

Abstract
Circulating and podocyte-bound urokinase receptor (uPAR) is a mediator of podocyte injury, proteinuria, and focal segmental glomerulosclerosis (FSGS) allowing pathological activation of the uPAR-β3 integrin signaling axis. Clinically, calcineurin inhibitors (e.g., cyclosporine A, CsA) are known to suppress T cells, yet are also being used to reduce proteinuria in FSGS, suggesting the possibility of signal cross talk between uPAR and calcineurin. Calcineurin is known to facilitate the nuclear translocation of the nuclear factor of activated T cells (NFAT). Accordingly, in vivo conditional NFATc1 activation in podocytes leads to proteinuria in mice, yet the downstream targets of NFAT remain unclear. Here, we show that inducible podocyte-specific expression of constitutively active NFATc1 increased podocyte uPAR expression by binding to the Plaur gene promoter (encoding uPAR) in chromatin immunoprecipitation assays. Pathological uPAR signals in podocytes are independent of T cells and affect cell motility via activation, but not expression, changes of the β3 integrin and can be blocked by CsA, NFAT-siRNA, or the cell-permeable NFAT inhibitor (11R-VIVIT) using rodent models of glomerular disease (LPS; 5/6 nephrectomized rats). Taken together, these findings identify podocyte uPAR as a downstream target of NFAT and provide further insights into the pathogenesis of FSGS.
Keywords: Podocyte, suPAR, uPAR, β3 integrin, Calcineurin, NFAT
Introduction
Proteinuria is the loss of plasma proteins into the urine and represents the cardinal feature of kidney filtration barrier (GFB) dysfunction; this condition constitutes a risk factor for both renal and extrarenal diseases [1]. Podocytes with their cellular extensions (foot processes), together with endothelial cells and the glomerular basement membrane, form the key components of the GFB [2].
Insults to podocytes lead to podocyte foot process fusion (effacement) and proteinuria and, if not resolved, to more sustained damage including loss of kidney function. An emerging concept in the development of podocyte foot process effacement and proteinuria is the pathological alteration of podocyte foot process motility [3, 4]. Urokinase receptor (uPAR), a molecule associated with cell motility [5], is highly expressed on motile cells. Recently, uPAR and its soluble form suPAR have been shown to be critical in the pathogenesis of proteinuria and focal segmental glomerulosclerosis (FSGS) [4, 6]. uPAR and β3 integrin co-localize in podocytes and form a lipid-dependent complex with β3 integrin, thereby causing a structural configuration change of β3 integrin, resulting in its activation with increased affinity for ligand binding [4, 6]. In vivo gene delivery of a structurally fixed and constitutively active β3 integrin is sufficient to induce proteinuria in mice; conversely, the inhibition of uPAR expression and β3 integrin activation improves podocyte foot process structure and has an anti-proteinuric effect [7].
The nuclear factor of activated T cells (NFAT) transcription factors are the most extensively studied calcineurin substrates and the major regulators of transcription in response to Ca2+/calcineurin signals [8]. The immunosuppressive action of the calcineurin inhibitor cyclosporine A (CsA) stems from the inhibition of NFAT signaling in T cells [8]. Clinically, CsA is used to reduce proteinuria in kidney diseases, such as nephrotic syndrome (especially FSGS). The effectiveness of CsA was initially attributed to its immunosuppressive effects, but the study of Faul et al. [9] suggested a novel role for CsA in inhibiting the dephosphorylation of podocyte synaptopodin by calcineurin, resulting in the protection of synaptopodin from cathepsin L-mediated degradation and stabilization of podocyte structure. Results from Wang et al. [10] and Nijenhuis et al. [11] suggested that conditional NFATc1 activation in podoyctes per se is sufficient to induce proteinuria in mice. Thus, both studies provide in vivo evidence that calcineurin pathways are important for podocyte function and disease.
In our study, we hypothesized that NFAT activation and uPAR might be functionally linked. We found that NFAT directly binds to the Plaur promoter, stimulating uPAR protein expression. In keeping with this finding, podocyte-specific and inducible NFATc1 mice display elevated glomerular uPAR expression levels that can be rescued by calcineurin/NFAT inhibition using CsA or the 11R-VIVIT. Both treatments lead to the inhibition of uPAR expression in cultured podocytes, thereby suppressing β3 integrin activation. Our findings suggest that uPAR-β3 integrin signaling is involved in calcineurin/NFAT-induced podocyte injury. The anti-proteinuric effect of CsA may be partially attributed to its inhibition of the uPAR-β3 integrin signaling axis in podocytes.
