

Abstract
Histone H3 lysine79 (H3K79) methyltransferase DOT1L plays an important role in the activation and maintenance of gene transcription. It is essential for embryonic development as well as normal functions of the hematopoietic system, heart and kidney in adults. DOT1L has been found to be a drug target for acute leukemia with mixed lineage leukemia (MLL) gene translocations. The rearranged onco-MLL can recruit DOT1L, causing aberrant H3K79 methylation, overexpression of leukemia relevant genes, and eventually leukemogenesis. Potent DOT1L inhibitors possess selective activity against this type of leukemia in cell-based and animal studies, with the most advanced compound being in clinical trials. In the medicinal chemistry point of view, we review the biochemistry, cancer biology and current inhibitors of DOT1L, as well as biophysical (including X-ray crystallographic) investigation of DOT1L-inhibitor interactions. Potential future directions in the context of drug discovery and development targeting DOT1L are discussed.
INTRODUCTION
The human genome is tightly packed into 23 pairs of chromosomes, which contain thousands of repetitive units called nucleosomes. A single nucleosome is composed of a short segment of DNA with ~146 base pairs in length winding around a disc-like histone octamer core, which consist of two copies each of histone H2A, H2B, H3 and H4 proteins, as schematically illustrated in Figure 1. Chromatin is classified into transcriptionally repressed heterochromatin and generally transcriptionally active euchromatin, mostly controlled by histone post-translational modifications.1 Histones are rich of basic amino acid residues lysine and arginine, which not only provide electrostatic/H-bond interactions with the negatively charged DNA chain for tight binding, but may be covalently modified. Histone methylation at the sidechain -NH2 of lysine and arginine is one of the most studied post-translational modifications.2 Histone methyltransferases (HMT) include a large family (>50) of histone lysine methyltransferases (HKMT) and histone/protein arginine methyltransferases (PRMT),3,4 many of which have been found to play important roles in cell differentiation, gene regulation, DNA recombination and damage repair.2,5 Therefore, small molecule inhibitors of HMTs are useful chemical probes for these biological studies as well as potential therapeutics. However, development of HMT inhibitors has been in its infancy: very few inhibitors of HKMT and PRMT have been discovered and developed.2,6 Considering the important roles of HMTs in normal physiology and the biology of diseases (e.g., cancer),7 this represents great opportunities to explore novel medicinal chemistry.
Figure 1.
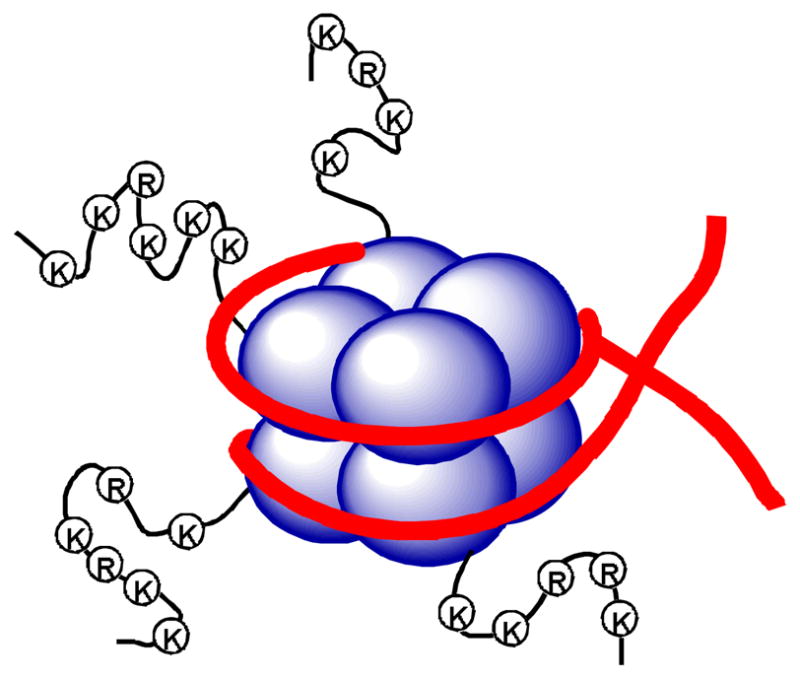
Illustration of the nucleosome structure, with histones shown as blue spheres and DNA as red strands.
This review is focused on histone H3 lysine 79 (H3K79) methyltransferase DOT1L in a medicinal chemistry perspective. DOT1L is a HMT sufficient and necessary for H3K79 methylation,8–12 playing important roles in gene regulation and the functions of many key organs.13 DOT1L has been found to be a drug target of mixed lineage leukemia (MLL) gene translocated acute leukemia. Several approaches have led to the discovery and development of potent and selective inhibitors of DOT1L with Ki values as low as 0.3 nM. These compounds demonstrate selective activity against MLL-rearranged leukemia, with the most advanced compound being in phase I clinical trials. The current problems, concerns and potential opportunities of DOT1L inhibitors as well as future inhibitor design are discussed.
WHAT IS DOT1L?
The full-length human DOT1L contains 1537 amino acids, with its N-terminal ~360 amino acids having a high homology to yeast DOT1 protein originally found in a genetic screen, which can disrupt telomeric silencing in budding yeast.14 This sequence was found to be highly conserved from yeast to mammals and identified to be an H3K79 methyltransferase.8 The remaining C-terminal part of mammalian DOT1L is involved in physical interactions with many transcription relevant proteins.15–19 Therefore, the general biological function of DOT1L is to methylate H3K79 as a member of a large protein complex, which can initiate and/or maintain an active transcription state. DOT1L is a unique HKMT, which belongs to the class I methyltransferase family,20 while all other known HKMTs are class V methyltransferases that possess a conserved SET (Su(var)3-9, Enhancer-of-zeste, Trithorax) domain with a distinct 3-dimentional structural feature.3,21 In addition, DOT1L’s substrate H3K79 is located in the ordered core structure of histone H3, while the substrates of all other HMTs are situated in the unordered histone tails.
Biochemistry
DOT1L catalyzes the methylation reaction of the ε-amino group of H3K79 up to trimethylation (H3K79Me3) using S-adenosyl-L-methionine (SAM) as the enzyme cofactor,20 producing the methylated substrate and S-adenosyl-L-homocysteine (SAH) as schematically illustrated in Figure 2a. In addition, studies show DOT1/DOT1L is the only enzyme that performs H3K79 methylation, since DOT1/DOT1L knockout in yeast, Drosophila and mouse led to a complete loss of H3K79 methylation.9,11,12 Although the sequence of human DOT1L(1-351) is conserved across H3K79 methyltransferases, the recombinant protein was found to be enzymatically inactive. The minimal length for an active human DOT1L is 1-416, with its C-terminal 391-416 being essential for the activity. This small segment of peptide is highly enriched of 15 basic Lys and Arg residues. In addition, as compared to other HMTs, a special feature of DOT1L is that it can only methylate nucleosomes, but not histone H3 and its shorter peptide. The strong electrostatic interactions between the condensed positive charges in residues 391-416 and the negative DNA chain in nucleosomes are essential for the enzyme-substrate recognition and binding. Post-translational modifications of histones, such as H2B ubiquitinylation,22 could be important for enzyme activity. A typical biochemical assay to study the enzyme kinetics of DOT1L is to use methyl-3H labeled SAM. Upon 3H-Me transfer, the substrate may be filtered, washed and the reaction rate measured by scintillation counting. The steady state kinetic parameters for human DOT1L(1-416) were determined to be kcat = 0.3/min, Km (SAM) = 670 nM, and Km (nucleosome) = 8.6 nM.23
Figure 2.

