains two high-scoring CatL cleavage sites30 (amino acids 422–426: ALGAE and amino acids 612–61: ALPRPS), the first of which predicts the generation of an N-terminal 44-kDa fragment (Fig. 1a). Therefore, we tested whether CatL can cleave purified Flag-synaptopodin. At pH 4.5, which reflects a lysosomal milieu, we found a concentration-dependent complete degradation of synaptopodin (Fig. 4a). At pH 6.5, we observed a dose-dependent accumulation of the 44-kDa fragment (Fig. 4a). At pH 7.0, which allows for cytosolic-like CatL activity29, we found a partial degradation of synaptopodin (Fig. 4a).
Figure 4.

14-3-3β, E64 and CsA block the CatL-mediated degradation of synaptopodin. (a) Dose-dependent degradation of purified Flag–Synpo-short by CatL at pH 4.5, 6.5 or 7.0 leads to the generation of a 44-kDa fragment. (b) H89-mediated reduction of stress fibers and synaptopodin abundance are prevented by the cathepsin inhibitor E64 (right, top). Similarly, E64 prevents the loss of synaptopodin expression and stress fibers caused by simultaneous inhibition of PKA and CaMKII (right, bottom). Scale bar, 25 μm. (c) Western blot analysis of synaptopodin steady-state abundance in differentiated WT podocytes. In control cells (Con) the 110-kDa full-length protein is visible. Inhibition of PKA and CaMKII (H89 + KN62) causes a partial degradation of full-length synaptopodin. Treatment with CsA, E64 or E64D blocks the H89- and KN62-mediated degradation of synaptopodin. Immunoblotting for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) shows equal protein loading. (d) Binding of 14-3-3β but not of α-actinin-4 partially protects synaptopodin against proteolytic processing by CatL (left). 14-3-3β (middle) or α-actinin-4 (right) are not cleaved by CatL. (e) Synaptopodin contains two evolutionarily conserved CatL cleavage sites. The first motif (ALGAE) is conserved from fish to humans (top). The second motif (ALPRPS) is preserved from mice to humans (bottom). (f) Site-directed mutagenesis of CatL cleavage sites separately (CM1 or CM2) or together (CM1+2) increases resistance of synaptopodin against CatL-mediated proteolytic processing. Flag-synaptopodin and its mutant forms were purified from HEK293 cells (left) and incubated with CatL at pH 7.0 (right).
To test whether decreased synaptopodin expression after protein kinase inhibition (Supplementary Fig. 1a) resulted from CatL-mediated cleavage, we repeated the kinase inhibition experiment in the presence of the cathepsin inhibitor E64, which prevented the H89-and KN62-induced loss of stress fibers and synaptopodin expression (Fig. 4b). Next, we monitored synaptopodin steady-state protein levels in the presence or absence of H89 and KN62 with or without CsA, E64 or E64D (a specific inhibitor of CatL). Full-length synaptopodin abundance was decreased by combined application of H89 and KN62 and could be restored by CsA, E64 or E64D (Fig. 4c).
In plants, 14-3-3 can increase the steady-state protein levels of its interacting partners by blocking their cleavage30. To test whether 14-3-3 binding protects synaptopodin from proteolytic degradation by CatL in a similar manner, we digested purified Flag–Synpo-short at pH 7.0 with CatL in the presence or absence of purified Flag–14-3-3β or Flag–α-actinin-4, another synaptopodin-binding protein6. The cleavage of synaptopodin by CatL resulted in a decrease in the amount of full-length synaptopodin and the appearance of the predicted 44-kDa fragment (Fig. 4d). We detected protection of full-length synaptopodin in the presence of 14-3-3β but not α-actinin-4 (Fig. 4d). To test whether 14-3-3β conferred true protection instead of simply competing with synaptopodin as a substrate of CatL, we separately incubated 14-3-3β and α-actinin-4 with CatL. Of note, neither 14-3-3β nor α-actinin-4 was cleaved by CatL (Fig. 4d).