Materials and methods
Animals
All animal experiments were performed with the approval of the GGH Animal Care and Use Committee in accordance with the NIH guidelines for the conduct of experiments on animals. The protocols and experimental procedures for NFATc1nuc transgenic mice were approved by the Institutional Animal Care and Use Committee at the University of Miami Miller School of Medicine. Sprague–Dawley rats and SCID mice were obtained from the Laboratory Animal Center, Sun Yat-sen University, China.
Animal models and treatment with the CsA or NFAT inhibitor (11R-VIVIT) were performed using the 5/6 nephrectomy FSGS model (NTX rats) which was induced in male Sprague–Dawley rats (initial weight, 250–300 g) by surgical resection. One week later, all animals were then randomized to (1) receive CsA at a lower dose (C3662, Sigma-Aldrich, St. Louis, MO) once daily by gastric gavage (1 mgkg−1day−1, NTX+CsA 1 group, n=13); (2) receive CsA at a higher dose once daily by gastric gavage (5 mg kg−1day−1, NTX+CsA 2 group, n=14); (3) or receive vehicle (equal volume, NTX group, n=14). The rats of the sham-operated group (Sham group, n=14) also received vehicle once daily by gastric gavage. We collected urine at time points of 2, 4, 8, and 12 weeks for Bradford protein analysis (Bradford protein assay kit, P0006, Beyotime, China). For each group, three rats were killed at 2, 4, and 8 weeks after randomization, respectively. The remaining rats (four for NTX+CsA1, five for NTX+CsA2, five for NTX, and five for Sham) were killed at 12 weeks.
For the lipopolysaccharide (LPS) mouse model of transient proteinuria (LPS mice) [4, 12], we injected SCID mice intraperitoneally with 200 μg LPS (L-2880, Sigma-Aldrich) in a total volume of 500 μl. After LPS injection, mice were gavaged with saline alone (LPS group, n=5) and CsA once daily (25 mgkg−1day−1, LPS+CsA group, n=5). Controls (Con group, n=5) received the same volume of sterile LPS-free saline.
For another experiment of a NFAT inhibitor (11R-VIVIT) treatment in LPS mice, we injected SCID mice intraperitoneally with saline alone (LPS group, n=5) and 11R-VIVIT once daily (5 mgkg−1day−1, LPS+11R-VIVIT(5) group, n= 5; 10 mgkg−1day−1, LPS+11R-VIVIT(10) group, n=5; 15 mgkg−1day−1, LPS+11R-VIVIT(15) group, n=5). Controls (Con group, n=5) received the same volume of sterile LPS-free saline.
Double transgenic doxycycline-inducible podocin-rtTA/TetO-HA-NFATc1nuc mice were described before [13]. Transgene expression was induced in podocytes by adding doxycycline (Dox; Sigma-Aldrich; 2 mg/ml in 7 % sucrose, pH 5) to the drinking water of 6- to 8-week-old double transgenic mice for 4 days [11]. Simultaneously, the mice were fed a special diet chow containing Dox (2,000 ppm). Control animals were single transgenic podocin-rtTA mice that also received Dox. For uPAR expression analysis, the glomeruli were isolated by a sieving technique [14]. Induction of NFATc1nuc expression in isolated glomeruli was monitored by RT-PCR using DNase-treated total RNA and NFATc1nuc-specific primers as described before [11].
Conditionally immortalized mouse podocyte cell lines were provided by Dr. P Mundel (Massachusetts General Hospital, Boston, USA) and cultured as reported previously [12]. To propagate podocytes, cells were cultivated on BD BioCoat Collagen I plates (BD Biosciences, USA) at 33 °C in the presence of 20 U/ml mouse recombinant IFN- γ (CYT-358, ProSpec-Tany TechnoGene Ltd., Israel) to enhance the expression of the thermosensitive T-antigen. To induce differentiation, podocytes were maintained at 37 °C without IFN-γ for at least 10–12 days. To quiescent the cells, differentiated podocytes were serum-starved overnight before the experiment.