(A) Mechanism of catalysis of DOT1L; (B) The overall structure of human DOT1L(1-416) in complex with SAM (ball & stick model with C atoms in green); (C) The close-up view of the active site of the DOT1L:SAM structure, with H-bonds shown as dotted lines; (D) The close-up view of the lysine binding channel, with the methyl group of SAM shown in ball & stick model.
X-ray structure of DOT1L-SAM complex
The crystal structure of DOT1L(1-416) in complex with SAM was initially determined to 2.5 Å,20 and later to 2.1 Å.23 The overall structure and a close-up view of the active site are shown in Figures 2b/c. Its seven-stranded β-sheet is a characteristic feature of the class I methyltransferases.3 The following five peptide segments of human DOT1L bind the enzyme cofactor SAM: 1) residues 161-169; 2) 186-191; 3) 239-245; 4) 133-139; and 5) 221-224, among which segments 1–3 represent consensus sequence motifs found in other SAM-dependent methyltransferases whereas segments 4 and 5 are unique to DOT1L. It is noted that the protein flexible loop including residues 133-139 acts as a cap, which opens and closes to allow SAM/SAH to enter/exit the enzyme. In addition to hydrophobic as well as π-π stacking interactions between the adenine ring with Phe223, the adenosine moiety of SAM has been specifically recognized by five H-bonds with DOT1L, including (a) adenine 6-NH2 with Asp222; (b) adenine N5 with Phe223; (c) adenine N3 with Lys187; (d/e) ribose 2′, 3′-diol with Glu186 (Figure 2c). SAM’s amino acid moiety forms additional five H-bonds with Gln168, Thr139, Asp161 and Gly163. The vast majority of the surface area of SAM is buried inside the protein. These structural information suggest SAM is tightly bound to DOT1L, which is also consistent with its Km value of ~700 nM.23,24 The methyl group of SAM is aligned with the “lysine binding channel” defined by hydrophobic residues Phe243, Ala244, Val135, Gly137 and Trp305, with an opening of ~4 Å wide that is sufficiently large to accommodate a trimethylated lysine (Figure 2d).
Although there is currently no structural information as to how the substrate nucleosome bind to DOT1L, a modeling study shows DOT1L has physical interactions with all four histones on the same side of nucleosome surface.20 These, in addition to the electrostatic DOT1L-DNA interactions, suggest nucleosome has a very high binding affinity to DOT1L. Indeed, our isothermal titration calorimetry (ITC) experiment demonstrated the Kd (dissociation constant) value between DOT1L and nucleosome is 14 nM,25 which is in line with the Km value of 8.6 nM.23
DOT1 is a distributive enzyme and H3K79 multiple methylation states have functional redundancy in yeast
Using mass spectrum (MS) analysis, DOT1, a yeast homolog of DOT1L, was found to be a distributive enzyme, which after one cycle of catalysis releases both products SAH and the methylated substrate.26 This results in ~22% H3K79Me1, 26% H3K79Me2 and 44% H3K79Me3 in yeast cells. This property is in contrast to a processive enzyme, including many other SET-domain HKMTs and PRMTs. The structures of SET-domain HKMTs allow the enzymes to release only the product SAH after one cycle of catalysis, while the methylated substrate remains bound in the active site and may be methylated for the second round.3 The degree of methylation is, however, dependent upon the property of the enzyme as well as associated factors.3,26 However, the structure of DOT1 might not allow the release of SAH with the methylated H3K79 bound, since SAM/SAH is deeply buried inside the enzyme and releasing it requires a large conformational change.20 The distributive mechanism of DOT1 is compatible with its structure.
It is known that different methylation states (i.e., Me1, Me2 or Me3) at other histone-lysine sites can have distinct biological functions.27 Using overexpressing of DOT1 and a less efficient mutant DOT1(G401A) in yeast, a study showed that Me1, Me2 and Me3 methylation states exert the same biological function.26 This suggests H3K79 only exhibits two functional states: methylated and unmethylated, with the former indicating active transcription. The model is consistent with the distributive mechanism of DOT1, since it can only generate H3K79 methylation states in a random manner. However, whether this model applies to higher organisms (e.g., human) is not clear. In addition, evidence shows the methylation status at H3K79 may be regulated by an α-ketoglutarate dependent histone demethylase in the JmjC family, although an H3K79 demethylase has not been identified to date.13 More studies are therefore needed to understand the biological role(s) of mono-, di- and tri-methylated H3K79 as well as their regulation in mammals.
BIOLOGY OF DOT1L
Molecular and cell biology of DOT1L and/or its yeast homolog DOT1 have been comprehensively reviewed in several recent articles.13,28,29 In general, DOT1L mediated H3K79 methylation serves as a biomarker for active gene transcription from yeast to mammals. The following serves as a brief summary of these findings.
Role of DOT1L in transcription
Genetic studies in yeast showed DOT1 links to an active transcription status. ~10% genome of yeast that is H3K79 hypomethylated is found to be located at transcriptionally inactive loci, while the other 90% with a methyl marker at H3K79 is actively transcribed.9,30 This was found to be the case in Drosophila and mammals.31,32 It has been generally accepted that H3K79 methylation is a biomarker for active gene transcription.
Several large DOT1L-associated protein complexes have been purified and identified using chromatin immunoprecipitation, including ENL-associated proteins (EAP), DOT1L-containing complex (DotCom) and Superelongation complex (SEC).15–19 Many member proteins, including P-TEFb kinase as well as transcription factors AF4, AF9, AF10, ENL, are repeatedly present in these complexes. P-TEFb is a cyclin-dependent kinase that can phosphorylates RNA polymerase II, which is required for transcription elongation. These studies support DOT1L play an important role in gene transcription elongation. It is also of interest that DOT1L-associated transcription factors AF4/9/10 and ENL are major fusion partners of onco-MLL gene, indicating DOT1L’s involvement in MLL rearranged acute leukemia (described below).33,34
Roles of DOT1L in normal physiology
DOT1L has been found to play an important role in embryonic development. In Drosophila and mouse, H3K79 methylation is absent in the very early stage of embryonic development, while increasing levels of H3K79Me2 can be observed in later stages, suggesting the DOT1L-mediated histone modification is critical to the important gene transcription for embryonic development.11,35 Germline knockout of mouse DOT1L causes embryonic lethality and examination of the knockout embryos indicated major defects in the cardiovascular system, showing the function of DOT1L is essential for embryonic development.12 In addition, H3K79 methylation has been found to be important for maintaining normal hematopoiesis in adult mice.36,37 Conditional knockout of DOT1L in bone marrow cells considerably reduces hematopoietic stem cells and progenitor cells in different lineages. Moreover, other molecular and cell biology investigation has demonstrated DOT1L plays roles in maintaining normal functions of heart and kidney.12,38–40 These studies have shown that DOT1L is also important to the normal physiology in adults.
