

Abstract
The mammalian kidney filtration barrier is a complex multicellular, multicomponent structure that maintains homeostasis by regulating electrolytes, acid–base balance, and blood pressure (via maintenance of salt and water balance). To perform these multiple functions, podocytes—an important component of the filtration apparatus—must process a series of intercellular signals. Integrating these signals with diverse cellular responses enables a coordinated response to various conditions. Although mature podocytes are terminally differentiated and cannot proliferate, they are able to respond to growth factors. It is possible that the initial response of podocytes to growth factors is beneficial and protective, and might include the induction of hypertrophic cell growth. However, extended and/or uncontrolled growth factor signalling might be maladaptive and could result in the induction of apoptosis and podocyte loss. Growth factors signal via the activation of receptor tyrosine kinases (RTKs) on their target cells and around a quarter of the 58 RTK family members that are encoded in the human genome have been identified in podocytes. Pharmacological inhibitors of many RTKs exist and are currently used in experimental and clinical cancer therapy. The identification of pathological RTK-mediated signal transduction pathways in podocytes could provide a starting point for the development of novel therapies for glomerular disorders.
Introduction
Podocytes are highly specialized cells with a unique structure and function. These cells, which are located adjacent to the glomerular capillaries and form part of the glomerular filtration barrier, have microtubule-based cellular extensions known as primary processes, and actin-based membrane extensions known as foot processes.1 The foot processes form distinct subcellular compartments within the podocyte and enable spatially and temporally distinct metabolic and signalling activities. As a result of their unique location at the interface of blood and urine, podocyte membranes comprise three distinct signalling platforms: the sole plate (baso-lateral membrane attached to the glomerular basement membrane [GBM]), slit diaphragm (cell-to-cell junction formed between adjacent podocytes), and apical membrane, which is bathed in urine (Figure 1).2 The existence of a ‘subpodocyte’ space within the glomerulus that might restrict fluid and solute movement across the glomerular capillary wall and generate concentration gradients of signalling molecules within the foot processes has also been suggested.3 Although both primary processes and foot processes receive signals, current research is mainly focused on signals that are generated at the foot processes. Podocytes express a number of signal transduction receptors, including receptor tyrosine kinases (RTKs), G-protein coupled receptors (GPCRs) and nuclear receptors.4,5 Integrins also have essential roles in podocytes, and mediate cell matrix adhesion as well as outside-in signalling, which has been reviewed previously.6 In this Review, we summarize the basic principles of signal transduction and discuss the physiological and pathological roles of RTK signalling in podocytes.
Figure 1.
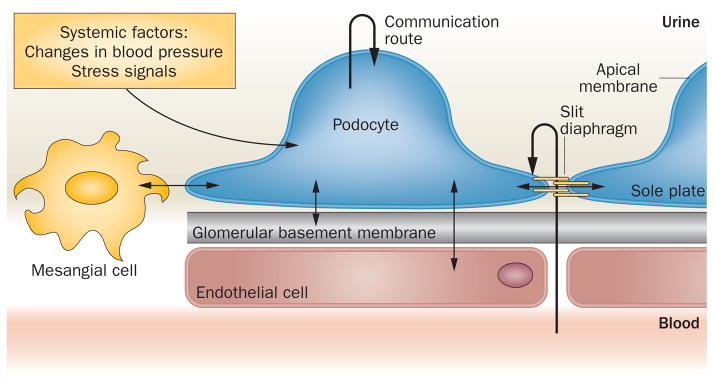
Podocytes form complex communication networks with their environment and are in constant contact with a variety of signal sources and receivers, including other glomerular cell types and the extracellular matrix. They also signal to neighbouring podocytes, enabling coordination of podocyte function in the glomerulus. Podocytes are exposed to a myriad of potential signalling molecules present in the circulation as well as biophysical cues, such as changes in blood pressure.
Signal transduction
First messengers
In the biological context, signal transduction refers to the mechanisms that permit external chemical signalling molecules—the first messengers—to direct cell activities. On a molecular level this process is difficult because the cell membrane, although very thin, is impermeable to ions and polar molecules, including amino acid derivatives, peptides and proteins. With a few exceptions (steroid hormones, thyroid hormones and prostaglandins), first messengers induce cellular changes without penetrating the target cell. They bind to receptors on the cell surface, thereby serving as extracellular soluble ligands of these receptors (Figure 2). The binding of only a few ligand molecules to cell-surface receptors might induce remarkable changes within the cell as it becomes activated. The evolution of receptors together with the first messengers (hormones, neurotransmitters, cytokines and growth factors) enabled membrane-impermeable external signals to influence cell behaviour and function and conferred high specificity and precise control in terms of the extent and duration of signalling.7 The concept of specific receptors for ligands predates the discovery of the first hormones and hormone receptors, and can be attributed to studies published in 1878 that identified the mutual antagonism of the poisons atropine and pilocarpine,8,9 which bind muscarinic cholinergic receptors.
Figure 2.

Cell signalling receptors differ in their mechanisms of activation and signal transmission, subcellular localization, and ligand binding. They can be broadly classified into three groups: GPCRs, receptors with enzyme-linked activities (such as receptor tyrosine kinases) and cytokine receptors. Some receptors mediate cell-to-cell or cell-to-matrix contact by binding transmembrane proteins or extracellular proteins, respectively, and link cell adhesion to outside-in and inside-out signalling. Ligand binding ultimately leads to the activation of second messengers, protein kinases and phosphatases that modify and determine the phosphorylation state of a variety of target proteins and thereby regulate all aspects of cell function and structure. Some ligands that induce signalling via receptor binding are transported across the cell membrane via the receptor, or induce transport of other extracellular molecules or ions that have intracellular effects, which might not involve a second messenger system. Lipophilic ligands that can cross the cell membrane by simple diffusion target intracellular receptors that act directly as transcriptional regulators. Abbreviation: GPCR, G-protein coupled receptor.
Second messengers
As first messengers cannot enter the cell, their receptors activate intracellular substitutes or second messengers, such as 3′ 5′ cyclic adenosine monophosphate, calcium ions, diacylglycerol, and phosphorylated inositol lipids, in response to ligand binding. These second messengers interact either directly or indirectly (via adaptor proteins) with a specific set of enzymes, and regulate enzymatic activity and access to substrates. As most of these enzymes are protein kinases or phosphatases, the majority of cellular responses to the stimulation of cell-surface receptors are eventually mediated by protein phosphorylation or dephosphorylation.10 The enzymes act directly on target structures or activate additional enzymes to generate a stepwise process of consecutive enzymatic activation, known as a signal transduction cascade. This cascade enables signals to be amplified (or alleviated) as they travel, and to branch out into many different signalling pathways. One first messenger can, therefore, stimulate many responses. Depending on the ultimate target of a particular signalling pathway (gene promoters and structural DNA components in the nucleus, the cytoskeleton, or metabolic factors in the cytoplasm, mitochondria and other organelles), a variety of cellular responses are possible.7
Receptors
Signal transduction receptors can be broadly divided into three classes (Figure 2). First, receptors that penetrate the plasma membrane and either have intrinsic enzymatic activity (such as RTKs) or are coupled to intracellular enzymatic activity (such as cytokine receptors). Second, receptors characterized by seven transmembrane spanning domains (also known as serpentine receptors or GPCRs) and coupled to intracellular proteins that bind and hydrolyze GTP. Third, nuclear receptors that bind to a ligand, translocate into the nucleus and directly affect gene transcription.