The first CatL cleavage site of synaptopodin (amino acids 422–426: ALGAE) is conserved phylogenetically from fish to humans, the second (amino acids 612–617: ALPRPS) is conserved among mammals (Fig. 4e). To explore the function of these sites, we mutated them individually (CM1: AQDSE; CM2: ARTEPS) or in combination (CM1+2) and determined the effect on the proteolytic processing by CatL (Fig. 4f). We detected no degradation in the absence of CatL (Fig. 4f). The mutation of each cleavage site individually conferred partial resistance against proteolysis, and the strongest protection was found for Synpo-CM1+2 (Fig. 4f). Therefore, we concluded that the mutation of the CatL cleavage sites stabilizes synaptopodin steady-state protein levels and hypothesized that the expression of such a protected synaptopodin mutant in podocytes would preserve stress fibers. In keeping with this hypothesis, Synpo-CM1+2 protected podocytes against H89- and KN62-induced or LPS-induced18 loss of stress fibers (Supplementary Fig. 2 online).
The degradation of synaptopodin by CatL is facilitated by the calcineurin-mediated dephosphorylation of synaptopodin, raising the possibility that synaptopodin is a direct target of the antiproteinuric effect of CsA15 and E6432. As shown before18, LPS (n = 6; 484.89 ± 205.46 micrograms albumin per milligram creatinine) but not PBS (n = 6; 74.67 ± 31.13 micrograms albumin per milligram creatinine) caused proteinuria in SCID mice that are devoid of T and B cells33 (P < 0.001; Fig. 5a). The LPS-induced proteinuria was significantly reduced by treatment with CsA (n = 10, 173.87 ± 74.24 micrograms albumin per milligram creatinine; P <0.001) or E64 (n = 8; 150.46 ± 67.27 micrograms albumin per milligram creatinine; P < 0.001) after LPS injection (Fig. 5a). Western blot analysis of isolated glomeruli showed that LPS caused a nearly complete loss of full-length synaptopodin and its downstream effector RhoA8, which could be blocked by CsA or E64 (Fig. 5b).
Figure 5.

CsA and E64 ameliorate LPS-induced proteinuria by blocking the CatL-mediated degradation of synaptopodin. (a) SDS-PAGE analysis of urines from control and LPS-injected mice (top). Quantitative analysis of albuminuria (bottom). (b) Western blot analysis of isolated glomeruli showing that LPS causes the degradation of endogenous synaptopodin and RhoA. CsA or E64 block the LPS-induced degradation of synaptopodin and RhoA. GAPDH shows equal protein loading. (c) Immunoblot analysis of Flag-synaptopodin proteins from isolated glomeruli (left) and liver extracts (right) 48 h after in vivo gene delivery. Glomerular RhoA levels are positively correlated with Flag-synaptopodin abundance. (d) Expression of Flag–Synpo-ED in podocytes after in vivo gene delivery as detected by double-labeling deconvolution microscopy with antibodies to Flag and the podocyte marker podocin. No Flag signal is seen in podocytes of delivery solution–injected mice (control). Scale bar, 50 μm. (e) Gene transfer of Synpo-CM1+2 or Synpo-ED but not WT synaptopodin, Synpo-AA or delivery solution (control) protects against LPS-induced proteinuria. NS, not significant. (f) Immunoblot analysis of synaptopodin protein expression after LPS injection and gene delivery. LPS causes degradation and decrease of synaptopodin (LPS + Con) when compared to PBS-injected mice (Con). High levels of synaptopodin can be found in LPS-injected mice after gene transfer of Synpo-CM1+2 or Synpo-ED but not of WT synaptopodin or Synpo-AA (left). Synaptopodin protein in the liver is not affected by LPS (right). Mice that did not receive synaptopodin cDNA (Con, Con + LPS) do not express synaptopodin in the liver. *P < 0.05; **P < 0.001; ***P < 0.0005.