DOT1L AND DISEASES
DOT1L is a drug target for acute leukemia with MLL translocations
MLL-rearranged leukemia accounts for ~75% of infant and ~10% child/adult acute leukemia,33,34,41 with the phenotype being either acute lymphocytic leukemia or acute myeloid leukemia. Despite the phenotypic difference, MLL-rearranged leukemias of both lineages overlap in their gene expression profiles.42 This type of acute leukemia has a particularly poor prognosis,43–45 with 5-year event-free survival with MLL-rearranged acute lymphocytic leukemia being only ~40%. There is therefore a pressing need to find new drugs.
MLL is a large protein (3969 amino acid residues) with multiple domains, as illustrated in Figure 3a. The biological functions of wild-type MLL as well as those of the oncogenic MLL-fusion genes have been recently reviewed.46–48 In brief, human (and mammalian) MLL is a homolog of trithorax in Drosophila. Of interest related to medicinal chemistry are the N-terminal AT hook and the C-terminal SET domain of MLL. The three AT hooks can bind to the minor groove of AT-rich DNA regions, while the SET domain is a histone H3 lysine4 (H3K4) methyltransferase.33 Studies have shown that the normal function of MLL is to bind to the promoter region of certain genes and methylate H3K4,49,50 as shown schematically in Figure 3b. Methylation of H3K4 represents an active transcription marker for gene expression. With this epigenetic modification, RNA polymerase II (Pol II) bound to the promoter region of a gene is able to proceed along the gene and start transcription.
Figure 3.

(A) Illustration of the wild-type MLL gene; (B) Function of wild-type MLL, which methylates H3K4 and initiates Pol II mediated gene transcription; (C) Illustration of onco-MLL fusion gene; (D) Function of onco-MLL protein is illustrated. The MLL protein complex involving AF4, AF9, AF10 and ENL recruits DOT1L, which methylates H3K79 and causes overexpression of leukemia-relevant genes.
MLL is frequently involved in a variety of translocations, leading to the initiation of acute leukemia. The rearranged MLL fusion protein loses the SET domain as well as the H3K4 methyltransferase ability (Figure 3c). More than 70 functionally diverse fusion partner genes have been documented, among which only a few genes are predominant, such as AF4, AF9, AF10 and ENL accounting for a total of ~70%.33,34 Many MLL fusion genes, including the above four, have been found to be sufficient and necessary to transform normal stem/progenitor cells to leukemic stem cells. Moreover, mouse models with knock-in MLL fusion genes (e.g., MLL-AF4 and -AF9) have also been found to lead to acute leukemia.34 These MLL-induced experimental leukemias with both technologies have been found to be able to closely recapitulate clinical leukemias.51
DOT1L has been found to be a target for leukemia with MLL translocations. Four most common MLL fusion partners, i.e., AF4, AF9, AF10 and ENL, were found to be able to physically bind to DOT1L as part of transcription protein complexes.15,17,40,51,52 These proteins, guided by the AT hook domain of the onco-MLL, were proposed to form a large complex that binds to the promoter regions of certain MLL targeted genes (e.g., Hoxa9).17 Onco-MLL fusion proteins without a SET domain at the C-terminal cannot methylate H3K4. Rather, the recruited DOT1L methylates H3K79, which can also initiate and maintain gene transcriptions. Such an aberrant epigenetic event dysregulates numerous MLL targeted genes, e.g., Hoxa9, Hoxa7 and Meis1, whose overexpression is known to cause leukemia.52 A model is schematically illustrated in Figure 3d to show how onco-MLL works with DOT1L and eventually leads to acute leukemia.
Another line of strong evidence is that aberrant H3K79 methylation is a signature of murine and human MLL-AF4 leukemia.51 Gene expression profiling of an MLL-AF4 knock-in mouse leukemia model shows significant overlaps with human MLL leukemia. Chromatin immunoprecipitation analysis demonstrated H3K79 methylation profiling of this mouse model correlates with that of the human disease. Since DOT1L is the only H3K79 methyltransferase, its involvement in MLL leukemogenesis is thus clear. In addition, an artificial fusion gene MLL-DOT1L was found to be able to transform normal progenitor cells to leukemic stem cells. However, MLL fused with a mutant DOT1L without methyltransferase activity failed to do so. This further demonstrated that the methyltransferase activity is required for this type of cancer.52 Moreover, a high level of DOT1L methyltransferase activity seems to be needed for the transformation and maintenance of MLL leukemia. A 50%–70% suppression of DOT1L expression by RNA interference was found to be able to abrogate the transforming ability of MLL-AF10 and -AF4.51,52 These findings have biologically validated that DOT1L plays a central role in leukemogenesis of MLL-rearranged leukemia and therefore represents a target for therapeutic intervention. Development of small molecule inhibitors of DOT1L is warranted. Not only can these compounds be further developed to become clinically useful therapeutics for MLL leukemia, they may also serve as powerful chemical probes to investigate the biology of DOT1L in cancer and other diseases.
Potential roles of DOT1L in other diseases or applications
To date, very few publications have linked DOT1L to other diseases. A recent paper describes deficiency of DOT1L blocks the proliferation of lung cancer cell lines A549 and NCI-H1299.53 H3K79 hypermethylation was observed for these two cells as well as lung cancer tissues. Using siRNA knocking down, the proliferation of A549/NCI-H1299 cells is inhibited due to induced cell senescence. Overexpression of DOT1L can eliminate the siRNA induced phenotypic changes. It is therefore implied that H3K79 methylation play an important role in the proliferation of lung cancer. No DOT1L inhibitors were, however, used in the study to confirm this finding.
Another interesting application targeting DOT1L is its role in cell reprogramming. Inhibition of DOT1L using RNA interference as well as a small molecule inhibitor was recently shown to considerably enhance the reprogramming efficiency to generate induced pluripotent stem cells (iPSC) from somatic cells.54 Given the potential roles of histone modifying enzymes for this purpose, a library of short hairpin RNAs (shRNA) targeting genes of DNA/histone methylation pathways were screened and reduction of mRNA of DOT1L exhibited the most pronounced effect to promote reprogramming. A specific inhibitor of DOT1L was found to be able to achieve the same effect, showing the potential application of DOT1L inhibitors in the field of regenerative medicine.
INHIBITORS OF DOT1L
Since the discovery of the critical role of DOT1L in MLL leukemia, efforts have been made to find selective inhibitors of DOT1L. In mid 2011, scientists from Epizyme Inc. reported the first DOT1L inhibitor as well as its selective activities against MLL-rearranged leukemia.55 Shortly after, we disclosed several potent and selective DOT1L inhibitors, using both ligand/structure and mechanism-based approaches.24 To date, three groups have published several dozens of DOT1L inhibitors in peer-reviewed journals,24,25,55–60 in addition to more in several patents.61–63 The structures of these inhibitors generally fall into four classes: SAH-like, mechanism-based, carbamate- and urea/benzimidazole-containing. Representative DOT1L inhibitors 1 – 24 are shown in Figures 4 and 5, together with their Ki or IC50 values against recombinant human DOT1L. The common substructure of these compounds is adenosine (or deaza-adenosine), which together with biophysical investigation show these compounds are competitive to the enzyme cofactor SAM, rather than the substrate nucleosome.