A flow cytometry assay was used to assess active or total beta3 integrin on the cell surface of podocytes during the treatment with LPS alone (50 μg/ml), LPS (50 μg/ml) plus CsA (0.25, 0.5, and 1 μg/ml), LPS (50 μg/ml) plus 11R-VIVIT (10 nm, 100 nM, and 1,000 nM; 480401, Merck, Germany) [15] or ionomycin (500 nM, 1 μM, and 2 μM; I0634, Sigma-Aldrich) [16]. After 24 h of these treatments, approximately 106/mlcells were trypsinized, washed with phosphate-buffered saline (PBS; Ca2+-free), and incubated with anti-mouse active β3 integrin (AP5 antibody, Genetic Testing Institute, USA) or anti-mouse total β3 integrin (PE Hamster Anti-Mouse CD61,553347, BD Pharmingen™, USA) for 20 min at 4 °C. The cells were then washed in PBS (Ca2+-free) and incubated with goat anti-mouse Alexa Fluor 488 (A11001, Invitrogen, USA) for 20 min at 4 °C. Data were collected using the Cell Lab Quanta™ SC Flow Cytometry System and analyzed using Cell Lab Quanta™ SC analyse (Beckman Coulter, Inc., USA).
For flow cytometry evaluation of uPAR, the podocytes were directly detached by a cell scraper instead of using trypsin since uPAR is sensitive to trypsin treatment [17]. The antibody against uPAR is phycoerythrin-conjugated rat monoclonal anti-mouse uPAR (FAB531P, R&D Systems, USA). To minimize the possibility of the uPAR antibody binding competitively to the natural ligand uPA and the possibility of the potential effect of uPA on uPAR, an anti-uPA antibody (uPA (M-20), sc-6831; Santa Cruz Biotech, Santa Cruz, CA) was used prior to podocyte uPAR staining [18].
For immunocytochemistry and immunoblotting, murine kidneys and cultured podocytes were harvested and snapfrozen. For immunofluorescent labeling, sections were washed once with PBS, permeabilized with 0.5 % Triton X-100 in PBS, and incubated with blocking solution (5 % BSA) for 20 min before further incubation with one of the primary antibodies (synaptopodin (N-14), sc-21536; uPAR (FL-290), sc-1085; total β3 integrin (H-96), sc-14009; nephrin (G-20), sc-32530; CD2AP (L-20), sc-34229, NFATc1, sc-13033; Santa Cruz Biotech; active β3 integrin (AP5 antibody, Genetic Testing Institute, USA)) for 2 h at room temperature. For double labeling, sections were washed three times with PBS for 5 min and one of the secondary antibodies (Alexa Fluor® 488 monkey anti-goat IgG (H+L), A11055; Alexa Fluor® 635 goat anti-mouse IgG (H+L), A31575; Alexa Fluor® 546 goat anti-rabbit IgG (H+L), A11010 (Invitrogen); rabbit anti-goat IgG-FITC, sc-2777 (Santa Cruz Biotech); NorthernLights™ anti-mouse IgG-NL637, NL008 (R&D Systems)) was applied for 2 h. Pictures were captured with confocal microscopy (Leica Microsystems).
Glomerular protein extracts were prepared as described before [11]. In brief, isolated glomeruli were lysed in a 20 mM Tris (pH 8.0) buffer containing 500 mM NaCl, 0.5 % 3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate, 1 % Triton X-100, and the Complete Mini cocktail of protease inhibitors (Roche Diagnostics). The proteins were resolved on a 4–12 % gel (NuPAGE, Invitrogen) and blotted to polyvinylidene difluoride membranes (Bio-Rad Laboratories). Blots were incubated with goat anti-uPAR (1:1,000, R&D Systems), goat polyclonal anti-synaptopodin (1:1,000, sc-21537, Santa Cruz Biotech), or mouse anti-GAPDH (1:10,000, Abcam) antibodies. Signals were quantified by densitometry using ImageJ software (http://rsbweb.nih.gov/ij/). Values were normalized to GAPDH signals, and protein extracts from two mice per group were analyzed.