Genetic overexpression of synaptopodin prevents proteinuria
To test whether the restoration of synaptopodin protein abundance in mice protects against LPS-induced proteinuria, we injected wild-type mice with LPS after in vivo gene delivery9,29 of Flag-synaptopodin. Delivery solution alone served as a negative control. First, we monitored Flag-synaptopodin protein abundance in the absence of LPS by immunoblotting (Fig. 5c). At 48 h after gene delivery, we detected the recombinant Flag-synaptopodin proteins in extracts from isolated glomeruli and livers. Whereas the protein abundance of wild-type synaptopodin and its mutant forms were comparable in the liver, they were markedly different in glomeruli (Fig. 5c). Synpo-CM1+2 and Synpo-ED showed the highest expression, whereas wild-type synaptopodin and Synpo-AA expression was much lower and comparable to endogenous synaptopodin in control mice (Fig. 5c). Notably, the delivered Flag-synaptopodin proteins were functional, because they increased glomerular amounts of RhoA (Fig. 5c). Hence, synaptopodin undergoes a constant physiological turnover via CatL-mediated degradation, even in the absence of LPS. In keeping with the biochemical results, we detected Flag–Synpo-ED in kidney cells including podocytes, as revealed by double-labeling deconvolution microscopy with the podocyte marker podocin34 (Fig. 5d).
Next, we conducted LPS studies after in vivo gene delivery of Flag-synaptopodin constructs or delivery solution serving as a negative control (Fig. 5e). The expression and activity of recombinant Flag-synaptopodin was monitored by immunoblotting (Fig. 5f). As expected9,18, no significant proteinuria was detected in PBS-injected control mice receiving delivery solution (n = 6; 0 h, 47.56 ± 8.55 micrograms albumin per milligram creatinine versus 48 h, 48.72 ± 11.89 micrograms albumin per milligram creatinine; not significant). In contrast, significant proteinuria developed in LPS-injected mice receiving delivery solution (n = 5, 400.44 ± 149.42 micrograms albumin per milligram creatinine; P < 0.0005). LPS-induced proteinuria was significantly reduced by gene transfer of Synpo-CM1+2 (n = 8, 165.04 ± 151.57 micrograms albumin per milligram creatinine; P < 0.05) or Synpo-ED (n = 6; 195.52 ± 126.90 micrograms albumin per milligram creatinine; P < 0.05) but not by wild-type synaptopodin (n = 7, 600.12 ± 280.35 micrograms albumin per milligram creatinine; not significant) or Synpo-AA (n = 8, 397.89 ± 107.77 micrograms albumin per milligram creatinine; P <0.05; not significant) (Fig. 5e). Biochemically, we detected comparable amounts of all Flag-synaptopodin proteins and endogenous RhoA in liver extracts (Fig. 5f). In contrast, in isolated glomeruli, expression of Synpo-CM1+2 and Synpo-ED was preserved after LPS treatment, whereas wild-type synaptopodin and Synpo-AA showed a nearly complete loss (Fig. 5f). The preservation of Synpo-CM1+2 or Synpo-ED expression was associated with the preservation of glomerular RhoA expression (Fig. 5f), thereby confirming that the delivered recombinant proteins were functional.