Figure 4.

Representative SAH-like, mechanism-based and carbamate-containing inhibitors of DOT1L. (Ki or IC50 values are from original references. IC50 values may not be used directly to compare the relative inhibitory potency, due to possibly different assay conditions.)
Figure 5.

Representative urea- and benzimidazole-containing inhibitors of DOT1L. (Ki or IC50 values are from original references. IC50 values may not be used directly to compare the relative inhibitory potency, due to different assay conditions.)
DOT1L inhibitors from Epizyme
Epizyme has disclosed 9 DOT1L inhibitors in journals as well as >300 in patents.55,56,60–63 While HTS did not yield good lead compounds for further development, a traditional, ligand based inhibitor design has been successful at Epizyme. SAH, the product of the DOT1L catalyzed reaction, was found to be a potent inhibitor of DOT1L. However, its amino acid moiety prevented it from entering cells. Compounds 4 and 5 (Figure 4) with a 5′-dimethylamino and isopropylmethylamino group, respectively, were designed and found to be weak inhibitors (Ki: 38 and 12 μM). Carbamate compound 14 with an Fmoc protecting group was unexpectedly found to be also an inhibitor with a Ki value of 20 μM, which eventually led to the finding of potent, urea-containing DOT1L inhibitors 21 (Ki: 13 nM) and 22 (EPZ004777, Ki = 0.3 nM).55 Modifications of 22 yielded compound 23 (EPZ-5676) which possesses improved enzyme activity (Ki: 0.08 nM) and biological activities.60 The -CH2CH2CH2- linker and the 4-tert-butylphenyl substituted urea moiety of 22 was replaced with a cis-ethylcyclobutane linker and a 5-tert-butylbenzimidazole ring. These changes were part of a plan to reduce the conformational flexibility as well as number of hydrogen bond donors in 22 to increase potency and bioavailability as well as reduce clearance from plasma. Epizyme has initiated the phase I clinical trials using compound 23 for patients with MLL-rearranged leukemia since September 2012. It is remarkable that this is the first HMT inhibitor to be evaluated for antitumor efficacy in the clinic.
Compounds 22 and 23 exhibit a very high enzyme selectivity for DOT1L, being essentially inactive against a panel of 8 HMTs (only weakly active against PRMT5). X-ray crystallographic studies indicated this high enzyme selectivity is due to a large protein conformational change induced by the hydrophobic urea sidechain of the inhibitor (described below).
DOT1L inhibitors from the Song laboratory
The inhibitor discovery and development targeting human DOT1L in our laboratory was also started from SAH, using a ligand/structure based approach. In addition to limited cell membrane permeability, another problem for SAH is its broad inhibitory activity against many other HMTs. For example, SAH strongly inhibits CARM1 (a histone arginine methyltransferase) and G9a (a SET-domain H3K9 methyltransferase) with Ki values of 400 and 570 nM. Selective DOT1L inhibitors are highly desirable in the context of drug discovery. From a comparative analysis between the X-ray structure of DOT1L:SAM and those of CARM1 and G9a in complex with SAH, N6-substituted SAH analogs, such as compounds 1 and 2 in Figure 4, were designed to selectively target DOT1L, since these N6-substitutents were predicted to disrupt key ligand-protein interactions with CARM1 and G9a, but not those with DOT1L.24 Indeed, compound 1 (or N6-methyl-SAH) is also a potent inhibitor of DOT1L with a Ki of 290 nM, while it is inactive against CARM1 and G9a as well as several other HMTs, showing a high selectivity for DOT1L. Compounds with the general structure of 2 having a variety of N6-substituents, including allyl, cyclopropyl, isopropyl, butyl, branched pentyls and benzyl, show weakened activity against DOT1L with Ki values of 1.1 – 8.9 μM.25
Next, a mechanism based approach was applied to find more potent inhibitors. Compounds 8 – 10, which may form an aziridinium intermediate (Figure 4) that mimics the methyl-sulfonium moiety of SAM (Figure 2a), were synthesized. While 8 exhibited a weak activity, compounds 9 and 10 are potent DOT1L inhibitors. The extra methylene in 9/10 was necessary to compensate for a loss in total molecule length when the sulfur (C-S bond length ~ 1.8 Å) atom in SAM was replaced with a nitrogen (C-N bond length ~ 1.5 Å).24
It turned out to be challenging to remove the amino acid moiety in SAH/1 to improve the cell membrane permeability while maintaining DOT1L activity.25 For example, compound 3 (or decarboxy-SAH) is almost inactive against DOT1L. Many other 5′-substituted adenosine compounds are inactive or only weakly active. Similar to the experience occurred at Epizyme, carbamate compound 11 (Figure 4), which is actually a benzyloxycarbonyl-protected intermediate for synthesizing a 5′-amino analog of 3, was surprisingly found to be a better inhibitor than 3. Due to the more hydrophobic nature of its sidechain, medicinal chemistry based on 11 was performed. Adding an additional isopropyl group onto 5′-N resulted in compound 12 showing an increased activity. Reversing the carbamate functionality or changing to an amide resulted in compounds 13a and b with slightly improved activity. A ~25-fold activity enhancement was observed for the urea compound 15 with a Ki of 1.9 μM, as compared to the carbamate 11. The phenyl urea compound 16a with a Ki of 550 nM was found to be superior to the benzyl analog 15. These results demonstrate both -NH- moieties of the urea group are important, with each one offering ~25 – >50-fold activity enhancement, as compared to less favored -O- and -CH2- groups (e.g., compound 15 vs. 11, as well as compound 16a vs. 13a/b). These two -NH- could serve as H-bond donors to increase the binding affinity to DOT1L. In addition, weaker activities of compounds 16b and c suggest the optimal linker between the urea group and the 5′-position is -CH2CH2CH2-. Additional SAR study revealed that an appropriate 4-substituent, such as 4-Cl, 4-tert-Bu and 4-CF3 in compounds 17a-c, is able to further improve the activity by up to 10-fold. Finally, replacing the -S- in 17b with an -N(i-Pr)-, which shows improved activity in compound 12, led to the finding of our most potent DOT1L inhibitors 18a and b (SYC-522 and -534, respectively) with Ki values of 0.5 and 0.8 nM. The large activity boost (as compared to that of 17b) might be due to coordinated protein conformational changes induced by co-binding of the N-(4-tert-butylphenyl)urea sidechain and the -N(i-Pr)- group.