Podocyte protein extracts were prepared using a RIPA reagent protocol and was quantified by a Bradford protein quantitation reagent. Samples of 30 μg of protein each were loaded on 8 % sodium dodecyl sulfate polyacrylamide gel and the bands transferred to a polyvinylidine difluoride membrane. The membranes were pre-incubated 2 h with 5 % nonfat milk in PBS containing 0.05 % Tween 20, incubated with 1 μg/ml of anti-NFATc1 antibody (purified mouse anti-NFATc1,556602, BD Pharmingen™) overnight, and washed in PBS containing 0.05 % Tween 20. Then, they were incubated with a 1:5,000 diluted peroxidase–goat anti-mouse IgG (BOSTER) and the immunoreactivity visualized by a Super ECL Plus detection reagent (Biocolor).
Real-time quantitative PCR for cultured podocytes and kidney glomeruli isolated from rodent proteinuric models was performed as recommended in PrimeScript™ RT reagent kit (Takara Bio Inc., Japan) using the following primers: mouse Itg beta3 (NM_016780.2); Plaur (NM_011113.3); NFATc1 (NM_001164109.1); 18S ribosomal RNA (NR_003278.2); and rat Plaur (forward: AGATGTGCTGGGAAACCG; reverse: CAGGGAGGCAATGAGGAT), yielding a 196-bp product.
ChIP analysis in podocytes using antibody to NFATc1 followed by qPCR using the plaur gene (uPAR) promoter-specific primer
Proteins were cross-linked to DNA with 1 % formaldehyde (final concentration) for 10 min at room temperature (RT); glycine (0.125 M) was then added for 5 min at RT to stop the reaction. After being washed twice with ice-cold PBS, the cells were resuspended with lysis buffer (10 mM Tris–HCl, pH 8.0; 100 mM NaCl; 1 mM EDTA, pH 8.0; 0.1 % Na-deoxycholate; and protease inhibitors) and incubated on ice for 30 min. Then, the cell suspension was sonicated for 10 min (30 s “on” and 30 s “off”) to reduce the DNA lengths to between 200 and 1,000 bp. The lysate was then divided into three fractions. The first lysate was incubated with anti-NFATc1 antibody (Santa Cruz Biotechnology) at 4 °C overnight. The second lysate was used for input control. The third lysate was used as the negative control. To collect the immunoprecipitated complexes, magnetic beads (Bangs Laboratories Inc.) were added and incubated for 1 h at 4 °C. Pellet beads were prepared by magnetic separation rack for 2 min at 4 °C before the magnetic beads were sequentially washed in low-salt, high-salt, LiCl salt, and TE buffers. The protein/DNA complexes were eluted and formaldehyde cross-links were reversed by heating the sample at 65 °C for 2 h. The samples were treated with RNase for 20 min at 37 °C, boiled for 10 min at 95 °C, and then prepared by magnetic separation rack for 2 min at 4 °C. DNA was extracted by the phenol/chloroform method, ethanol-precipitated, and resuspended in 10 mM Tris–HCl. The DNA pool from chromatin immunoprecipitation (ChIP), input control, and negative control was used for quantitative PCR (qPCR). PCR amplification was performed on an ABI PRISM7900 system (Applied Biosystems, Foster City, CA). The PCR conditions were an initial step of 10 min at 95 °C, followed by 40 cycles of 10 s at 95 °C, 60 s at 60 °C, and 15 s at 95 °C. The primers were designed by Primer 5.0 system. The amplified Plaur gene promoter sequence is designed at the region 455–572 bp upstream of TSS (chr7:25246946+25247063; F: 5′CCCTCTAGTTTGGCAAGTTAGT3′, R: 5′AACCTCCTGCCTCAGTCTCC3′, 118 bp). Data were analyzed using the 2−ΔΔCT method.
The small interfering RNA (siRNA) sequences that target NFATc1 and control siRNA were synthesized by GenePharma: NFATc1 siRNA sequence (sense: 5′-GCCAUAACUUUCUGCAAGATT-3′; antisense: 5′-UCUUGCAGAAAGUUAUGGCTT-3′); control siRNA sequence (sense: 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense: 5′-ACGUGACACGUUCGGAGAATT-3′).