The large GTPase dynamin is another target of CatL, and gene transfer of CatL-resistant dynamin also protects against LPS-induced proteinuria29, suggesting a functional link between synaptopodin and dynamin. Therefore, we analyzed the above-mentioned protein extracts (Fig. 5b,f) for dynamin. CsA and E64 preserved not only synaptopodin and RhoA (Fig. 5b) but also dynamin steady-state protein levels (Supplementary Fig. 3a online). Similar to RhoA (Fig. 5f), Synpo-CM1+2 or Synpo-ED gene transfer was also sufficient to stabilize dynamin (Supplementary Fig. 3b). In light of another report suggesting that CsA interferes with the expression of the tight junction protein ZO-1 in podocytes35, we analyzed the protein extracts for ZO-1 (Fig. 5f). CsA and E64 (Supplementary Fig. 3a) or gene transfer of Synpo-CM1+2 and Synpo-ED stabilized ZO-1 (Supplementary Fig. 3b), thereby suggesting that the effect of CsA on ZO-1 expression is indirect via the stabilization of synaptopodin. In keeping with the biochemical results (Fig. 4d), the expression of α-actinin-4 was not affected by LPS, CsA, E64 or gene transfer (Supplementary Fig. 3). Taken together, expression of calcineurin-resistant Synpo-ED or CatL-resistant Synpo-CM1+2 stabilizes glomerular RhoA, dynamin and ZO-1 steady-state levels, thereby protecting against LPS-induced proteinuria. These experiments also show that synaptopodin protein levels in the liver are stable and not affected by LPS.
To further confirm the results of the gene delivery studies in another in vivo setting (Fig. 5), we created a podocyte-specific and tetracycline-inducible transgenic mouse line expressing GFP–Synpo-CM1+2 by crossing podocin-rtTA mice36 with a tetO-GFP-Synpo-CM1+2 strain (Fig. 6a). We gave the resulting double-transgenic offspring doxycycline or vehicle solution for 7 d followed by LPS injection36. Biochemically, synaptopodin remained detectable after LPS treatment (Fig. 6b). The preservation of synaptopodin expression was associated with the preservation of endogenous RhoA, dynamin and ZO-1 protein levels after LPS treatment (Fig. 6b) and protected against LPS-induced proteinuria (Fig. 6c). Significant proteinuria developed in vehicle-treated control mice (n = 4; 0 h, 41.23 ± 15.66 micrograms albumin per milligram creatinine versus 48 h, 327.65 ± 114.65 micrograms albumin per milligram creatinine; P < 0.005) but not in doxycycline-treated, Synpo-CM1+2–expressing mice (n = 5; 0 h, 36.54 ± 57.95 micrograms albumin per milligram creatinine versus 48 h, 62.00 ± 54.78 micrograms albumin per milligram creatinine; not significant). At 48 h, we also detected a significant difference (P < 0.005) between both groups (Fig. 6c).
Figure 6.

Expression of Synpo-CM1+2 in podocytes protects against proteinuria, whereas activation of calcineurin in podocytes causes proteinuria. (a) Schematic of constructs used for the generation of Synpo-CM1+2–transgenic mice. rTetR, reverse Tet repressor; VP16, herpes simplex VP16 protein; tetO, tet operator sequences; pCMV, cytomegalovirus promoter. (b) Immunoblot analysis of isolated glomeruli from doxycycline (Dox)- or vehicle (Veh)-treated double-transgenic mice. After LPS injection, synaptopodin protein abundance remains stable in Dox-treated but not in Veh-treated mice. Protein abundance of RhoA, dynamin and ZO-1 are positively correlated with synaptopodin abundance. GAPDH shows equal protein loading. (c) GFP–Synpo-CM1+2 (Dox +LPS) protects against LPS-induced proteinuria. (d) Schematic of constructs used for the generation of aCnA-transgenic mice. (e) Immunoblot analysis of isolated glomeruli showing reduced steady-state protein levels of synaptopodin, RhoA, dynamin and ZO-1 but not of α-actinin-4 in GFP-aCnA–expressing (Dox) mice. (f) Detection of proteinuria in GFP-aCnA–expressing (Dox) but not in control (Veh) mice. (g) Model for the regulation of podocyte function by calcineurin. Phosphorylation of synaptopodin by PKA or CaMKII promotes 14-3-3 binding, which protects synaptopodin against CatL-mediated cleavage, thereby contributing to the intact glomerular filtration barrier. Dephosphorylation of synaptopodin by calcineurin (CaN) abrogates the interaction with 14-3-3. This renders the CatL cleavage sites of synaptopodin accessible and promotes the degradation of synaptopodin. CsA and E64 safeguard against proteinuria by stabilizing synaptopodin steady-state protein levels in podocytes. *P < 0.005.