A problem with compounds 22 and 23, as well as other ribose-containing DOT1L inhibitors, in animal studies is their metabolic instability, resulting in a poor pharmacokinetics, e.g., a short half-life in plasma. Many human enzymes can recognize adenosine or deaza-adenosine moiety, leading to rapid cleavage of adenine and/or 5′-substituent. Our solution is to replace the ribofuranose ring of these compounds with a 5-membered carbocycle. Compound 19 with a cyclopentane ring was efficiently synthesized in 20 steps with an overall yield of 19%, starting from readily available D-ribose.57 In addition, a cyclopentene analog, compound 20, was also synthesized from D-ribose. Hydrogenation of compound 20 produced epi-19 with a reversed asymmetric center at 4′-position. Compounds 19 and 20 were found to possess comparable DOT1L inhibitory activity with Ki values of 1.1 and 1.3 nM, respectively. But epi-19 is essentially inactive against DOT1L, showing its adeninylcyclopentane and N-tert-butylphenyl urea moieties cannot be located in the favored positions. Cyclopentane-containing DOT1L inhibitor 19 was found to exhibit a high metabolic stability (CLint = 0.36 μL/min/mg protein) in human plasma and liver microsomes, the latter of which are mainly responsible for drug metabolism. However, like its deaza-analog 22, ribose-containing inhibitor 18a can be quickly degraded in the presence of human liver microsomes with an intrinsic clearance value (CLint) of 24 μL/min/mg protein. Cyclopentene compound 20 also shows a poor metabolic stability, likely due to its C=C double bond that can be oxidized by cytochrome P450 enzymes in liver microsomes. These results suggest cyclopentane-containing DOT1L inhibitors have improved metabolic stability and represent a direction for future drug development.
DOT1L inhibitors from Structural Genomics Consortium
Scientists from the Structural Genomics Consortium at University of Toronto found 7-substituted adenosine derivatives are potent inhibitors of DOT1L.58,59 Using HTS, compound 6 (Figure 4) was identified to be a weak inhibitor of DOT1L with an IC50 of 18 μM. This finding has led to the discovery of compound 7, an analog of SAH with a 7-bromo group, to be a potent inhibitor of DOT1L (IC50 = 77 nM), ~8× more active than SAH. This shows a bulky 7-halo substituent is favored for DOT1L inhibition. In addition, compound 7 was found to exhibit a good selectivity profile, being less active against PRMTs (Ki ~ 1 μM) and inactive against SET-domain HMTs. Compound 24, which is the 7-bromo substituted compound 22, was also synthesized and found to exhibit a comparable inhibitory activity (Ki = 0.3 nM) against DOT1L.
Biophysical investigation of DOT1L-inhibitor interactions
The large hydrophobic, urea-containing sidechains of the DOT1L inhibitors in Figure 5 provide greatly enhanced activity as well as high selectivity for DOT1L. It is therefore of interest to find where these urea sidechains bind in DOT1L. Molecular modeling (docking) was explored to predict the binding site of these groups, using the crystal structure of the DOT1L:SAM complex as the docking template. The computational studies suggested all of these urea-containing groups are predicted to extend into the nucleosome (substrate) binding pocket of DOT1L through the “lysine binding channel”.25 The bulky and lipophilic aromatic groups are predicted to be located in a mainly hydrophobic pocket on the nucleosome binding surface. Isothermal titration calorimetry (ITC) experiments were next performed to test if the modeling results are real. The ITC results show the binding of nucleosome to DOT1L, however, increases the binding affinities of inhibitors 18a and b to the protein, suggesting these two urea-containing compounds do not compete with the substrate nucleosome.25 In addition, ITC experiments confirmed compounds 18a compete with the enzyme cofactor SAM/SAH, since the binding affinity of 18a to DOT1L decreases in the presence of increasing concentrations of SAH in a linear fashion. The docking studies using the DOT1L:SAM structure failed to predict the binding site of the urea sidechain, because, as described below, the flexible loops of DOT1L that is involved in the binding of the amino acid group of SAM undergo large conformational changes.
Surface plasmon resonance (SPR) was used to study the binding kinetics of DOT1L inhibitors.56,58 The results show despite a large difference in inhibitory activity/binding affinity, all of the DOT1L inhibitors tested, including the natural ligand SAH, have a relatively slow association rate constants (kon) in a narrow range of 1.2 – 12×106 M−1 s−1. This suggests that binding of these inhibitors requires a protein conformational change. Indeed, the crystal structures of the DOT1L:SAM and :SAH complexes show these ligands are deeply bound inside the protein without an obvious channel to allow them to enter or exit the protein. A backbone conformational change of the flexible loop 130–139 is required for these ligands to enter or exit the enzyme. The large binding affinity differences (Kd: 0.1 – 170 nM) are mostly due to the dissociation rate constants (koff) ranging from 0.0003 – 0.2 s−1. As a result, highly potent DOT1L inhibitors, such as compound 22, can be described as “slow on, extremely slow off”. For example, 22 was found to have a residence time of >1 h in DOT1L.
More knowledgeable insight into the DOT1L-inhibitor interactions came from X-ray crystallographic studies.24,56,58,59 The currently available DOT1L inhibitors modify three positions of SAH, i.e., the N6-, 7- and 5′-positions of adenosine. X-ray crystallography has been used to investigate how these inhibitors interact with DOT1L, especially with respect to the binding sites of these three modifications. Since these interactions are characteristic to DOT1L, the structural information obtained could be useful for future structure based inhibitor design specifically targeting DOT1L. The crystal structures of human DOT1L in complex with ~10 inhibitors, including several SAH-like (e.g., SAH, 1, 4, 6 and 7) and urea-containing (e.g., 18a, 21, 22 and 24) inhibitors, have been determined. A common feature among these structures is the binding mode of the adenosine (or deaza-adenosine) moiety, which closely mimics the binding conformation of the adenosyl of SAM (Figure 6a).20 All of the 5 H-bonds (3 for adenine and 2 for ribose) as well as the hydrophobic/π-π interactions described for the crystal structure of DOT1L:SAM remain intact in these X-ray structures, showing a strong recognition of adenosine by DOT1L. There are several cavities near the adenosine that could be exploited to potentially enhance the binding affinity.
Figure 6.

(A) Superimposed crystal structures of SAM (C atoms in green), 1 (in pink), 7 (in purple) and 18a (in orange) in DOT1L, showing the binding poses of the adenosine moieties are very similar; (B) The structure of DOT1L:1 showing the binding pocket of the N6-Me (pink sphere); (C) The structure of DOT1L:7 showing the binding pocket of the 7-Br (brown sphere); (D) The structure of DOT1L:22 (with C atoms in orange) showing the binding pocket of the 4-tert-butylphenyl urea group of 22 (in ball & stick model). Together superimposed is the structure of DOT1L:SAM (for clarity only) showing SAM (in green lines) as well as residues Phe239 and Thr139 (in light blue lines). It is noted that these two residues undergo considerable conformational changes to allow the binding of 22. The corresponding Thr139 in DOT1L:22 is largely unordered (with B factors >120) and therefore not shown; (E) Superimposed structures of DOT1L:SAM (in green) and :22 (in orange), highlighting that the flexible loops 302–312 (left, shown as a tube model) and 128–140 (right) undergo considerable conformational changes. Both loops (in green) in the DOT1L:SAM structure are ordered, while the loop 128–140 (in orange) in the DOT1L:22 structure is largely unordered. For clarity, other protein 3-D structures are shown as curved lines.