Before transfection, podocytes were cultured to a density of 70–80 % at 37 °C as described above and then were transfected with the siRNA using lip 2000 (Invitrogen) transfection reagent protocols. Cells were harvested 48 h after siRNA treatment for RT-PCR and Western blot analyses.
For cell motility experiments, cultured differentiated podocytes (each 1×105ml−1) were seeded overnight on vitronectin-coated coverslips in six-well plates. Each coverslip was then scratched with a sterile 200-μl pipette tip, washed with PBS, and placed into fresh medium. After 24 h, cells were fixed with cold methanol, permeabilized with 0.5 % Triton X-100 in PBS, and the cell nuclei stained with DAPI (Roche Diagnostics). Pictures were captured by phase-contrast microscopy under a ×10 objective on a microscope (Leica Microsystems) at 0 and 24 h after scratching.
For statistical analysis, we assessed significance using one-way ANOVA followed by LSD test for comparison between two groups. A value of p<0.05 was considered significant. All values are expressed as the mean±SD.
Results
CsA induces remission of proteinuria and restoration of glomerular podocyte slit diaphragm in 5/6 nephrectomy rats and LPS-induced proteinuric SCID mice
At first, we tested the anti-proteinuric effect of CsA in 5/6 nephrectomy FSGS rats (NTX rats), a well-characterized non-immune model in which critical nephron loss leads to proteinuria, podocyte dysfunction, glomerulosclerosis, and progressive renal dysfunction [19, 20]. As expected, vehicle-treated NTX rats developed heavy proteinuria. In contrast, NTX rats treated with CsA showed a reduced proteinuria at time points of 8 and 12 weeks (p<0.05 and p<0.01, respectively; Fig. 1a).
Fig. 1.
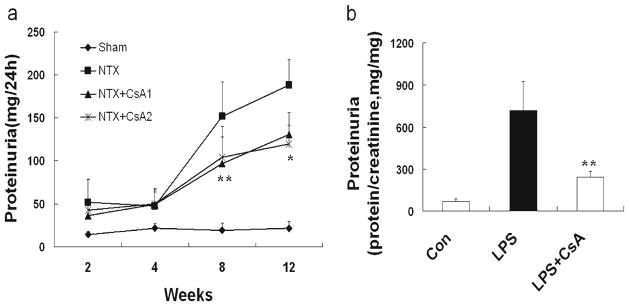
CsA reduces proteinuria in 5/6 nephrectomy rats (NTX rats) and in LPS-induced proteinuric SCID mice (LPS mice). a At time points of 8 and 12 weeks, proteinuria in CsA-treated NTX rats ( at a dose of 1 or 5 mgkg−1day−1) was significantly lower than in untreated NTX rats(p <0.05 and p<0.01, respectively). Sham-operated rats (Sham) served as the control. b Compared with untreated LPS mice, CsA-treated LPS mice showed a significant reduction in proteinuria (p<0.05). All values are expressed as the means±SD. *p<0.01, **p<0.05 versus untreated NTX rats or untreated LPS mice
We next wondered whether CsA exerts the anti-proteinuric effect via its actions on podocytes. Thus, we used CsA in LPS-induced proteinuric SCID mice (LPS mice) that are devoid of T and B cells [21]. In this model, LPS causes proteinuria by targeting podocytes and not other cell types in the kidney, which is in keeping with the observation that podocyte-specific expression of cathepsin L-resistant dynamin [22] or synaptopodin [9] is sufficient to safeguard against LPS-induced proteinuria [23]. As expected, LPS but not PBS caused proteinuria in SCID mice (Fig. 1b). CsA significantly reduced LPS-induced proteinuria (p<0.05; Fig. 1b). Some of the salutary effects of CsA treatment lie in the stabilization of the podocyte slit diaphragm as CsA prevented the loss of nephrin and CD2AP in both the NTX rats and LPS mice (Electronic supplementary material (ESM) Supplementary figure).
All together, our results suggest that CsA is anti-proteinuric in NTX rats and LPS SCID mice because of the inhibition of podocyte uPAR expression in vivo and in vitro.