Podocyte-specific expression of aCnA causes proteinuria
The inhibition of calcineurin by CsA protected against LPS-induced proteinuria (Fig. 5a) and prevented the degradation of synaptopodin (Fig. 5b). Additionally, calcineurin caused the dephosphorylation of synaptopodin and the loss of 14-3-3 binding (Fig. 3). Therefore, we hypothesized that the activation of calcineurin in podocytes should have an LPS-like effect leading to the reduction of synaptopodin steady-state protein levels and to the disruption of glomerular barrier function. To test this hypothesis, we created a tetracycline-inducible36 transgenic mouse line expressing aCnA22, a constitutively active form of calcineurin, as a GFP fusion protein specifically in podocytes in a manner similar to the experiments described in the previous paragraph (Fig. 6d). The resulting double-transgenic offspring were administered doxycycline or vehicle36. Similarly to LPS injection (Fig. 5 and Supplementary Fig. 3), the induction of GFP-aCnA expression in podocytes caused the degradation of endogenous synaptopodin and the downregulation of RhoA, dynamin and ZO-1 (Fig. 6e). Most notably, the mice expressing the transgene (doxycycline-treated) but not the control mice (vehicle-treated) developed significant proteinuria (vehicle-treated: n = 10; 51.40 ± 19.47 micrograms albumin per milligram creatinine versus doxycycline-treated: n = 15; 275.62 ± 191.42 micrograms albumin per milligram creatinine; P < 0.005) (Fig. 6f).
DISCUSSION
Our data unveil a new calcineurin signaling pathway that operates in podocytes and is essential for the maintenance of kidney filter function (Fig. 6g). In contrast to most other calcineurin-controlled signaling events10–13, the antiproteinuric effect of CsA described here does not result from the inhibition of NFAT signaling. Instead, CsA blocks the calcineurin-mediated dephosphorylation of the actin-organizing protein synaptopodin6,8,9. Our data also show that activation of calcineurin in the podocyte is sufficient to cause proteinuria via the degradation of synaptopodin. Our findings also identify synaptopodin as a substrate of PKA and CaMKII. Together with calcineurin, these kinases control the phosphorylation state of synaptopodin, thereby regulating 14-3-3 binding and CatL-mediated proteolysis (Fig. 6g). Although it was previously shown that 14-3-3 binding can increase protein steady-state levels31,37, to our knowledge, the present study is the first to provide mechanistic insight into how 14-3-3 binding can stabilize target proteins. Furthermore, our results unveil a new role of cytoplasmic CatL under not only pathological29 but also physiological conditions. Collectively, our data show that the expression of CatL-resistant synaptopodin by in vivo gene transfer or by an inducible transgene in podocytes is sufficient to protect against LPS-induced proteinuria. Finally, our study has uncovered a mechanism underlying the antiproteinuric effect of CsA, an established drug in the treatment of proteinuric human kidney diseases, in particular in cases of MCD or FSGS that are resistant to treatment with corticosteroids or that relapse after treatment15. Our findings suggest that the antiproteinuric effect of CsA is independent of its immunosuppressive effect and instead stems from a direct effect on synaptopodin in podocytes (Fig. 6g).
It is possible that, similar to the activation of calcineurin by transient receptor potential channel-6 (TRPC6) in the heart38, increased TRPC6-mediated calcium influx may cause FSGS39,40 by enhancing calcineurin activation in podocytes, which, in turn, would lead to the loss of synaptopodin via the above-described mechanism. This idea is supported by the observation that overexpression of TRPC6 in podocytes causes the loss of stress fibers41, thereby pheno-copying the loss of synaptopodin8. A role for reduced synaptopodin expression as contributing factor in the pathogenesis of FSGS is further supported by the observation that heterozygosity for synaptopodin is sufficient to cause proteinuria and FSGS-like disease7.