The crystal structure of the DOT1L:1 complex shows the binding pocket for the N6-substituent,24 as shown in Figure 6b. The wall of the channel-like pocket is composed of mostly hydrophobic residues Leu224, Val249, Lys187 and Pro133, with the other end open to the solvent. The channel is ~4 Å wide and ~5 Å deep. The small N6-methyl group of compound 1 is comfortably situated in the pocket with favorable interactions with the hydrophobic wall. However, the bulkier and longer alkyl groups, e.g., isopropyl and isopentyl, of 2 cannot bind well, as it is either too large for the channel or their hydrophobic tails expose to the solvent water, resulting in reduced binding affinities. The binding site for the 7-substituents (e.g., -Br) in the crystal structure of the DOT1L:7 complex is shown in Figure 6c.59 Different from the N6-pocket, this pocket is buried inside the protein, defined by Phe245, Val249, Leu224, Lys187, Pro133 and Glu134. Favorable hydrophobic interactions between these residues and the -Br result in an enhanced binding affinity. It is noteworthy that the binding pockets for the N6- and 7-substituents exist in the structure of DOT1L:SAH and binding of compounds with these two substituents does not cause a major conformation change of the protein.
However, the binding of urea-containing inhibitors leads to a series of considerable protein conformational changes.56,58 In the crystal structures of DOT1L:18a, :21, :22 and :24, the 4-tert-butylphenyl-substituted urea group is located in a newly formed pocket, which includes the amino acid binding pocket of SAM that connects to a newly opened hydrophobic cavity, as shown in Figure 6d. The urea functionality is recognized by forming three H-bonds with DOT1L. The carbonyl O atom has one H-bond with the sidechain of Asn241. The two -NH- moieties of the urea of these compounds forms two H-bonds with the negatively charged carboxy group of Asp161. This explains the SAR showing each of the -NH- can provide >25-fold activity improvements. Without an amino acid in place, the sidechains of Phe239 and Thr139, which constitute the bottom end of the amino acid binding pocket in DOT1L:SAM, move sideways (Figure 6d) and open up to form a big, mostly hydrophobic pocket, together with Asn241, Ser269, V267, Tyr312, Met147, Leu143, Val144 and Val169. This new room contains and has favorable van der waals interactions with the 4-tert-butylphenyl group. The alterations in amino acid sidechains are accompanied by major backbone conformational changes of two flexible loops (128–140 and 302–312), induced by the binding of the urea-containing inhibitors (Figure 6e). With the natural ligands (i.e., SAM or SAH) bound, these two loops are fully ordered. However, the binding of a urea-containing inhibitor causes the two loops to become partially or fully unordered. This series of conformational changes induced by the urea sidechain seem to be DOT1L-specific, since these DOT1L inhibitors do not bind to other HMTs. Consequently, the crystal structures, especially the newly observed urea-binding pocket can be further exploited for rational drug design. For example, compound 23 without a urea functionality was found to have even more potent activity.
BIOLOGICAL ACTIVITY OF DOT1L INHIBITORS
Cellular H3K79 methylation inhibition
Several highly potent urea containing compounds, i.e., 18a, 22-24, were found to inhibit H3K79 methylation in a variety of cells with the reported EC50 values ranging from 9 – 200 nM.55,57,58,60 As compared to their extremely low enzyme Ki/IC50 values (0.08 – 1 nM), the seemingly reduced cellular activity should be mainly due to a higher cellular concentration of SAM of ~300 μM64 (as compared to that used in a typical DOT1L inhibition assay of 0.7 μM, the Km value for SAM). As competitive inhibitors against SAM, an elevated inhibitor concentration is needed to block the enzyme activity of DOT1L. In addition, consistent with their selective activity against DOT1L, these compounds do not affect the methylation states at H3K4, H3K9, H3R17, H3K27, H3K36 as well as H4R3 and H4K20 sites,55 showing these compounds are very useful probes to investigate the biological functions of DOT1L. H3K79 methylation inhibition by these compounds was found to be time-dependent, which can be observed after incubation for 1 day but the maximal effect is shown in ~4 days. The slow kinetics suggests the reduced H3K79 methylation is due to histone protein turnover. However, the study is complicated by whether there is an H3K79 demethylase. Although not been identified, there is evidence that H3K79 methylation could also be regulated by a JmjC family demethylase.13
Activity against MLL-rearranged leukemia cells
Compounds 18a, 22 and 23 were reported to possess strong activity against the proliferation of leukemia cell lines having an MLL translocation, such as MV4-11 (containing MLL-AF4), MOLM-13 (MLL-AF9) and THP1 (MLL-AF9), with EC50 values ranging from 0.004 – 11 μM.25,55,60 However, these compounds are considerably less active against non-MLL leukemia cells, showing a selectivity of >10-fold. Of interest is the time-dependent antiproliferative activity of DOT1L inhibitors with the maximum potency shown after ~15 days, while these compounds are typically devoid of any activity within 3 days. These features are different from those of a conventional chemotherapeutic or cytotoxic agent that targets non-specifically any rapidly proliferating cells. The slow action of DOT1L inhibitors is likely due to a long time required for cellular events that lead to cell growth arrest, including blocked histone methylation, followed by decreased levels of mRNA expression for the targeted genes, as well as ultimately the depletion of the gene products (proteins) key to the proliferation of MLL leukemia cells. In addition to the MLL leukemia cell lines, compounds 22 and 24 were shown activity against the proliferation of human cord blood cells transformed by MLL-AF9 oncogene, while they exhibited no activity against these cells transformed by a non-MLL oncogene, showing again the selectivity against MLL leukemia.58
Molecular and cell biology studies have been performed to find possible mechanisms by which DOT1L inhibitors block the proliferation of MLL-leukemia cells.55 First, incubation of compound 22 with these cells can inhibit the expression of key onco-MLL targeted genes, including Hoxa9 and Meis1 whose overexpression were found to be a hallmark of this type of leukemia. In addition, the gene expression inhibition was seen in a dose-dependent manner with EC50 values of ~0.7 μM. Second, compound 22 was found to be able to induce differentiation and apoptosis of MV4-11 and MOLM-13 cells. Flow cytometry was used to monitor cells expressing CD14, which is a cell surface marker of more differentiated monocytes/macrophages in the myeloid lineage. Upon treatment with 22, CD14-expressing cell population is considerably expanded. Moreover, Annexin-V staining experiments also showed compound 22 is able to induce apoptosis of MV4-11 (~40%) and MOLM-13 (~20%) cells, although at a relatively high concentration (17 and 4-fold above the corresponding anti-proliferative EC50 values, respectively). Third, gene profiling indicated that the gene expression pattern of cells treated with 22 overlaps significantly with that using DOT1L genetic knocking-down, suggesting the observed cell activities of 22 are caused by DOT1L inhibition. More potent DOT1L inhibitor 23 possesses even more pronounced cell activity than compound 22.60
These cell based studies show DOT1L inhibitors represent promising drug candidates to treat MLL-rearranged leukemia. Different from chemotherapeutic drugs, these compounds do not have general cytotoxicity. Rather, they potently inhibit H3K79 methylation, giving considerably lowered levels of onco-MLL targeted gene expression as well as the downstream effector proteins. Relatively low activity against cells without an MLL rearrangement, even after an extended period of treatment, suggests these compounds have a low toxicity. Mechanistic studies indicated that the anti-proliferative activity of these compounds is mainly mediated by inducing cell differentiation and/or apoptosis.