Receptor tyrosine kinases
Erickson and colleagues’ discovery that v-Src (the protein encoded by the transforming gene of Rous sarcoma virus) is a protein kinase was a landmark event in the late 1970s.11 In 1980, Hunter and Sefton reported that v-Src phosphorylates tyrosine residues in substrate proteins and v-Src became the first tyrosine kinase to be identified.12 In the same year, epidermal growth factor receptor (EGFR) was shown to be a protein tyrosine kinase that is activated when bound to epidermal growth factor (EGF),13 and became the first RTK to be identified. The insulin receptor was identified as a RTK a couple of years later.14
The discovery that many growth factor receptors are protein tyrosine kinases stimulated the search for physiological substrates. Surprisingly, the most prominent substrates seemed to be the receptors themselves.15 This finding made more sense in the late 1980s, when Pawson and colleagues showed that proteins containing Src homology 2 (SH2) domains interact directly with activated RTKs16 by binding phosphorylated tyrosine residues within a specific amino acid motif.17 Receptor autophosphorylation is critical to induce binding sites for cytoplasmic proteins with SH2 domains, which then stimulate downstream signalling pathways.17 Many SH2-proteins also contain Src homology 3 (SH3) domains, which bind proline-rich motifs in target proteins and thereby serve as a molecular link between tyrosine phosphorylation mediated by receptor and nonreceptor tyrosine kinases and downstream signalling molecules.18 About 100 SH2 and 250 SH3 domains are encoded in the human genome, underlining their importance in the assembly of specific protein complexes into signalling modules.19,20 Since the discovery of SH2 and SH3 domains, the number of protein–protein and protein–phospholipid interaction signatures found in proteins that are part of signal transduction pathways, and link a particular upstream effector containing a particular binding domain to a specific downstream target protein that has a compatible acceptor motif, has exponentially increased.21
Structure
The two main groups of tyrosine kinases—cytoplasmic nonreceptor tyrosine kinases and RTKs—share the same tyrosine kinase module.22 RTKs are present in all metazoans, but are not found in lower eukaryotic organisms, such as yeast and bacteria.22 The number and complexity of RTKs increased during evolution, possibly in association with the acquisition of new cellular functions or regulatory processes.7 In humans, the RTK family is comprised of 58 members, which are encoded by separate genes.22 All RTKs share a common overall composition comprising an extracellular ligand-binding region, a single-pass transmembrane domain, and an intracellular kinase domain, which is sometimes split into two parts.15,22 The RTKs have been subdivided into 20 subfamilies based on the presence of various extracellular domains, including immunoglobulin-like and cysteine-rich domains.23
Activation
RTKs exist as monomers, and ligand binding induces receptor dimerization—the first step in receptor activation.15 Several growth factors (including vascular endothelial growth factor [VEGF] and platelet-derived growth factor [PDGF]) exist as homodimers, whereas others (including EGF and fibroblast growth factors [FGFs] act as monomers and form a 2:2 ligand to receptor stoichiometry.23 In many cases, ligand binding requires specific coreceptors (transmembrane proteins or components of the extracellular matrix) and receptor activation leads to the formation of a high-molecular-weight signalling complex at the cell surface. Receptor dimerization results in the transphosphorylation of several tyrosine residues in the intracellular part of the RTK, which is catalysed by the kinase domain.14 By contrast, the most complex members of the RTK family, the insulin and insulin-like growth factor (IGF) receptors, exist as disulphide-linked heterotetramers in the inactive state.24 Phosphorylated tyrosine residues in RTKs serve as docking sites for adaptor proteins (such as growth factor receptor-bound protein 2 and SHC-transforming protein 1) and enzymes (such as phospholipase C-γ, Ras-associated GTPase activating protein 1 and phosphoinositide 3-kinase) that contain SH2 domains. These RTK-binding proteins carry the signal forward, leading to activation of specific downstream signalling cascades.15
Key roles
The majority of growth factors induce their cellular effects by binding RTKs.15 Many growth factors are quite versatile, whereas others are specific to particular cell types. This specificity is explained by the patterns of expression of their RTKs. Growth factors control several cellular processes, including cell proliferation, differentiation, survival and migration. RTKs, therefore, have key roles in maintaining tissue homeostasis during the development and adult life of multicellular organisms.23 Unsurprisingly, deregulated RTK signalling has been linked to various human diseases, including cancer.25 Mechanisms that might contribute to aberrant RTK activation in carcinogenesis include gene amplification, chromosomal translocation, point mutation, autocrine activation and impaired receptor downregulation.26 RTKs that are altered in human cancer include members of the EGFR, PDGF receptor (PDGFR), VEGF receptor (VEGFR) and FGF receptor (FGFR) families.27 Growth factors and their receptors participate in promoting several steps of carcinogenesis, including clonal cancer cell expansion, epithelial–mesenchymal transition (EMT), invasion and angiogenesis.25
Receptor tyrosine kinases in podocytes
Mature podocytes are post-mitotic cells that cannot undergo cell division.28 The effects of growth factors on podocytes must, therefore, be different from their ‘classic’ effects in other cell types and must not involve the induction of cell proliferation. Hypertrophic growth of podocytes accompanied by mitosis serves as a compensatory mechanism in situations of podocyte injury and loss.29 However, the activation of mitogenic programmes in podocytes is thought to lead to aberrant mitosis and cell death, a mechanism termed mitotic catastrophe.30 As growth factors are mitogenic, they might be involved in the induction and fine regulation of mitotic events in podocytes, and the potential beneficial versus pathological effects of these events.
To date, at least 15 RTKs have been implicated in podocyte biology (Table 1). Expression of most of these RTKs in podocytes was confirmed by determining the effects of treatment with the respective ligands on cultured podocytes or mouse glomeruli. Only a few RTKs have been shown to be present in podocytes using immunohistological analyses. Prominent examples of RTKs that have a role in podocytes include members of the VEGFR, FGFR, EGFR, and PDGFR families, hepatocyte growth factor (HGF) and the insulin receptor.
Table 1.
Roles of receptor tyrosine kinases in podocytes
| Receptor tyrosine kinase | Ligand | Physiological role | Pathological role | References |
|---|---|---|---|---|
| Tie-1 Tie-2* |
ANG 1 ANG 2 |
Mediates crosstalk between podocytes and endothelial cells | Induces apoptosis of glomerular endothelial cells and proteinuria | 145,146 |
| Unknown‡ | ANGPTL3 ANGPTL4 |
Regulates cell motility, promotes cell adhesion and protects against injury-induced cell detachment | Induces proteinuria and MCD-like injury | 147–149 |
| Unknown§ | CTGF | Maintains actin cytoskeleton and cell morphology under stress | Promotes podocyte injury and loss in diabetes | 150–153 |
| DDR1 | Type IV collagen | Maintains structural integrity of the GBM and preserves foot process and slit diaphragm structure | Serves as positive feedback loop with inflammatory response in glomerulonephritis | 154,155 |
| EGFR | EGF HB-EGF TGFα |
Mediates proliferation and differentiation during development and hypertrophic growth of mature cells | Promotes angiotensin II-mediated injury, dedifferentiation and conversion into a proliferative and/or migratory phenotype in rapidly progressive glomerulonephritis | 135–137, 141 |
| EphB | Ephrin B1 Ephrin B2|| |
Mediates maintenance of the slit diaphragm and prosurvival signalling during transient capillary collapse | Unknown | 155,156 |
| FGFR | FGF2 FGF4 |
Mediates proliferation and differentiation (epithelial–mesenchymal transition) during development and regeneration after injury | Increases nephrin ubiquitination and internalization and induces actin rearrangement, cell hypertrophy, mitosis (resulting in aneuploidy), foot process effacement and focal segmental glomerulosclerosis-like injury | 57,58,66, 70,71 |
| HGFR (MET) | HGF | Mediates antiapoptotic and protective effects and preserves the structure of the actin cytoskeleton, slit diaphragm and foot processes | Unknown | 110,115–117 |
| IGF-IR | IGF-I IGF-II |
Promotes podocyte survival and outgrowth, maintains glomerular filtration barrier | Unknown | 157–160 |
| IGF-IIR (M6P) | α-GalA | Mediates endocytotic uptake of α-GalA. | Mediates accumulation of glycosphinglolipid deposits in podocytes in Fabry disease | 161 |
| Insulin receptor | Insulin | Promotes glucose uptake into podocytes, maintains the glomerular filtration barrier and regulates expression of VEGF-A and TRPC6 | Induces proteinuria in diabetic nephropathy | 40,163,164 |
| NTR¶ | Neurotrophins (NGF, BDNF) | Mediates antiapoptotic effects and might protect against calcium-induced mitochondrial swelling | SNPs of neurotrophins and NTRs are associated with susceptibility, pathological advancement, podocyte foot process effacement, and development of proteinuria in children with IgAN | 165,166 |
| PDGFR | PDGF | Unknown | Induces podocyte dedifferentiation and loss, and foot process effacement | 101,102,105 |
| Ret | GDNF | Mediates prosurvival effects and serves as adaptive response for remodelling and repair, also involved in podocyte development | Unknown | 167,168 |
| VEGFR1# VEGFR2** |
VEGF | Mediates crosstalk between podocytes and endothelial cells and maintains the integrity of the glomerular endothelium | Increases in ligand levels results in various types of podocyte injury (including MCD-like and collapsing glomerulopathy) | 48–50,169 |
Tie-1 and Tie-2 are expressed in glomerular endothelial cells, whereas ANG 1 and ANG 2 are expressed by podocytes.
ANGPTLs do not bind to Tie-1 or Tie-2. Their receptors are not well described but might include integrins.
Several RTKs, including PDGFR, VEGFR and NTR, function as CTGF receptors in other cell types. Nonreceptor tyrosine kinases can also mediate CTGF effects in other cell types.
Ephrin ligands are membrane bound and can function as signalling receptors (reverse signalling).
Seems to be expressed only in mitochondria of undifferentiated, proliferating podocytes.
Also exists in a truncated soluble form that might serve as decoy receptor for VEGF and PlGF and can contribute to endothelial dysfunction and proteinuria in pre-eclampsia.169
In the glomerulus, podocytes seem to be the main source of VEGF, whereas endothelial cells receive the signal via VEGFR2. VEGF might also bind neuropilin-1 and neuropilin-2.