Altogether, the antiproteinuric effect of CsA results, at least in part, from the maintenance of synaptopodin protein abundance in podocytes, which, in turn, is sufficient to maintain the integrity of the glomerular filtration barrier and to safeguard against proteinuria. These findings should open new avenues for the development of antiproteinuric drugs that directly and selectively target the podocyte, thereby avoiding the serious side effects of prolonged NFAT inhibition caused by long-term CsA treatment16,42.
METHODS
Cathepsin L digestion
For the in vitro digestion studies, we added 28, 70, or 140 ng human CatL (Sigma-Aldrich) to 1 μg purified Flag-synaptopodin in reaction buffer (100 mM sodium acetate pH 4.5 or 100 mM sodium phosphate monobasic pH 6.5 or 7.0, 1 mM EDTA, and 2 mM dithiothreitol) in a total volume of 50 μl29,43. In some experiments, we added 1 μg purified Flag–14-3-3β or Flag–α-actinin-4 before the digest. After incubation on ice for 30 min, we added 84 ng CatL and 10 μl reaction buffer and further incubated the proteins at 37 °C for 30 min. We stopped the reaction by boiling in sample buffer. After resolving the samples by 8–12% gradient SDS-PAGE, we analyzed them by immunoblotting.
Lipopolysaccharide-induced proteinuria
All animal studies were approved by the Mount Sinai School of Medicine Animal Institute Committee. We induced proteinuria in female SCID mice (n = 6–10 per group) by LPS injection as described before9,18. We injected the mice with ultrapure LPS (300 μg intraperitoneally (i.p.); InvivoGen) and started treatment with CsA (25 mg per kilogram body weight i.p.) or E64 (3 mg per kilogram body weight i.p.) 1 h after disease induction. We repeated the LPS, CsA and E64 injections at 24 h and killed the mice after 48 h. At 12 and 36 h, we injected all mice with 0.8 ml of isotonic saline (i.p.) to prevent hypovolemia. To quantify the level of proteinuria, we diluted 5 μl of urine with 5 μl of double-distilled water, boiled the diluted urine in sample buffer and analyzed the samples by SDS-PAGE followed by Coomassie blue staining. We ran BSA standards (0.1, 0.5, 1.0 and 5.0 μg) on the same gel and used them to identify and quantify urinary albumin bands. We quantified the albumin bands with ImageJ software (US National Institutes of Health) and determined the creatinine abundance from the same urine samples with a commercial kit (Exocell) according to the manufacturer’s protocol. We calculated albuminuria as micrograms albumin per milligram creatinine.
In vivo gene delivery
We achieved in vivo hydrodynamic gene delivery by tail vein injection as previously described9, with some modifications. We determined baseline urinary protein excretion of 8-week-old female mice as described above. We then injected the mice (n = 5–8 per group) with 50 μg of Flag-synaptopodin cDNA (wild-type, Synpo-CM1+2, Synpo-AA or Synpo-ED) in 2 ml of isotonic saline. In control experiments, we injected the mice with 2 ml of isotonic saline only. We then inoculated the mice i.p. with 300 μg LPS at 1 h and 24 h after gene delivery. Forty-eight hours after gene delivery, we measured proteinuria again as described above. We killed the mice and monitored the efficacy of the gene transfer by western blot analysis of synaptopodin in protein extracts from livers and isolated glomeruli and by double-labeling immunofluorescence microscopy for Flag and the podocyte marker podocin34.