In vivo antileukemia efficacy
Pharmacokinetic studies of compounds 22 and 23 in mice and rats show the compounds are poor drug candidates with a short half-life in plasma, preventing them from being used in animal studies with conventional dosing methods, such as intraperitoneal and oral administration.55,60 Instead, these two compounds were continuously infused for in vivo studies using an osmotic mini-pump subcutaneously implanted in experimental animals. With this drug delivery route, 22/23 were found to be able to inhibit H3K79 methylation of MV4-11 tumors subcutaneously transplanted in immunosuppressed mice or rats. With a dose of 70 mg/kg/day for 21-day continuous infusion, compound 23 is able to cause a complete regression of subcutaneously transplanted MV4-11 tumor and, after drug treatment, suppress the tumor regrowth for >30 days. Shortening the treatment time to 14 days or reducing the dose to 35 mg/kg/day for 21 days can also inhibit tumor growth, though with less efficacies.60 In a more clinically relevant mouse model using intravenously transplanted MV4-11 leukemia, a 14-day treatment with compound 22 (150 mg/mL in a mini-pump) resulted in a prolonged survival of experimental mice with statistic significance.55
Toxicological studies
In vivo toxicological studies were performed to determine if treatment with 22 and 23 causes acute or chronic toxicities, especially in the hematopoietic system, since DOT1L plays an important role in normal hematopoiesis and homeostasis. A 14- or 21-day continuous administration of 22 (150 mg/mL in a mini-pump) or 23 (70 mg/kg/day) was found to be well tolerated and cause no overt toxicities in experimental animals.55,60 For example, after treatment with 22, the colony forming ability of bone marrow cells are preserved and further flow cytometry analysis showed the hematopoietic stem cell population is not significantly changed. While a statistically significant reduction of the myeloid and erythroid progenitor cells was observed, the granulocyte/monocyte progenitor cells are not affected by the drug treatment. These results suggest selective, pharmacological inhibition of DOT1L may be tolerable and, combined with the in vivo efficacy data, DOT1L inhibitors represent novel drug candidates for treating MLL-rearranged leukemia.
CONCLUSION AND PERSPECTIVES
DOT1L is a unique HMT for several reasons. First, DOT1L is the only HKMT that belongs to the class I methyltransferase family, with a distinct structure from other SET-domain HKMTs. Second, it only binds to nucleosome and methylates H3K79 which is located in the globular histone core. However, other histone methylation sites are located in the unordered N-terminal tails of H3 and H4. Histone or even shorter histone peptides may serve as substrates of other HMTs. Third, DOT1L is a distributive enzyme and able to perform mono-, di- and tri-methylation of H3K79. Molecular and cell biology studies have shown H3K79 methylation is normally associated with active gene transcription and elongation. DOT1L play an important role in embryonic development as well as maintaining normal functions of the hematopoietic system, heart and kidney in adults. Of particular interest to medicinal chemists is that DOT1L has been validated to be a drug target for MLL-rearranged leukemia, which currently has no targeted therapies and carry a poor prognosis. Very potent and selective inhibitors of DOT1L have been discovered and developed. Several of these compounds were found to have activity against this disease in cell-based assays as well as in animal models, with the most advanced compound being in clinical trials. Given the importance of DOT1L in normal physiology, there are concerns in targeting DOT1L for leukemia treatment. However, MLL leukemia cells seem to be much more sensitive to DOT1L inhibition, while at the treatment concentrations the adverse effects to normal tissues are minimal or reversible. This hopefully provides a safe therapeutic window.
There are still several challenges in medicinal chemistry targeting DOT1L, which represent possible future directions for this research. First, more metabolically stable inhibitors of DOT1L are needed, given the poor pharmacokinetic properties of current adenosine-containing inhibitors. The cyclopentane-containing inhibitor 19 shows improved in vitro stability in liver microsomes and plasma, but its in vivo pharmacokinetic properties as well as antileukemia activity remain to be investigated. The available crystal structures of the DOT1L:inhibitor complexes should provide useful tools for future design of more stable and/or potent inhibitors. For example, since the adenosine moiety is attributed to the weak pharmacokinetics, replacing it with a more drug-like fragment could be a viable strategy. However, it is important that the new fragment molecule must occupy the adenosine binding site of DOT1L. Second, there is a need to perform deeper mechanistic studies as to how exactly DOT1L causes MLL leukemia. It has been known that H3K79 hypermethylation leads to overexpression of certain leukemia relevant genes such as Hoxa9 and Meis1. However, what are the downstream molecular events or effector proteins that cause the leukemia? To this end, highly potent and selective DOT1L inhibitors could be explored as a chemical probe for these studies. More knowledge about this point could provide additional targets for MLL leukemia treatment. In addition, compounds targeting a downstream protein could provide a desired synergistic effect once combined with a DOT1L inhibitor. Finally, it has been known that many diseases including cancer are caused by aberrant epigenetic modifications. Given the crucial role of DOT1L or H3K79 methylation in gene regulation, it is anticipated that more diseases or medical applications may be connected to dysfunction of DOT1L. However, except for MLL-rearranged leukemia, there have been very few studies providing compelling evidence showing potential medical applications of targeting DOT1L. More investigation is therefore needed and again, the available DOT1L-specific inhibitors may serve as useful probes for this purpose.
Acknowledgments
This work was supported by a grant (RP110050) from Cancer Prevention and Research Institute of Texas (CPRIT) to Y.S. J.L.A. was supported by a training fellowship from the Keck Center of the Gulf Coast Consortia, on the Pharmacological Sciences Training Program, National Institute of General Medical Sciences (NIGMS) T32GM089657.
ABBREVATIONS
- DOT1
disruptor of telomeric silencing 1
- DOT1L
DOT1-like
- H3K4
histone H3-lysine4
- H3K79
histone H3-lysine79
- HKMT
histone lysine methyltransfers
- HMT
histone methyltransfers
- HTS
high throughput screening
- ITC
isothermal titration calorimetry
- MLL
mixed lineage leukemia
- PRMT
histone/protein arginine methyltransferases
- SAM
S-adenosyl-L-methionine
- SAH
S-adenosyl-L-homocysteine
- SAR
structure activity relationship
- SET
(Su(var)3-9, Enhancer-of-zeste, Trithorax)
- SPR
surface plasmon resonance
Biographies
Justin L. Anglin was born in the Texas Medical Center at the Women’s Hospital of Texas on June 1, 1987. He received his B.Sc. degree in Chemistry from the University of Texas at Dallas. Currently he is working towards his Ph.D. as a graduate student in the laboratory of Dr. Yongcheng Song. Mr. Anglin has worked on rational design, synthesis and biological activity evaluation of DOT1L inhibitors and is looking forward towards completing his thesis in the near future.