Recent evidence suggests that the increased uPAR expression in podocytes or the binding of circulating suPAR to podocytes causes proteinuria and FSGS [4, 6]. We investigated the effect of CsA on the expression of uPAR in podocytes in NTX rats and LPS mice. uPAR was localized in podocytes, as indicated by co-labeling with synaptopodin, a marker of this cell type [4]. As expected, the expression of uPAR was substantially increased in podocytes in both proteinuric models (Fig. 2a, b), which is consistent with the recently published evidence [4, 6]. Interestingly, we found that CsA treatment could substantially inhibit uPAR expression in both NTX rats (Fig. 2a) and LPS mice (Fig. 2b). Synaptopodin staining in the glomerulus appeared near normal despite it being cleaved by cytosolic cathepsin L [9]. The reason is the antibody against synaptopodin which recognizes also the cleavage fragment, resulting in a staining of synaptopodin that appears to be unaltered or only slightly reduced (Fig. 2a, b). In contrast, the reported Western blots from cultured podocytes and isolated glomeruli show a reduction in the total synaptopodin protein content and the appearance of a cleavage fragment [9, 24].
Fig. 2.

CsA inhibits podocyte uPAR expression in vivo and in vitro. a Double immunofluorescence staining for uPAR (red) and the podocyte marker synaptopodin (synpo, green) in glomeruli from NTX rats treated with or without CsA, as evaluated by confocal microscopy. The podocytes of untreated NTX rats showed an increased expression of uPAR protein. CsA substantially inhibited uPAR induction. b Same as in Fig. 2a, but now in the LPS mice. LPS substantially enhanced podocyte uPAR induction. CsA significantly inhibited uPAR induction. c Quantitative real-time RT-PCR was performed on kidney glomerulus isolated from NTX rats. PLAUR mRNA (encoding uPAR) was upregulated in untreated NTX rats. CsA inhibited PLAUR mRNA expression (p<0.01). d Same as in Fig. 2c, but now in the LPS mice. Untreated LPS mice showed an increased PLAUR mRNA expression. CsA inhibited PLAUR mRNA expression (p<0.01). e, f Flow cytometry for uPAR cell surface expression showed the development of a high uPAR population after LPS treatment. CsA (0.25–1 μg/ml) reduced uPAR cell surface expression (p<0.01). g Quantitative real-time RT-PCR was performed on cultured differentiated podocytes. PLAUR mRNA expression in LPS-treated podocytes was upregulated. Treatment with CsA (0.25–1 μg/ml) inhibited PLAUR mRNA expression (p <0.01). All values are expressed as the means±SD. *p<0.01 versus untreated NTX rats or LPS mice or podocytes treated with LPS only
We also analyzed PLAUR mRNA (encoding uPAR) in samples from NTX rats (Fig. 2c) or LPS mice (Fig. 2d). NTX rats and LPS mice had a significant increase in PLAUR mRNA expression. When treated with CsA, NTX rats and LPS mice showed a reduced PLAUR mRNA expression (p< 0.01; Fig. 2c, d).
LPS-treated cultured podocytes showed a significant induction of uPAR cell surface expression (Fig. 2e, f) and PLAUR mRNA expression (Fig. 2g). In contrast, podocytes treated with CsA showed a significantly decreased uPAR cell surface expression (p<0.01; Fig. 2e, f) and decreased PLAUR mRNA(p<0.01; Fig. 2g). We also noted a reduction in Plaur mRNA in cultured podocytes after CsA treatment, albeit weaker when compared with in vivo mRNA analysis. All together, it suggests that there is a tight regulation of uPAR expression, in line with previous reports comparing uPAR mRNA levels in normal human and FSGS glomeruli [4].
CsA suppresses β3 integrin activation, but not its surface expression
Because recent evidence suggests that the activation of β3 integrin in podocytes mediates uPAR-induced cellular events leading to proteinuria [4, 6, 25], we next analyzed the alteration in the activity of β3 integrin in podocytes treated with CsA. We used AP5 antibody to detect the active configuration of the β3 integrin [4, 26]. In NTX rats and LPS mice, active β3 integrin was strongly induced (Fig. 3a, b). Of interest is that the administration of CsA largely prevented β3 integrin activation in vivo. Similarly, flow cytometry analysis showed that the activity of β3 integrin was blocked by CsA (p<0.01; Fig. 3e).