Abbreviations: α-GalA, α-galactosidase A; ANG, angiopoietin; ANGPTL, ANG-related protein; BDNF, brain derived neurotrophic factor; CTGF, connective tissue growth factor; DDR1, discoidin domain receptor tyrosine kinase 1; EGF, epidermal growth factor; EGFR, EGF receptor; EphB, ephrin type B receptor; FGF, fibroblast growth factor; FGFR, FGF receptor; GBM, glomerular basement membrane; GDNF, glial cell-line derived neurotrophic factor; HB-EGF; heparin-binding EGF-like growth factor; HGF, hepatocyte growth factor; HGFR, HGF receptor; IgAN, IgA nephropathy; IGF-I, insulin-like growth factor 1; IGF-II, insulin-like growth factor II; IGF-IR, IGF-I receptor; IGF-IIR, IGF-II receptor; M6P, mannose 6-phosphate receptor; MCD, minimal change disease; NGF, β-nerve growth factor; NTR, neurotrophin receptor (also known as trkA and p75NTR); PDGF, platelet-derived growth factor; PDGFR, PDGF receptor; PlGF, placental growth factor; SNP, single nucleotide polymorphism; TGFα, transforming growth factor-α; TRPC6, transient receptor potential cation channel 6; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
Insulin
The effects of insulin and IGF on podocytes and glomerular filtration are discussed briefly here and in detail elsewhere.31–34 Podocytes were identified as a direct target for insulin in 2005.35 Glomerular dysfunction in insulin-resistant patients was initially thought to be related to the actions of insulin on the vasculature.36,37 However, the effects of insulin on podocytes,35 combined with increasing evidence that podocyte dysfunction has an important role in the pathogenesis of diabetic nephropathy,38 indicates that insulin signalling is crucial for the regulation of podocyte function. Insulin stimulates nephrin-dependent prosurvival cascades that involve activation of AKT,39,40 and mice with podocyte-specific deletion of the insulin receptor develop features of diabetic nephropathy, including albuminuria, podocyte apoptosis and glomerulosclerosis.40 These findings support the hypothesis that impaired insulin signalling in podocytes has a causative role in renal pathology in patients with diabetes.
The precise functions of insulin signalling in podocytes are unclear, but most likely involve regulation of the actin cytoskeleton.40 Reversible effects of insulin on podocyte morphology might explain why insulin infusions cause transient proteinuria in healthy individuals.41 Interestingly, insulin also increases the cell-surface localization of glucose transporter type 1 and glucose transporter type 4, resulting in increased glucose uptake.35 This finding indicates that podocytes can respond to insulin in a similar way to the ‘classic’ insulin target tissues, such as skeletal muscle, liver and fat. Whether glucose uptake into podocytes is physiologically relevant (for example, during the postprandial period), and whether it has a role in insulin-mediated changes in podocyte morphology, remains unclear.
Vascular endothelial growth factor
VEGFR–VEGF signalling has been intensively studied and most likely serves as an important communication route to coordinate endothelial and podocyte function within the glomerulus.42,43 The precise source of the ligand and localization of the receptor are still under debate, and results of experimental studies are contradictory. Expression of VEGFR1 and/or VEGFR2 in podocytes44–46 and direct effects of VEGF on cultured podocytes, including positive effects on cell survival,47 have been reported. In addition, mice with podocyte-specific overexpression of VEGF undergo pathological alterations, including foot process effacement,48,49 further indicating that podocytes respond to VEGF and might use VEGF signalling as an autocrine regulatory system.
By contrast, podocyte-specific deletion of VEGFR2 in mice did not alter glomerular function or ameliorate the glomerular phenotype induced by VEGF over-expression.50 As global deletion of VEGFR2 results in vascular defects, including defects in the glomerular microvasculature,50 this finding suggests that VEGF signalling has paracrine functions within the glomerulus, with the podocyte as the source of the ligand, and the endothelium as the target. Clearly, more research is needed to understand the role of VEGF signalling in podocytes and determine whether endothelial cells also signal to podocytes via VEGF. Most likely, VEGF affects multiple cell types and exerts multiple effects on patho-physiological processes in the glomerulus. The current controversies regarding VEGF signalling in podocytes illustrate the complexity of this field and the limitations of experimental in vitro and in vivo models to determine precise communication routes between cells and the localization of individual signalling events. The analyses of other ligand and receptor systems, including the ones discussed below, face similar challenges.
Fibroblast growth factors
Physiological role
The FGF family consists of 22 members51,52 with various functions depending on their target tissues, including regulation of cell proliferation, survival, migration and differentiation.53 Their biological effects on target cells are mediated by interaction with one of four widely expressed FGFRs (FGFR1, FGFR2, FGFR3 and FGFR4).54 FGFR signalling is critical for the growth and patterning of all renal lineages during the early and late stages of kidney development.55 A soluble dominant-negative receptor that binds several FGF isoforms can abolish renal development,56 indicating that FGF–FGFR signalling is essential for the process. Mature podocytes express FGFR1 and FGFR2,57,58 as well as several FGF family members (including FGF1, FGF2, FGF7 and FGF10).57,59–61 Within the glomerulus, FGF2 has mitogenic effects on podocytes,60 mesangial cells62,63 and endothelial cells.64 As mesangial cells also express FGF2,62 autocrine and paracrine FGFR signalling is likely to be an important regulatory component of podocyte function and form part of an intraglomerular communication system. As for other secreted factors and signalling mediators, to experimentally determine the exact communication routes and to separate the FGF source from its target, whose identity and degree of involvement might vary dependent on the physiological or pathological context, will be challenging.
Current understanding of the physiological role of FGF–FGFR signalling in the regulation of podocyte and overall glomerular function is limited. FGFs are thought to be required for proper podocyte differentiation and to be involved in podocyte recovery after glomerular injury.57,65 FGF2 is upregulated during podocyte differentiation and remains highly expressed in mature podocytes.65 Murine podocytes that lack FGF2 do not undergo EMT; their post-mitotic differentiation is blocked, they cannot upregulate the expression of key regulators of podocyte differentiation and function (such as synaptopodin and Wilms’ tumor suppressor gene [WT-1]), and they fail to reorganize their actin cytoskeleton into a stress-fibre pattern and extend cellular process.57
Although several FGFs bind FGFR1 and FGFR2, which are expressed in podocytes, surprisingly few studies have investigated the roles of FGFs other than FGF2 in these cells. FGF4 can induce internalization and enrichment of nephrin in intracellular vesicles, and accelerates ubiquitination of nephrin and podocin in murine podocytes that lack CD2-associated protein (CD2AP).66 FGF4-induced activation of mitogen- activated protein kinase (MAPK) and AKT signalling is decreased in podocytes that lack CD2AP, suggesting that this adaptor protein is important for the activity of RTKs such as FGFR.67
Pathological role
Given the mitogenic nature of FGFs, it is not surprising that this family of growth factors has also been linked to the induction of injury in the mature kidney. FGF2 is involved in tubulointerstitial damage by increasing the proliferation of tubular epithelia cells68 and renal fibroblasts.69 Furthermore, several animal studies have shown that FGF2 can induce glomerular injury. In rats, long-term treatment with FGF2 results in conspicuous structural changes in the glomeruli, consisting of hypertrophy, widespread vascular degeneration,70 and the development of a focal segmental glomerulosclerosis (FSGS)-like phenotype.71 In response to FGF2 treatment, rat podocytes seem to re-enter the cell cycle and undergo mitosis, but are unable to complete cytokinesis. The process, therefore, results in the generation of multi-nucleated cells and might lead to podocyte degeneration.71 The majority of podocytes in FGF2-treated rats exhibit degenerative changes including cell body attenuation, extensive pseudocyst formation, foot process effacement and detachment from the GBM. Interestingly, the glomerular pathology in these rats includes hypertrophic growth of podocytes.70,71 Our unpublished work indicates that in podocytes, FGF2 can activate nuclear factor of activated T cells (NFAT), which is a potent inducer of cellular hypertrophy in other post-mitotic cells, such as cardiac myocytes.72 It is intriguing to speculate that FGF2-induced activation of the calcineurin–NFAT signalling axis might be an important mechanism of FSGS-like injury, a view that is supported by the finding that podocyte-specific activation of NFAT causes podocyte damage and proteinuria.73,74
Injection studies in rats with passive Heymann nephritis (PHN), a model of membranous glomerulonephritis, have shown that FGF2 can also augment pre-existing podocyte damage and accelerate glomerulosclerosis.58,75 In these rats, FGF2 increased podocyte mitosis and ploidy by changing expression of cell-cycle regulators, such as p21, and eventually induced podocyte apoptosis.75 FGF2 injections also increase podocyte injury in models of other diseases, including diabetic nephropathy.76 In rats treated with puromycin aminonucleoside (PAN), a model of foot process effacement and minimal change disease (MCD)-like alterations, expression of FGF2 and the four FGFR isoforms were increased in podocytes.77,78 Interestingly, FGF2 injection in this model markedly increased podocyte damage and proteinuria, whereas injection of an FGF2-neutralizing antibody suppressed podocyte injury and reduced foot process effacement.77,78 These studies suggest that FGF2 accelerates podocyte injury by inducing mitosis.