Generation of podocyte-specific and tetracycline-inducible transgenic mice
We created the cDNA constructs for the generation of the tetO–GFP–Synpo-CM1+2 and tetO–GFP-aCnA–transgenic mice using the BD Creator Cloning System (BD Biosciences, Clontech). We confirmed the proper orientation of the cDNA by restriction enzyme digest mapping. We then transferred the tetO–GFP–Synpo-CM1+2 and tetO–GFP-aCnA cDNAs from the donor vector to pLP-Tre2, an inducible tetracycline-responsive expression vector, using Cre-loxP site-specific recombination. We released the inducible tetO–GFP–Synpo-CM1+2 and tetO–GFP-aCnA cDNA constructs from the vector backbone by enzymatic digest and purified them by gel extraction (QIAquick Gel Exctraction Kit, Qiagen) before injecting them into pronuclei of fertilized oocytes of FvB/NJ mice. We identified transgenic founder mice by PCR and mated them to podocin-rtTA mice on an FvB/NJ background36 to generate double-transgenic doxycycline-inducible podocin-rtTA–tetO–GFP–Synpo-CM1+2 and podocin-rtTA–tetO–GFP-aCnA mice, respectively. We identified double-transgenic mice by PCR36. We mated podocin-rt-TA–tetO–GFP–Synpo-CM1+2 or podocin-rtTA–tetO–GFP-aCnA F1 littermates to obtain F2 double-transgenic mice for experimental procedures. To induce transgene expression in podocytes, we administered doxycycline (Sigma-Aldrich) at a concentration of 2 mg ml–1 in 7% sucrose (pH ~5) to double-transgenic mice in the drinking water for 7 d36. Simultaneously, we fed the mice a special chow diet containing doxycycline (2,000 p.p.m.; Research Diets). We fed the control mice (vehicle) normal chow and 7% sucrose in the drinking water. We treated five podocin-rTA–tetO–GFP–Synpo-CM1+2 mice with doxycycline and four with vehicle. We also treated 15 podocin-rtTA–tetO–GFP-aCnA mice with doxycycline and 10 with vehicle.
Statistical analyses
The results of all animal studies were assessed by Student’s t-test and are expressed as means ± s.d.
Additional methods
Detailed methodology is described in Supplementary Methods online.
Acknowledgments
We thank C. Chiu and S. Ratner for excellent technical assistance and T. Reinheckel for the analysis of CatL cleavage sites. We thank the Mount Sinai School of Medicine Mouse Genetics Research Facility for performing pronuclear injections. We also thank J.B. Kopp (US National Institutes of Health) for providing the podocin-rtTA mice, H. Fu (Emory University) for yellow fluorescence protein–tagged difopein and E.N. Olson (The University of Texas Southwestern Medical Center at Dallas) for wild-type and constitutively active calcineurin cDNA constructs. Y.H.C. was supported by a research fellowship from the Albert Einstein College of Medicine, S.F. was supported by Karger Stiftung and J.D. was supported by the Deutsche Forschungsgemeinschaft. This work was supported by US National Institutes of Health grants DA18886, DK57683 and DK062472 and the George M. O’Brien Kidney Center grants DK064236 (to P.M.) and DK073495 (to J.R.).