Fig. 3.

CsA suppresses β3 integrin activation, but not its cell surface expression. Double immunofluorescence staining for active β3 integrin (red) and the podocyte marker synaptopodin (synpo, green) in glomeruli from NTX rats (a) and LPS mice (b) treated with or without CsA, as evaluated by confocal microscopy. AP5 antibody was used to detect active β3 integrin since the binding ability of AP5 to active β3 integrin has been tested [4, 24]. AP5 labeling was strongly induced in podocytes form untreated NTX rats (a) or LPS mice (b). When treated with CsA, NTX rats (a) or LPS mice (b) showed a substantial reduction of AP5 labeling in podocytes. However, the total expression of β3 integrin (red) remained unchanged in glomeruli from NTX rats (c) and LPS mice (d) treated with or without CsA. (e) Flow cytometry for the AP5 antibody binding to cultured differentiated podocytes showed a high activated β3 integrin population after LPS treatment. CsA (0.25–1 μg/ml) reduced the activation of β3 integrin (p<0.01). However, the total expression of β3 integrin protein (f) and mRNA (g) was unresponsive to LPS or CsA treatment (p>0.05). All values are expressed as the means±SD. *p<0.01 versus podocytes treated with LPS only
We next studied the cell surface expression of total β3 integrin in kidney glomeruli from animal models. The expression of total β3 integrin in podocytes remained unchanged in NTX rats and LPS mice with or without CsA administration (Fig. 3c, d). β3 integrin protein and the ITGB3 gene (encoding β3 integrin) also remained unchanged in cultured podocytes stimulated with LPS alone or in the presence of CsA (p> 0.05; Fig. 3f, g). These data indicate that CsA affects β3 integrin activation, but not its expression.
Calcineurin inhibition reduces podocyte motility in a NFATc1-dependent manner
uPAR is a molecule that is associated with cell motility in cancer cells and podocytes, and its expression is elevated in highly motile cells [3–5]. To study whether CsA affects cellular dynamics in podocytes, we employed podocyte motility studies in vitro. Podocyte motility was analyzed using a scrape-wound assay [27]. LPS treatment for 24 h significantly promoted the motility of podocytes (Fig. 4a, b). In contrast, CsA significantly decreased the number of migrating podocytes (p<0.01; Fig. 4a, b). These results show that CsA inhibits LPS-induced podocyte motility and suggest a link to previous studies where the inactivation of NFAT blocked the invasion and motility of breast cancer cells [28]. Next, we hypothesized whether CsA inhibits podocyte motility in a NFAT-dependent manner. To test this idea, we first evaluated the nuclear translocation of NFATc1 in response to LPS in podocytes (Fig. 4c). Like in the established model of ionomycin-induced NFAT translocation, nuclear import of NFATc1 in podocytes was induced by LPS and blocked by CsA (Fig. 4c). As shown in Fig. 4a, b, 11R-VIVIT (a cell-permeable NFAT inhibitor) inhibited podocyte motility. To address the endogenous role of NFATc1 in podocyte motility, NFATc1-siRNA was used. As shown in Fig. 4a, b, NFATc1-siRNA inhibited LPS-induced podocyte motility, indicating that NFATc1 may mediate LPS-induced podocyte motility.
Fig. 4.