Few human studies have investigated the potential association between FGF2 expression and glomerular disease. The lack of clinical studies might be related to the paracrine nature of FGF2 signalling; local changes in the expression and/or release of FGF2 (rather than increases in plasma levels that can easily be detected by ELISA) are expected to underlie tissue injury.79 Interestingly, in a clinical study in which patients with angina pectoris received a single intracoronary injection of recombinant FGF2, the participants developed proteinuria that correlated with the FGF2 dose.80 In addition, increased FGF2 activity has been reported in plasma from diabetic patients with overt proteinuria,81 and increased glomerular accumulation of FGF2 has been shown in patients with HIV-associated haemolytic uraemic syndrome.82
These findings indicate that FGF2 can induce podocyte injury, and it is likely that FGF2 signalling in podocytes regulates actin dynamics and consequently cell morphology and function. Both FGF2 deficiency and elevated FGF2 signalling might be detrimental, as podocytes that lack FGF2 fail to develop stress fibres,57 and rats treated with PAN show increased FGF2 expression and podocyte injury.77,78 The release of FGF2 from glomerular sources (including podocytes) during injury might represent an important mechanism by which podocyte damage is enhanced or becomes self-sustained. This pathological effect might involve an increase in FGFR1 expression in podocytes.58 Unlike the majority of secreted proteins (such as hormones and growth factors), FGF2 lacks a classic peptide signal sequence for endoplasmic reticulum targeting and secretion via the Golgi system.84 Therefore, it was initially assumed that cytosolic FGF2 can only be released by plasma membrane disruption and serves as an endogenous amplifier of cytotoxic damage following immune-mediated injury to cells, such as mesangial injury in rats with anti-Thy1.1 glomerulonephritis.84 However, a specific FGF2-release mechanism that is independent of the endoplasmic reticulum has now been identified, indicating that FGF2 can also be released from living cells as part of normal cell-to-cell communication.85
The extracellular matrix (ECM) has a key role in storing and sequestering FGFs and in concentrating FGFs around their receptors, thereby regulating FGF signalling. Heparin sulphate and heparin sulphate proteoglycans (HSPGs) are essential coreceptors for FGF-induced FGFR activation.86 It has been postulated that FGF2 produced by podocytes is stored in the GBM, and might stimulate podocyte proliferation in response to podocyte injury and loss.60 Furthermore, WT-1 regulates expression of 6-O-endosulphatases, which are critically involved in the maintenance of the glomerular filtration barrier by modulating the bioavailability of signalling molecules including FGF2.87 Mice that are deficient in the extracellular sulphatases Sulf1 and Sulf2, which remodel the heparan sulphate 6-O-sulfation pattern in the ECM, show increased FGF2-induced activation of the MAPK pathway and develop age-dependent proteinuria as a result of ultrastructural abnormalities in podocytes and endothelial cells.87 This phenotype is similar to that observed in children with WT1 mutations, which are associated with the severe, early-onset nephrotic syndrome, Denys-Drash Syndrome (DDS).88 The possibility exists that FGF2 signalling in podocytes might also mediate the glomerular phenotype in children with DDS.
A genome-wide association study, which included 195 patients with biopsy-proven nephrotic syndrome, showed that single nucleotide polymorphisms in the GPC5 gene, which encodes glypican-5, were associated with the disease.89 Glypicans belong to the family of cell surface HSPGs and bind a multitude of growth factors, including FGF2.90 Glypican-5 enhances FGF2 signalling in podocytes and promotes their dedifferen-tiation.89 In proteinuric mice, Gpc5 knockdown using systemic injections of short-interfering RNA ameliorated proteinuria and reduced pathological changes, even after the development of nephrotic syndrome, indicating a role of glypican-5 in FGF2-induced podocyte injury in vivo.89 Interestingly, combined injection of PAN and FGF2 is a fast and homogenous way of inducing podocyte injury and nephrotic syndrome-range proteinuria in mice.89 These mice develop massive proteinuria within 5 days after combined injection, but this does not occur if only one of these substances is injected. Proteinuria lasts for at least 10 days and mice develop an FSGS-like histological phenotype with extensive foot process effacement and tubulointerstitial damage. This mouse model demonstrates pathological effects of FGF2 on the glomerular filter, at least in the context of co-existing pathological stimuli, and could be used as a model of nephrotic syndrome to study FSGS-like mechanisms.
Platelet-derived growth factors
The four PDGF family members, (PDGF-A, PDGF-B, PDGF-C and PDGF-D) act as disulphide-linked dimeric growth factors. The PDGFRs exist as α-chains and β-chains that can form homodimers and heterodimers.91 In mesangial cells, PDGFs are autocrine regulators of cell proliferation and migration as well as ECM production, and are involved in mesangial expansion in renal disease.92 The expression of PDGF-B and PDGFR-β is increased in glomerular lesions in patients with diabetic nephropathy or IgA nephropathy, and in rodent models of renal ablation and glomerulonephritis.93–96 PDGF-D can induce mesangial cell proliferation in vitro and is a mediator of mesangial-proliferative nephritis in vivo.97–99
As they express PDGF-D100 but not PDGFR-β,101,102 which is required for PDGF-D responsiveness,103 podocytes seem to function as a source but not a target of PDGF-D. In transgenic mice, podocyte-specific overexpression of PDGF-D induces proliferative glomerulonephritis, widespread glomerulosclerosis, tubulointerstitial injury and proteinuria.104 Interestingly, podocyte injury in these mice includes foot process effacement, podocyte dedifferentiation (indicated by reduced expression of nephrin and podocin) and podocyte loss.104 Two possibilities exist, podocytes might respond directly to PDGF-D in an autocrine fashion, or PDGF-D expressed by podocytes might act on mes-angial cells, which then respond by sending out a different pathological signal that induces podocyte injury. To date, very little is known about a potential podocyte– mesangial crosstalk, and these transgenic mice might provide the first evidence that podocyte-specific over-expression of a growth factor is capable of inducing paracrine cell proliferation in the glomerular tuft upstream of the filtration flow. Podocytes might locally alter their PDGF release in response to injury, and this alteration might induce intraglomerular damage, such as the mesangial expansion observed in FSGS.
Hepatocyte growth factor
HGF was originally identified as a potent mitogen for hepatocytes.105,106 However, HGF acts on various cell types and exhibits pleiotropic activities during embryogenesis and tissue repair.107 The mitogenic and morphogenic properties of HGF suggest that this growth factor participates in tissue regeneration not only by stimulating cell proliferation, but also by promoting cell motility and spatial organization.107 The biological activities of HGF are mediated by a single HGF receptor (HGFR, also known as MET),108 which is predominantly expressed in epithelial cells in various organs, whereas HGF is primarily derived from the mesenchyme.109 This separation of ligand and receptor suggests paracrine roles of HGF.
In the kidney, HGF expression is limited to non-epithelial cells, whereas HGFR is ubiquitously expressed.110 HGF has antifibrotic effects and stimulates tubular cell mitogenesis and morphogenesis, preventing the onset and progression of a variety of progressive kidney diseases, including diabetic nephropathy.111,112 Glomerular effects of HGF are supported by evidence from in vivo studies in which administration of recombinant HGF protein or cDNA ameliorated proteinuria in mouse models of diabetic and PAN nephropathy.113–115 Furthermore, injection of HGF suppressed foot process effacement and attenuated albuminuria in mice with lipopolysaccharide-induced podocyte injury.116 Interestingly, mice with podocyte-specific deletion of HGFR develop normally without pathological lesions or proteinuria,117 indicating that HGF signalling is not required for podocyte maturation, survival and function under normal conditions. However, after doxorubicin treatment, HGF-knockout mice developed more-severe podocyte injury and albuminuria than did their wild-type littermates, indicating that HGF has protective functions in podocytes.117 Furthermore, ectopic HGF expression protected wild-type mice from the detrimental effects of doxorubicin, but did not protect mice with podocyte-specific HGFR depletion. The protective role of HGF in podocytes seems to include antiapoptotic effects,113,117,118 preservation of the actin cytoskeleton,115 and maintenance of nephrin and synaptopodin expression.116,117 As HGF regulates EMT in other renal cell types, such as tubular epithelial cells,119,120 and podocytes undergo EMT after injury,121,122 HGF might also have a role in preserving podocyte structure and function after injury, thereby preventing the development of proteinuria.