Yongcheng Song obtained his Ph.D. degree in Chemistry from Department of Chemistry at National University of Singapore. Supported by a JSPS (Japan Society for Promotion of Science) fellowship, he did a two-year postdoctoral training with Professor Fumie Sato at Tokyo Institute of Technology. He then worked with Professor Eric Oldfield at University of Illinois at Urbana-Champaign in drug discovery. In 2008, Dr. Song joined the faculty of Baylor College of Medicine as an Assistant Professor of Pharmacology as well as a member of Dan L. Duncan Cancer Center. The research interests in his group are to discover and develop small-molecule inhibitors of biologically important proteins targeting cancer and infectious diseases, using a combination of rational drug design, X-ray crystallography, chemical synthesis and biological activity testing.
References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 3.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 6.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 9.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 10.Lacoste N, Utley RT, Hunter JM, Poirier GG, Cote J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002;277:30421–30424. doi: 10.1074/jbc.C200366200. [DOI] [PubMed] [Google Scholar]
- 11.Shanower GA, Muller M, Blanton JL, Honti V, Gyurkovics H, Schedl P. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics. 2005;169:173–184. doi: 10.1534/genetics.104.033191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, Gonzalo S, Zhang Y, Li E, Chen T. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, Slany RK. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7:e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 18.Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, Shilatifard A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes Dev. 2010;24:574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4: a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 21.Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee C, McGinty RK, Fierz B, Muir TW. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nat Chem Biol. 2010;6:267–269. doi: 10.1038/nchembio.315. [DOI] [PubMed] [Google Scholar]
- 23.Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 24.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BV, Song Y. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anglin JL, Deng L, Yao Y, Cai G, Liu Z, Jiang H, Cheng G, Chen P, Dong S, Song Y. Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J Med Chem. 2012;55:8066–8074. doi: 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, van Leeuwen F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol. 2008;15:550–557. doi: 10.1038/nsmb.1432. [DOI] [PubMed] [Google Scholar]
- 27.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barry ER, Corry GN, Rasmussen TP. Targeting DOT1L action and interactions in leukemia: the role of DOT1L in transformation and development. Expert Opin Ther Targets. 2010;14:405–418. doi: 10.1517/14728221003623241. [DOI] [PubMed] [Google Scholar]
- 29.Bernt KM, Armstrong SA. A role for DOT1L in MLL-rearranged leukemias. Epigenomics. 2011;3:667–670. doi: 10.2217/epi.11.98. [DOI] [PubMed] [Google Scholar]
- 30.Laurenson P, Rine J. Silencers, silencing, and heritable transcriptional states. Microbiol Rev. 1992;56:543–560. doi: 10.1128/mr.56.4.543-560.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 34.Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15:175–188. doi: 10.1016/j.semcancer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Ooga M, Inoue A, Kageyama S, Akiyama T, Nagata M, Aoki F. Changes in H3K79 methylation during preimplantation development in mice. Biol Reprod. 2008;78:413–424. doi: 10.1095/biolreprod.107.063453. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen AT, He J, Taranova O, Zhang Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011;21:1370–1373. doi: 10.1038/cr.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, Fields TA, Vivian JL, Fields PE. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010;116:4483–4491. doi: 10.1182/blood-2010-03-276501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen AT, Xiao B, Neppl RL, Kallin EM, Li J, Chen T, Wang DZ, Xiao X, Zhang Y. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25:263–274. doi: 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reisenauer MR, Wang SW, Xia Y, Zhang W. Dot1a contains three nuclear localization signals and regulates the epithelial Na+ channel (ENaC) at multiple levels. Am J Physiol Renal Physiol. 2010;299:F63–76. doi: 10.1152/ajprenal.00105.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–18068. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–6068. doi: 10.1182/blood-2008-12-197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, Ribeiro RC, Rubnitz JE, Girtman K, Williams WK, Raimondi SC, Liang DC, Shih LY, Pui CH, Downing JR. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 43.Chen CS, Sorensen PH, Domer PH, Reaman GH, Korsmeyer SJ, Heerema NA, Hammond GD, Kersey JH. Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood. 1993;81:2386–2393. [PubMed] [Google Scholar]
- 44.Felix CA, Hosler MR, Winick NJ, Masterson M, Wilson AE, Lange BJ. ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood. 1995;85:3250–3256. [PubMed] [Google Scholar]
- 45.Mrozek K, Heinonen K, Lawrence D, Carroll AJ, Koduru PR, Rao KW, Strout MP, Hutchison RE, Moore JO, Mayer RJ, Schiffer CA, Bloomfield CD. Adult patients with de novo acute myeloid leukemia and t(9; 11)(p22; q23) have a superior outcome to patients with other translocations involving band 11q23: a cancer and leukemia group B study. Blood. 1997;90:4532–4538. [PubMed] [Google Scholar]
- 46.Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 47.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 48.Butler LH, Slany R, Cui X, Cleary ML, Mason DY. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood. 1997;89:3361–3370. [PubMed] [Google Scholar]
- 49.Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 51.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, Kutok JL, Kung AL, Armstrong SA. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 53.Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem. 2012;287:5588–5599. doi: 10.1074/jbc.M111.328138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, Lander ES, Armstrong SA, Daley GQ. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basavapathruni A, Jin L, Daigle SR, Majer CR, Therkelsen CA, Wigle TJ, Kuntz KW, Chesworth R, Pollock RM, Scott MP, Moyer MP, Richon VM, Copeland RA, Olhava EJ. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80:971–980. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 57.Deng L, Zhang L, Yao Y, Wang C, Redell MS, Dong S, Song Y. Synthesis, activity and metabolic stability of non-ribose containing inhibitors of histone methyltransferase DOT1L. Med Chem Commun. 2013;4:822–826. doi: 10.1039/C3MD00021D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, Marcellus R, Iacob RE, Engen JR, Griffin C, Aman A, Wienholds E, Li F, Pineda J, Estiu G, Shatseva T, Hajian T, Al-Awar R, Dick JE, Vedadi M, Brown PJ, Arrowsmith CH, Bradner JE, Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 59.Yu W, Smil D, Li F, Tempel W, Fedorov O, Nguyen KT, Bolshan Y, Al-Awar R, Knapp S, Arrowsmith CH, Vedadi M, Brown PJ, Schapira M. Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg Med Chem. 2013;21:1787–1794. doi: 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, Waters NJ, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM. Potent inhibition of DOT1L as treatment for MLL-fusion leukemia. Blood. 2013 doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chesworth R, Kuntz KW, Olhava EJ, Patane MA. Preparation of nucleosides as modulators of histone methyltransferase. WO 2011-US63309 PCT Int Appl. 2012
- 62.Chesworth R, Kuntz KW, Olhava EJ, Patane MA. 7-Deazapurines as Histone methyltransferase modulators. WO 2011-US63314 PCT Int Appl. 2012
- 63.Olhava EJ, Chesworth R, Kuntz KW, Richon VM, Pollock RM, Daigle SR. Preparation of substituted purine and 7-deazapurine compounds as modulators of epigenetic enzymes. WO 2011-US63044 PCT Int Appl. 2012
- 64.Chiba P, Wallner C, Kaiser E. S-adenosylmethionine metabolism in HL-60 cells: effect of cell cycle and differentiation. Biochim Biophys Acta. 1988;971:38–45. doi: 10.1016/0167-4889(88)90159-0. [DOI] [PubMed] [Google Scholar]