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 2.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 3.Tryggvason K, Pikkarainen T, Patrakka J. Nck links nephrin to actin in kidney podocytes. Cell. 2006;125:221–224. doi: 10.1016/j.cell.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Mundel P, et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asanuma K, et al. Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huber TB, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn and synaptopodin. J Clin Invest. 2006;116:1337–1345. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asanuma K, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 9.Yanagida-Asanuma E, et al. Synaptopodin protects against proteinuria by disrupting Cdc42-IRSp53-Mena signaling complexes in kidney podocytes. Am J Pathol. 2007;171:415–427. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343–348. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109 (Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 12.Heit JJ, et al. Calcineurin-NFAT signalling regulates pancreatic beta cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 13.Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132:299–310. doi: 10.1016/j.cell.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koga T, et al. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–885. doi: 10.1038/nm1270. [DOI] [PubMed] [Google Scholar]
- 15.Meyrier A. Treatment of focal segmental glomerulosclerosis. Expert Opin Pharmacother. 2005;6:1539–1549. doi: 10.1517/14656566.6.9.1539. [DOI] [PubMed] [Google Scholar]
- 16.Charbit M, et al. Cyclosporin therapy in patients with Alport syndrome. Pediatr Nephrol. 2007;22:57–63. doi: 10.1007/s00467-006-0227-y. [DOI] [PubMed] [Google Scholar]
- 17.Chen D, et al. Cyclosporine A slows the progressive renal disease of alport syndrome (X-linked hereditary nephritis): results from a canine model. J Am Soc Nephrol. 2003;14:690–698. doi: 10.1097/01.asn.0000046964.15831.16. [DOI] [PubMed] [Google Scholar]
- 18.Reiser J, et al. Induction of B7–1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 20.Yaffe MB, et al. The structural basis for 14-3-3–phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 21.Faul C, Dhume A, Schecter AD, Mundel P. Protein kinase A, Ca2+-calmodulin–dependent kinase II and calcineurin regulate the intracellular trafficking of myopodin between the Z-disc and the nucleus of cardiac myocytes. Mol Cell Biol. 2007;27:8215–8227. doi: 10.1128/MCB.00950-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK-506– and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 23.Yaffe MB. How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002;513:53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 24.Faul C, et al. Promotion of importin α-mediated nuclear import by the phosphorylation-dependent binding of cargo protein to 14-3-3. J Cell Biol. 2005;169:415–424. doi: 10.1083/jcb.200411169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 26.Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- 27.Masters SC, Fu H. 14-3-3 proteins mediate an essential anti-apoptotic signal. J Biol Chem. 2001;276:45193–45200. doi: 10.1074/jbc.M105971200. [DOI] [PubMed] [Google Scholar]
- 28.Reiser J, et al. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and α3 integrin. J Biol Chem. 2004;279:34827–34832. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 29.Sever S, et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007;117:2095–2104. doi: 10.1172/JCI32022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohmuller T, et al. Toward computer-based cleavage site prediction of cysteine endopeptidases. Biol Chem. 2003;384:899–909. doi: 10.1515/BC.2003.101. [DOI] [PubMed] [Google Scholar]
- 31.Cotelle V, et al. 14-3-3s regulate global cleavage of their diverse binding partners in sugar-starved Arabidopsis cells. EMBO J. 2000;19:2869–2876. doi: 10.1093/emboj/19.12.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baricos WH, et al. Evidence suggesting a role for cathepsin L in an experimental model of glomerulonephritis. Arch Biochem Biophys. 1991;288:468–472. doi: 10.1016/0003-9861(91)90222-5. [DOI] [PubMed] [Google Scholar]
- 33.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 34.Schwarz K, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim BS, et al. Impact of cyclosporin on podocyte ZO-1 expression in puromycin aminonucleoside nephrosis rats. Yonsei Med J. 2005;46:141–148. doi: 10.3349/ymj.2005.46.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shigehara T, et al. Inducible podocyte-specific gene expression in transgenic mice. J Am Soc Nephrol. 2003;14:1998–2003. doi: 10.1681/ASN.V1481998. [DOI] [PubMed] [Google Scholar]
- 37.Zheng W, et al. Cellular stability of serotonin N-acetyltransferase conferred by phosphonodifluoromethylene alanine (Pfa) substitution for Ser-205. J Biol Chem. 2005;280:10462–10467. doi: 10.1074/jbc.M412283200. [DOI] [PubMed] [Google Scholar]
- 38.Kuwahara K, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MP, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 40.Reiser J, et al. TRPC6 is a glomerular slit diaphragm–associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moller CC, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 42.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 43.Goulet B, et al. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14:207–219. doi: 10.1016/s1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]