Epidermal growth factor
EGFRs are widely expressed and their activation is involved in the regulation of many cellular processes, including cell proliferation, differentiation and survival.123 Overexpression or overactivation of EGFRs is associated with various cancers, and genetic alterations in erbB2 (also known as HER2), a member of the EGFR subfamily of RTKs, have been implicated in about 30% of all epithelial cancers.124 EGFRs are promising targets for an expanding class of anticancer therapies, including the FDA approved drugs trastuzumab, erlotinib and gefitinib, and many other agents that are currently in development or clinical trials.125,126
EGF and related growth factor peptides, such as heparin-binding EGF-like growth factor (HB-EGF) and transforming growth factor-α (TGFα), are transmembrane proteins, which are cleaved and transformed into soluble active ligands by metalloproteinases.123 EGF has a number of key roles in the kidney, contributing to cell proliferation and survival, renal metabolism and renal development.127,128 Some EGFR ligands have been implicated in experimental models of progressive kidney injury.129–131 EGFRs are expressed in podocytes132–134 and EGF seems to trigger the proliferation and differentiation of podocyte precursor cells in isolated glomeruli.135
HB-EGF–EGFR signalling might be involved in angiotensin II-mediated effects on podocytes. Angiotensin II induces the release of HB-EGF from podocytes, which in turn activates MAPK signalling pathways in an autocrine fashion via EGFR activation.136 Angiotensin II might also transactivate EGFR signalling.136 Furthermore, treatment of angiotensin II-infused rats with pharmacological EGFR inhibitors reduced glomerular cell proliferation without lowering the animals’ elevated blood pressure.137 EGFR has been suggested to have a pivotal role in the induction of renal hypertrophy, by regulating cell growth and proliferation and mediating the actions of angiotensin II.138 This hypothesis is supported by findings in experimental models of diabetes with angiotensin II-dependent hypertension in which EGFR inhibition attenuated glomerular enlargement in association with podocyte preservation and a reduction in albuminuria.139
Interestingly, in a rat model of anti-GBM disease, HB-EGF expression was increased in podocytes and application of anti-HB-EGF blocking antibody reversed acute glomerular injury.140 A subsequent study confirmed de novo expression of HB-EGF in podocytes and parietal epithelial cells in mice with rapidly progressive glomerulonephritis (RPGN) and in biopsy samples from patients with RPGN.141 HB-EGF is expressed in tubular cells, but is not present in normal glomerular epithelial cells.142 However, HB-EGF is expressed during proliferation and dedifferentiation of podocytes in glomerulonephritis.140 In a mouse model of anti-GBM disease, activation of EGFR in podocytes by HB-EGF resulted in the development and progression of RPGN, and in cultured podocytes, EGFR activation caused cell dedifferentiation and conversion to a proliferative and migratory phenotype.141 Pharmacological blockade of EGFR improved the course of RPGN in mice, prevented infiltration of inflammatory cells, and suppressed proteinuria. Most importantly, podocyte-specific, temporally controlled ablation of either EGFR or HB-EGF reduced the severity of glomerular injury and prevented renal failure and death.141 As selective deletion of HB-EGF in haematopoietic cells did not alter the course of the disease, HB-EGF expression in podocytes, but not in T cells or macrophages, seems to induce and drive the glomerular pathogenesis. Also, as other EGFR ligands (TGFα and epiregulin) failed to induce podocyte injury in vitro, HB-EGF seems to be the primary causative factor. This elegant study indicates that interference of EGF–EGFR signalling in podocytes might serve as a new therapeutic avenue to target cresentic and immune-mediated glomerular disease. In addition, treatment with the anti-EGFR small-molecule inhibitor erlotinib was able to rescue the glomerular phenotype of mice 4 days after induction of injury, suggesting that treatment of patients with manifested RPGN might be possible. It will be interesting to investigate if EGFR inhibitors are effective in the treatment of human RPGN, and if EGFR signalling is involved in the development of other types of glomerular disorders.
Conclusions
Podocytes are perhaps the most important contributor to a properly functioning glomerular filtration barrier, and the signals that converge on these post-mitotic cells are numerous and complex. A series of receptors— including RTKs—on the cell surface receive and process signals that enable podocytes to adapt to their environment and respond to chemical and biophysical cues. This knowledge has changed the anatomical view of the filter barrier from a rigid sieve to a physiological multicellular circuit that is highly dynamic and modifiable,4 and has resulted in the identification of RTKs as promising drug targets for glomerular diseases. In the past two decades, a myriad of cell signalling pathways and networks of extreme complexity, with multiple interconnections and feedback loops have been discovered. This complexity is impressively illustrated by the EGFR signalling node, which includes 211 distinct reactions and 322 components.143 However, what determines the hierarchy of signals received by podocytes, and how these cells determine which signals do not require a response, is not well understood. To better understand these phenomena and unravel the multiple signalling pathways that lead to podocyte injury and recovery, a combination of genetic, in vitro and biochemical studies is required. Such an approach should help to decipher the roles of podocytes and RTK signalling in health and disease as well as increase our understanding of how these cells integrate a plethora of converging signals into a hierarchical response that regulates proper kidney filtration.
Key points.
Podocytes respond to hormones and growth factors that are present in the circulation or locally produced in the glomerulus
A variety of receptors on the cell surface are thought to enable podocytes to respond to these external stimuli
The podocyte response to growth factors does not involve cell proliferation, but does include mitosis and hypertrophic cell growth, eventually leading to pathological alterations
Growth factors signal via receptor tyrosine kinases (RTKs), which are promising targets for cancer therapy
To understand and interfere with the pathological effects of growth factors on podocytes and the glomerular filter, their precise receptors must be identified and characterized
RTK inhibitors that are already used in cancer therapy might be promising new treatment options for proteinuric kidney diseases
Review criteria.
A search for original articles published between 1990 and 2013 and focusing on podocyte biology and signal transduction was performed in MEDLINE and PubMed. The search terms used were “podocyte”, “receptor tyrosine kinases”, “growth factors” and “signal transduction”, alone and in combination. All articles identified were English-language, full-text papers. We also searched the reference lists of identified articles for further relevant papers.
Acknowledgments
J. Reiser’s research is supported by NIH grants DK073495, DK089394, DK093773 and DK101350. S. Sever’s research is funded by NIH grant DK087985. C. Faul’s research is funded by a Carl W. Gottschalk Research Scholar Grant from the American Society of Nephrology.
Footnotes
Competing interests
J. Reiser has intellectual property related to this topic. See the article online for full details. S. Sever and C. Faul declare no competing interests.
Author contributions
J. Reiser and C. Faul researched the data and wrote the article. All authors made a substantial contribution to discussions of the content and reviewed and/or edited the manuscript before submission.666
Contributor Information
Jochen Reiser, Department of Medicine, Rush University Medical Center, 1735 West Harrison Street, Cohn Building, Suite 724, Chicago, IL 60612, USA.
Sanja Sever, Department of Medicine, Division of Nephrology, Massachusetts General Hospital and Harvard Medical School, 149 13th Street, Charlestown, MA 02129, USA.
Christian Faul, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 North West 10th Avenue (R-762), Batchelor Building 626, Miami, FL 33136, USA.
References
- 1.Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl) 1995;192:385–397. doi: 10.1007/BF00240371. [DOI] [PubMed] [Google Scholar]
- 2.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Neal CR, et al. Glomerular filtration into the subpodocyte space is highly restricted under physiological perfusion conditions. Am J Physiol Renal Physiol. 2007;293:F1787–F1798. doi: 10.1152/ajprenal.00157.2007. [DOI] [PubMed] [Google Scholar]
- 4.Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khurana S, Bruggeman LA, Kao HY. Nuclear hormone receptors in podocytes. Cell Biosci. 2012;2:33. doi: 10.1186/2045-3701-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sachs N, Sonnenberg A. Cell-matrix adhesion of podocytes in physiology and disease. Nat Rev Nephrol. 2013;9:200–210. doi: 10.1038/nrneph.2012.291. [DOI] [PubMed] [Google Scholar]
- 7.Gomperts BD, Tatham PER, Kramer IM. Signal Transduction. 2. Elsevier Academic Press; 2009. [Google Scholar]
- 8.Langley JN. On the physiology of the salivary secretion: part II. On the mutual antagonism of atropin and pilocarpin, having especial reference to their relations in the sub-maxillary gland of the cat. J Physiol. 1878;1:339–369. doi: 10.1113/jphysiol.1878.sp000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davenport HW. Early history of the concept of chemical transmission of the nerve impulse. Physiologist. 1991;34:178–190. [PubMed] [Google Scholar]
- 10.Cohen P. The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 11.Collett MS, Erikson RL. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc Natl Acad Sci USA. 1978;75:2021–2024. doi: 10.1073/pnas.75.4.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ushiro H, Cohen S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J Biol Chem. 1980;255:8363–8365. [PubMed] [Google Scholar]
- 14.Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science. 1982;215:185–187. doi: 10.1126/science.7031900. [DOI] [PubMed] [Google Scholar]
- 15.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 16.Sadowski I, Stone JC, Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol. 1986;6:4396–4408. doi: 10.1128/mcb.6.12.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson D, et al. Binding of SH2 domains of phospholipase C γ1, GAP, and Src to activated growth factor receptors. Science. 1990;250:979–982. doi: 10.1126/science.2173144. [DOI] [PubMed] [Google Scholar]
- 18.Pawson T, Schlessingert J. SH2 and SH3 domains. Curr Biol. 1993;3:434–442. doi: 10.1016/0960-9822(93)90350-w. [DOI] [PubMed] [Google Scholar]
- 19.Liu BA, et al. The human and mouse complement of SH2 domain proteins—establishing the boundaries of phosphotyrosine signaling. Mol Cell. 2006;22:851–868. doi: 10.1016/j.molcel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Karkkainen S, et al. Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 2006;7:186–191. doi: 10.1038/sj.embor.7400596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 22.Grassot J, Mouchiroud G, Perriere G. RTKdb: database of receptor tyrosine kinase. Nucleic Acids Res. 2003;31:353–358. doi: 10.1093/nar/gkg036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward CW, et al. The insulin and EGF receptor structures: new insights into ligand-induced receptor activation. Trends Biochem Sci. 2007;32:129–137. doi: 10.1016/j.tibs.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010;25:85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haglund K, Rusten TE, Stenmark H. Aberrant receptor signaling and trafficking as mechanisms in oncogenesis. Crit Rev Oncog. 2007;13:39–74. doi: 10.1615/critrevoncog.v13.i1.20. [DOI] [PubMed] [Google Scholar]
- 27.Giamas G, et al. Kinases as targets in the treatment of solid tumors. Cell Signal. 2010;22:984–1002. doi: 10.1016/j.cellsig.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Kriz W, et al. The role of podocytes in the development of glomerular sclerosis. Kidney Int Suppl. 1994;45:S64–S72. [PubMed] [Google Scholar]
- 29.Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol. 2013;304:F333–F347. doi: 10.1152/ajprenal.00478.2012. [DOI] [PubMed] [Google Scholar]
- 30.Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol. 2013;183:1364–1374. doi: 10.1016/j.ajpath.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coward RJ, Saleem MA. Podocytes as a target of insulin. Curr Diabetes Rev. 2011;7:22–27. doi: 10.2174/157339911794273883. [DOI] [PubMed] [Google Scholar]
- 32.Jauregui A, Mintz DH, Mundel P, Fornoni A. Role of altered insulin signaling pathways in the pathogenesis of podocyte malfunction and microalbuminuria. Curr Opin Nephrol Hypertens. 2009;18:539–545. doi: 10.1097/MNH.0b013e32832f7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stieger N, Worthmann K, Schiffer M. The role of metabolic and haemodynamic factors in podocyte injury in diabetes. Diabetes Metab Res Rev. 2011;27:207–215. doi: 10.1002/dmrr.1164. [DOI] [PubMed] [Google Scholar]
- 34.Hale LJ, Coward RJ. The insulin receptor and the kidney. Curr Opin Nephrol Hypertens. 2013;22:100–106. doi: 10.1097/MNH.0b013e32835abb52. [DOI] [PubMed] [Google Scholar]
- 35.Coward RJ, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095–3102. doi: 10.2337/diabetes.54.11.3095. [DOI] [PubMed] [Google Scholar]
- 36.Anfossi G, Russo I, Doronzo G, Trovati M. Contribution of insulin resistance to vascular dysfunction. Arch Physiol Biochem. 2009;115:199–217. doi: 10.1080/13813450903136791. [DOI] [PubMed] [Google Scholar]
- 37.Ritchie SA, Ewart MA, Perry CG, Connell JM, Salt IP. The role of insulin and the adipocytokines in regulation of vascular endothelial function. Clin Sci (Lond) 2004;107:519–532. doi: 10.1042/CS20040190. [DOI] [PubMed] [Google Scholar]
- 38.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 39.Coward RJ, et al. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes. 2007;56:1127–1135. doi: 10.2337/db06-0693. [DOI] [PubMed] [Google Scholar]
- 40.Welsh GI, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mogensen CE, Christensen NJ, Gundersen HJ. The acute effect of insulin on heart rate, blood pressure, plasma noradrenaline and urinary albumin excretion. The role of changes in blood glucose. Diabetologia. 1980;18:453–457. doi: 10.1007/BF00261700. [DOI] [PubMed] [Google Scholar]
- 42.Fogo AB, Kon V. The glomerulus—a view from the inside—the endothelial cell. Int J Biochem Cell Biol. 2010;42:1388–1397. doi: 10.1016/j.biocel.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Advani A. Vascular endothelial growth factor and the kidney: something of the marvellous. Curr Opin Nephrol Hypertens. 2014;23:87–92. doi: 10.1097/01.mnh.0000437329.41546.a9. [DOI] [PubMed] [Google Scholar]
- 44.Ku CH, et al. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57:2824–2833. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guan F, Villegas G, Teichman J, Mundel P, Tufro A. Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol. 2006;291:F422–F428. doi: 10.1152/ajprenal.00448.2005. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, et al. Podocyte-derived vascular endothelial growth factor mediates the stimulation of α3(IV) collagen production by transforming growth factor-β1 in mouse podocytes. Diabetes. 2004;53:2939–2949. doi: 10.2337/diabetes.53.11.2939. [DOI] [PubMed] [Google Scholar]
- 47.Foster RR, et al. Functional evidence that vascular endothelial growth factor may act as an autocrine factor on human podocytes. Am J Physiol Renal Physiol. 2003;284:F1263–F1273. doi: 10.1152/ajprenal.00276.2002. [DOI] [PubMed] [Google Scholar]
- 48.Veron D, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010;77:989–999. doi: 10.1038/ki.2010.64. [DOI] [PubMed] [Google Scholar]
- 49.Veron D, et al. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid-resistant nephrotic syndrome. Am J Pathol. 2010;177:2225–2233. doi: 10.2353/ajpath.2010.091146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sison K, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–1701. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Itoh N, Ornitz DM. Evolution of the FGF and FGFR gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 52.White KE, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 53.Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- 54.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Bates CM. Role of fibroblast growth factor receptor signaling in kidney development. Pediatr Nephrol. 2007;22:343–349. doi: 10.1007/s00467-006-0239-7. [DOI] [PubMed] [Google Scholar]
- 56.Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G. Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J. 1998;17:1642–1655. doi: 10.1093/emboj/17.6.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davidson G, Dono R, Zeller R. FGF signalling is required for differentiation-induced cytoskeletal reorganisation and formation of actin-based processes by podocytes. J Cell Sci. 2001;114:3359–3366. doi: 10.1242/jcs.114.18.3359. [DOI] [PubMed] [Google Scholar]
- 58.Floege J, et al. Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J Clin Invest. 1995;96:2809–2819. doi: 10.1172/JCI118351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cauchi J, et al. Light-microscopic immunolocalization of fibroblast growth factor-1 and -2 in adult rat kidney. Cell Tissue Res. 1996;285:179–187. doi: 10.1007/s004410050635. [DOI] [PubMed] [Google Scholar]
- 60.Takeuchi A, et al. Basic fibroblast growth factor promotes proliferation of rat glomerular visceral epithelial cells in vitro. Am J Pathol. 1992;141:107–116. [PMC free article] [PubMed] [Google Scholar]
- 61.Floege J, et al. Localization of fibroblast growth factor-2 (basic FGF) and FGF receptor-1 in adult human kidney. Kidney Int. 1999;56:883–897. doi: 10.1046/j.1523-1755.1999.00637.x. [DOI] [PubMed] [Google Scholar]
- 62.Floege J, et al. Rat glomerular mesangial cells synthesize basic fibroblast growth factor. Release, upregulated synthesis, and mitogenicity in mesangial proliferative glomerulonephritis. J Clin Invest. 1992;90:2362–2369. doi: 10.1172/JCI116126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Floege J, et al. Infusion of platelet-derived growth factor or basic fibroblast growth factor induces selective glomerular mesangial cell proliferation and matrix accumulation in rats. J Clin Invest. 1993;92:2952–2962. doi: 10.1172/JCI116918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ballermann BJ. Regulation of bovine glomerular endothelial cell growth in vitro. Am J Physiol. 1989;256:C182–C189. doi: 10.1152/ajpcell.1989.256.1.C182. [DOI] [PubMed] [Google Scholar]
- 65.Dono R, Zeller R. Cell-type-specific nuclear translocation of fibroblast growth factor-2 isoforms during chicken kidney and limb morphogenesis. Dev Biol. 1994;163:316–330. doi: 10.1006/dbio.1994.1151. [DOI] [PubMed] [Google Scholar]
- 66.Tossidou I, et al. CIN85/RukL is a novel binding partner of nephrin and podocin and mediates slit diaphragm turnover in podocytes. J Biol Chem. 2010;285:25285–25295. doi: 10.1074/jbc.M109.087239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tossidou I, et al. CD2AP/CIN85 balance determines receptor tyrosine kinase signaling response in podocytes. J Biol Chem. 2007;282:7457–7464. doi: 10.1074/jbc.M608519200. [DOI] [PubMed] [Google Scholar]
- 68.Ray PE, et al. bFGF and its low affinity receptors in the pathogenesis of HIV-associated nephropathy in transgenic mice. Kidney Int. 1994;46:759–772. doi: 10.1038/ki.1994.331. [DOI] [PubMed] [Google Scholar]
- 69.Strutz F, et al. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000;57:1521–1538. doi: 10.1046/j.1523-1755.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 70.Mazue G, Bertelero F, Garofano L, Brughera M, Carminati P. Experience with the preclinical assessment of basic fibroblast growth factor (bFGF) Toxicol Lett. 1992;64–65:329–338. doi: 10.1016/0378-4274(92)90205-x. [DOI] [PubMed] [Google Scholar]
- 71.Kriz W, Hähnel B, Rösener S, Elger M. Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int. 1995;48:1435–1450. doi: 10.1038/ki.1995.433. [DOI] [PubMed] [Google Scholar]
- 72.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109 (Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–1666. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nijenhuis T, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shankland SJ, et al. Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int. 1997;52:404–413. doi: 10.1038/ki.1997.347. [DOI] [PubMed] [Google Scholar]
- 76.Riley SG, Steadman R, Williams JD, Floege J, Phillips AO. Augmentation of kidney injury by basic fibroblast growth factor or platelet-derived growth factor does not induce progressive diabetic nephropathy in the Goto Kakizaki model of non-insulin-dependent diabetes. J Lab Clin Med. 1999;134:304–312. doi: 10.1016/s0022-2143(99)90211-1. [DOI] [PubMed] [Google Scholar]
- 77.Sasaki T, Hatta H, Osawa G. Cytokines and podocyte injury: the mechanism of fibroblast growth factor 2-induced podocyte injury. Nephrol Dial Transplant. 1999;14 (Suppl 1):33–34. doi: 10.1093/ndt/14.suppl_1.33. [DOI] [PubMed] [Google Scholar]
- 78.Sasaki T, Jyo Y, Tanda N, Tamai H, Osawa G. The role of basic fibroblast growth factor (FGF2) in glomerular epithelial cell injury. Contrib Nephrol. 1996;118:68–77. doi: 10.1159/000425078. [DOI] [PubMed] [Google Scholar]
- 79.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Unger EF, et al. Effects of a single intracoronary injection of basic fibroblast growth factor in stable angina pectoris. Am J Cardiol. 2000;85:1414–1419. doi: 10.1016/s0002-9149(00)00787-6. [DOI] [PubMed] [Google Scholar]
- 81.Zimering MB, Eng J. Increased basic fibroblast growth factor-like substance in plasma from a subset of middle-aged or elderly male diabetic patients with microalbuminuria or proteinuria. J Clin Endocrinol Metab. 1996;81:4446–4452. doi: 10.1210/jcem.81.12.8954057. [DOI] [PubMed] [Google Scholar]
- 82.Ray PE, Liu XH, Xu L, Rakusan T. Basic fibroblast growth factor in HIV-associated hemolytic uremic syndrome. Pediatr Nephrol. 1999;13:586–593. doi: 10.1007/s004670050749. [DOI] [PubMed] [Google Scholar]
- 83.Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 84.Floege J, et al. Endogenous fibroblast growth factor-2 mediates cytotoxicity in experimental mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 1998;9:792–801. doi: 10.1681/ASN.V95792. [DOI] [PubMed] [Google Scholar]
- 85.Nickel W. Pathways of unconventional protein secretion. Curr Opin Biotechnol. 2010;21:621–626. doi: 10.1016/j.copbio.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 86.Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays. 2000;22:108–112. doi: 10.1002/(SICI)1521-1878(200002)22:2<108::AID-BIES2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 87.Schumacher VA, et al. WT1-dependent sulfatase expression maintains the normal glomerular filtration barrier. J Am Soc Nephrol. 2011;22:1286–1296. doi: 10.1681/ASN.2010080860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pelletier J, et al. Germline mutations in the Wilm’s tumor suppressor gene are associated with abnormal urogential development in Denys-Drash syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 89.Okamoto K, et al. Common variation in GPC5 is associated with acquired nephrotic syndrome. Nat Genet. 2011;43:459–463. doi: 10.1038/ng.792. [DOI] [PubMed] [Google Scholar]
- 90.Bernfield M, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 91.Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 92.Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- 93.Floege J, et al. Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int. 1992;41:297–309. doi: 10.1038/ki.1992.42. [DOI] [PubMed] [Google Scholar]
- 94.Matsuda M, et al. Gene expression of PDGF and PDGF receptor in various forms of glomerulonephritis. Am J Nephrol. 1997;17:25–31. doi: 10.1159/000169067. [DOI] [PubMed] [Google Scholar]
- 95.Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular expression of platelet-derived growth factor (PDGF)-A, -B chain and PDGF receptor-α, -β in human diabetic nephropathy. Clin Exp Nephrol. 2004;8:36–42. doi: 10.1007/s10157-003-0265-8. [DOI] [PubMed] [Google Scholar]
- 96.Iida H, et al. Platelet-derived growth factor (PDGF) and PDGF receptor are induced in mesangial proliferative nephritis in the rat. Proc Natl Acad Sci USA. 1991;88:6560–6564. doi: 10.1073/pnas.88.15.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Roeyen CR, et al. Biological responses to PDGF-BB versus PDGF-DD in human mesangial cells. Kidney Int. 2006;69:1393–1402. doi: 10.1038/sj.ki.5000332. [DOI] [PubMed] [Google Scholar]
- 98.Ostendorf T, et al. A fully human monoclonal antibody (CR002) identifies PDGF-D as a novel mediator of mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2003;14:2237–2247. doi: 10.1097/01.asn.0000083393.00959.02. [DOI] [PubMed] [Google Scholar]
- 99.Hudkins KL, et al. Exogenous PDGF-D is a potent mesangial cell mitogen and causes a severe mesangial proliferative glomerulopathy. J Am Soc Nephrol. 2004;15:286–298. doi: 10.1097/01.asn.0000108522.79652.63. [DOI] [PubMed] [Google Scholar]
- 100.Changsirikulchai S, et al. Platelet-derived growth factor-D expression in developing and mature human kidneys. Kidney Int. 2002;62:2043–2054. doi: 10.1046/j.1523-1755.2002.00662.x. [DOI] [PubMed] [Google Scholar]
- 101.Gesualdo L, et al. Expression of platelet-derived growth factor receptors in normal and diseased human kidney. An immunohistochemistry and in situ hybridization study. J Clin Invest. 1994;94:50–58. doi: 10.1172/JCI117348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int. 1993;43:286–294. doi: 10.1038/ki.1993.45. [DOI] [PubMed] [Google Scholar]
- 103.Bergsten E, et al. PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 104.van Roeyen CR, et al. Induction of progressive glomerulonephritis by podocyte-specific overexpression of platelet-derived growth factor-D. Kidney Int. 2011;80:1292–1305. doi: 10.1038/ki.2011.278. [DOI] [PubMed] [Google Scholar]
- 105.Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–1459. doi: 10.1016/0006-291x(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 106.Nakamura T, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 107.Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:588–610. doi: 10.2183/pjab.86.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park M, et al. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci USA. 1987;84:6379–6383. doi: 10.1073/pnas.84.18.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sonnenberg E, Meyer D, Weidner KM, Birchmeier C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol. 1993;123:223–235. doi: 10.1083/jcb.123.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang X, et al. Sp1 and Sp3 transcription factors synergistically regulate HGF receptor gene expression in kidney. Am J Physiol Renal Physiol. 2003;284:F82–F94. doi: 10.1152/ajprenal.00200.2002. [DOI] [PubMed] [Google Scholar]
- 111.Mizuno S, Matsumoto K, Nakamura T. HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci. 2008;13:7072–7086. doi: 10.2741/3211. [DOI] [PubMed] [Google Scholar]
- 112.Mizuno S, et al. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J Clin Invest. 1998;101:1827–1834. doi: 10.1172/JCI1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dai C, et al. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J Am Soc Nephrol. 2004;15:2637–2647. doi: 10.1097/01.ASN.0000139479.09658.EE. [DOI] [PubMed] [Google Scholar]
- 114.Cruzado JM, et al. Regression of advanced diabetic nephropathy by hepatocyte growth factor gene therapy in rats. Diabetes. 2004;53:1119–1127. doi: 10.2337/diabetes.53.4.1119. [DOI] [PubMed] [Google Scholar]
- 115.Bu X, et al. Systemic administration of naked plasmid encoding HGF attenuates puromycin aminonucleoside-induced damage of murine glomerular podocytes. Am J Physiol Renal Physiol. 2011;301:F784–F792. doi: 10.1152/ajprenal.00210.2011. [DOI] [PubMed] [Google Scholar]
- 116.Kato T, Mizuno S, Nakamura T. Preservations of nephrin and synaptopodin by recombinant hepatocyte growth factor in podocytes for the attenuations of foot process injury and albuminuria in nephritic mice. Nephrology (Carlton) 2011;16:310–318. doi: 10.1111/j.1440-1797.2010.01392.x. [DOI] [PubMed] [Google Scholar]
- 117.Dai C, Saleem MA, Holzman LB, Mathieson P, Liu Y. Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int. 2010;77:962–973. doi: 10.1038/ki.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fornoni A, Li H, Foschi A, Striker GE, Striker LJ. Hepatocyte growth factor, but not insulin-like growth factor I protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol. 2001;158:275–280. doi: 10.1016/S0002-9440(10)63966-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96–107. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- 120.Zhang J, Yang J, Liu Y. Role of Bcl-xL induction in HGF-mediated renal epithelial cell survival after oxidant stress. Int J Clin Exp Pathol. 2008;1:242–253. [PMC free article] [PubMed] [Google Scholar]
- 121.Yamaguchi Y, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54:653–664. doi: 10.1053/j.ajkd.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 122.Li Y, et al. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 124.Zhang H, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Takeuchi K, Ito F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol Pharm Bull. 2011;34:1774–1780. doi: 10.1248/bpb.34.1774. [DOI] [PubMed] [Google Scholar]
- 126.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10:111–126. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- 127.Nowak G, Schnellmann RG. Integrative effects of EGF on metabolism and proliferation in renal proximal tubular cells. Am J Physiol. 1995;269:C1317–C1325. doi: 10.1152/ajpcell.1995.269.5.C1317. [DOI] [PubMed] [Google Scholar]
- 128.Pugh JL, Sweeney WE, Jr, Avner ED. Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture. Kidney Int. 1995;47:774–781. doi: 10.1038/ki.1995.118. [DOI] [PubMed] [Google Scholar]
- 129.Zeng F, Singh AB, Harris RC. The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp Cell Res. 2009;315:602–610. doi: 10.1016/j.yexcr.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lautrette A, et al. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med. 2005;11:867–874. doi: 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- 131.Pillebout E, et al. JunD protects against chronic kidney disease by regulating paracrine mitogens. J Clin Invest. 2003;112:843–852. doi: 10.1172/JCI17647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coaxum SD, Garnovskaya MN, Gooz M, Baldys A, Raymond JR. Epidermal growth factor activates Na+/H+ exchanger in podocytes through a mechanism that involves Janus kinase and calmodulin. Biochim Biophys Acta. 2009;1793:1174–1181. doi: 10.1016/j.bbamcr.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Harris RC, Hoover RL, Jacobson HR, Badr KF. Evidence for glomerular actions of epidermal growth factor in the rat. J Clin Invest. 1988;82:1028–1039. doi: 10.1172/JCI113659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Adler S, Eng B. Reversal of inhibition of rat glomerular epithelial cell growth by growth factors. Am J Pathol. 1990;136:557–563. [PMC free article] [PubMed] [Google Scholar]
- 135.Tassin MT, et al. Effects of epidermal growth factor on calf renal glomerular cells in vitro. Growth Factors. 1992;6:243–254. doi: 10.3109/08977199209026931. [DOI] [PubMed] [Google Scholar]
- 136.Flannery PJ, Spurney RF. Transactivation of the epidermal growth factor receptor by angiotensin II in glomerular podocytes. Nephron Exp Nephrol. 2006;103:e109–e118. doi: 10.1159/000092196. [DOI] [PubMed] [Google Scholar]
- 137.Suzuki H, Yamamoto T, Fujigaki Y, Eguchi S, Hishida A. Comparison of ROCK and EGFR activation pathways in the progression of glomerular injuries in AngII-infused rats. Ren Fail. 2011;33:1005–1012. doi: 10.3109/0886022X.2011.618923. [DOI] [PubMed] [Google Scholar]
- 138.Chen J, Chen JK, Neilson EG, Harris RC. Role of EGF receptor activation in angiotensin II-induced renal epithelial cell hypertrophy. J Am Soc Nephrol. 2006;17:1615–1623. doi: 10.1681/ASN.2005111163. [DOI] [PubMed] [Google Scholar]
- 139.Advani A, et al. Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology (Carlton) 2011;16:573–581. doi: 10.1111/j.1440-1797.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 140.Feng L, et al. Heparin-binding EGF-like growth factor contributes to reduced glomerular filtration rate during glomerulonephritis in rats. J Clin Invest. 2000;105:341–350. doi: 10.1172/JCI2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bollee GM, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17:1242–1250. doi: 10.1038/nm.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sakai M, et al. Production of heparin binding epidermal growth factor-like growth factor in the early phase of regeneration after acute renal injury. Isolation and localization of bioactive molecules. J Clin Invest. 1997;99:2128–2138. doi: 10.1172/JCI119386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100014. 2005.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Satchell SC, et al. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J Am Soc Nephrol. 2002;13:544–550. doi: 10.1681/ASN.V132544. [DOI] [PubMed] [Google Scholar]
- 145.Davis B, et al. Podocyte-specific induced overexpression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 146.Gao X, et al. Angiopoietin-like protein 3 regulates the motility and permeability of podocytes by altering nephrin expression in vitro. Biochem Biophys Res Commun. 2010;399:31–36. doi: 10.1016/j.bbrc.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 147.Jia R, Hong X, Li S, Haichun Y, Chuanming H. Expression of angiopoietin-like 3 associated with puromycin-induced podocyte damage. Nephron Exp Nephrol. 2010;115:e38–e45. doi: 10.1159/000313829. [DOI] [PubMed] [Google Scholar]
- 148.Clement LC, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–122. doi: 10.1038/nm.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–3368. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]
- 150.Yokoi H, et al. Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int. 2008;73:446–455. doi: 10.1038/sj.ki.5002722. [DOI] [PubMed] [Google Scholar]
- 151.Fuchshofer R, et al. Connective tissue growth factor modulates podocyte actin cytoskeleton and extracellular matrix synthesis and is induced in podocytes upon injury. Histochem Cell Biol. 2011;136:301–319. doi: 10.1007/s00418-011-0844-9. [DOI] [PubMed] [Google Scholar]
- 152.Dai HY, et al. The roles of connective tissue growth factor and integrin-linked kinase in high glucose-induced phenotypic alterations of podocytes. J Cell Biochem. 2012;113:293–301. doi: 10.1002/jcb.23355. [DOI] [PubMed] [Google Scholar]
- 153.Gross O, et al. DDR1-deficient mice show localized subepithelial GBM thickening with focal loss of slit diaphragms and proteinuria. Kidney Int. 2004;66:102–111. doi: 10.1111/j.1523-1755.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- 154.Kerroch M, et al. Genetic inhibition of discoidin domain receptor 1 protects mice against crescentic glomerulonephritis. FASEB J. 2012;26:4079–4091. doi: 10.1096/fj.11-194902. [DOI] [PubMed] [Google Scholar]
- 155.Hashimoto T, et al. Ephrin-B1 localizes at the slit diaphragm of the glomerular podocyte. Kidney Int. 2007;72:954–964. doi: 10.1038/sj.ki.5002454. [DOI] [PubMed] [Google Scholar]
- 156.Wnuk M, et al. Podocyte EphB4 signaling helps recovery from glomerular injury. Kidney Int. 2012;81:1212–1225. doi: 10.1038/ki.2012.17. [DOI] [PubMed] [Google Scholar]
- 157.Hale LJ, et al. Insulin-like growth factor-II is produced by, signals to and is an important survival factor for the mature podocyte in man and mouse. J Pathol. 2013;230:95–106. doi: 10.1002/path.4165. [DOI] [PubMed] [Google Scholar]
- 158.Fujinaka H, et al. Expression and localization of insulin-like growth factor binding proteins in normal and proteinuric kidney glomeruli. Nephrology (Carlton) 2010;15:700–709. doi: 10.1111/j.1440-1797.2010.01285.x. [DOI] [PubMed] [Google Scholar]
- 159.Bridgewater DJ, Ho J, Sauro V, Matsell DG. Insulin-like growth factors inhibit podocyte apoptosis through the PI3 kinase pathway. Kidney Int. 2005;67:1308–1314. doi: 10.1111/j.1523-1755.2005.00208.x. [DOI] [PubMed] [Google Scholar]
- 160.Bridgewater DJ, Dionne JM, Butt MJ, Pin CL, Matsell DG. The role of the type I insulin-like growth factor receptor (IGF-IR) in glomerular integrity. Growth Horm IGF Res. 2008;18:26–37. doi: 10.1016/j.ghir.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 161.Prabakaran T, et al. Receptor-mediated endocytosis of α-galactosidase A in human podocytes in Fabry disease. PLoS ONE. 2011;6:e25065. doi: 10.1371/journal.pone.0025065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hale LJ, et al. Insulin directly stimulates VEGF-A production in the glomerular podocyte. Am J Physiol Renal Physiol. 2013;305:F182–F188. doi: 10.1152/ajprenal.00548.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol. 2012;302:F298–F307. doi: 10.1152/ajprenal.00423.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Carito V, et al. Localization of nerve growth factor (NGF) receptors in the mitochondrial compartment: characterization and putative role. Biochim Biophys Acta. 2012;1820:96–103. doi: 10.1016/j.bbagen.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 165.Hahn WH, Suh JS, Cho BS. Linkage and association study of neurotrophins and their receptors as novel susceptibility genes for childhood IgA nephropathy. Pediatr Res. 2011;69:299–305. doi: 10.1203/PDR.0b013e31820b9365. [DOI] [PubMed] [Google Scholar]
- 166.Tsui CC, Shankland SJ, Pierchala BA. Glial cell line-derived neurotrophic factor and its receptor ret is a novel ligand-receptor complex critical for survival response during podocyte injury. J Am Soc Nephrol. 2006;17:1543–1552. doi: 10.1681/ASN.2005080835. [DOI] [PubMed] [Google Scholar]
- 167.Benz K, et al. Early glomerular alterations in genetically determined low nephron number. Am J Physiol Renal Physiol. 2011;300:F521–F530. doi: 10.1152/ajprenal.00490.2009. [DOI] [PubMed] [Google Scholar]
- 168.Eremina V, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maynard SE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]