Calcineurin inhibition reduces podocyte motility in a NFATc1-dependent manner. a, b Wound healing assay of podocytes grown on vitronectin. Twenty-four hours after scraping of the podocyte cell layer, cells have started to migrate into the wound track. Treatment with LPS significantly enhanced directed podocyte motility. CsA reduced directed podocyte motility (p<0.01). The wound healing assay showed that 11R-VIVIT (NFAT inhibitor) and NFATc1-siRNA inhibited the podocyte number of directed migration (p<0.01). c Nuclear translocation of NFATc1 in response to CsA in podocytes. NFATc1 (green, arrow) was detected by immunocytochemistry. Ionomycin activation of NFAT was used as a positive control. Podocytes were treated with LPS, alone or in the presence of CsA, respectively. Like in the established model in T cells, both ionomycin and LPS induced NFATc1 translocation in podocytes. As expected, nuclear import of NFATc1 in podocytes was blocked by CsA. All values are expressed as the means±SD. *p< 0.01 versus LPS-treated podocytes
NFATc1 mediates the regulation of uPAR-β3 integrin signaling
Given that CsA inhibited podocyte motility in a NFATc1-dependent manner, it is possible that NFAT may mediate the regulation of uPAR in podocytes. To test whether NFAT is able to mediate uPAR cell surface expression, 11R-VIVIT, a NFAT inhibitor, was used. As shown in Fig. 5a–c, ionomycin (a NFAT activator) induced the expression of uPAR in a dose-dependent manner. In contrast, 11R-VIVIT inhibited LPS-induced uPAR expression in a dose-dependent manner (Fig. 5d–f). To show that endogenous NFAT is able to mediate uPAR expression, NFATc1-siRNA was used (p< 0.01; Fig. 5g–i). As shown in Fig. 5j–l, NFATc1-siRNA-treated podocytes showed a significant reduction of uPAR expression compared to control cells (p<0.01). Based on these findings, it is likely that NFATc1 is involved in the regulation of uPAR expression.
Fig. 5.

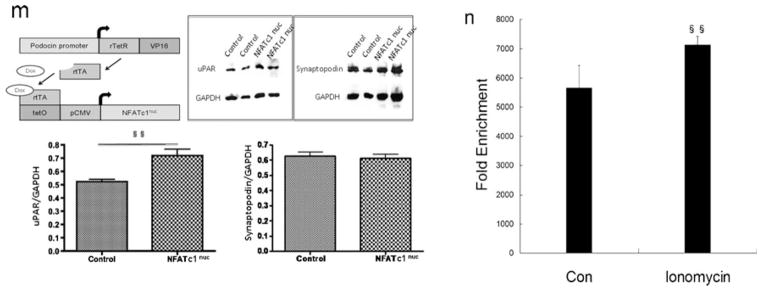
NFATc1 mediates the regulation of uPAR expression in podocytes. a Ionomycin activation of NFAT was used as a positive control. Ionomycin (0.5–2 μM) induced the expression of Plaur mRNA (encoding uPAR) in a dose-dependent manner (p<0.05). b, c Flow cytometry of uPAR expression showed an ionomycin-induced uPAR cell surface expression (p<0.01). d–f Same as ionomycin, LPS induced uPAR expression in podocytes. In contrast, 11R-VIVIT (NFAT inhibitor, 10–1,000 nM) inhibited the LPS-induced expression of Plaur mRNA (encoding uPAR (d); p<0.05 and p<0.01, respectively) and uPAR protein (e, f) (p<0.01) in a dose-dependent manner. NFATc1 was silenced using NFATc1-siRNA. Differentiated podocytes were identified by synaptopodin, a marker of this cell type (g). h, i Western blotting and quantitative real-time RT-PCR analysis showed that NFATc1-siRNA inhibited the expression of the NFATc1 protein (h) and mRNA (i) (p<0.01). j–l NFATc1-siRNA-treated podocytes showed a substantial reduction of Plaur mRNA (encoding uPAR (j); p<0.01) and uPAR protein (k, l) (p<0.01). m Schematic of constructs used for the generation of podocyte-specific, Dox-inducible NFATc1-nuc-transgenic mice. rTetR reverse Tet repressor, rtTA reverse tetracycline-controlled transactivator, VP16 herpes simplex VP16 protein, tetO tet operator sequences, pCMV cytomegalovirus promoter. Western blot analysis of isolated glomeruli from double-transgenic NFATc1nuc mice treated with Dox for 4 days revealed an increase in uPAR protein levels when compared to Dox-treated single transgenic rtTA mice used as the negative control. The protein levels of synaptopodin were not affected by NFATc1nuc transgene expression. The intensity of immunoblot signals was quantified by densitometry and the uPAR protein levels are depicted relative to the GAPDH levels in arbitrary units. n ChIP analysis in podocytes using antibody to NFATc1, followed by qPCR using the Plaur gene promoter-specific primer. The Amplified promoter sequence is designed at the region 455–572 bp upstream of TSS (chr7:25246946+25247063). Fold enrichment=[%(ChIP/Input)]/[%(Negative control/Input)]. All values are expressed as the means±SD. §p<0.01, §§p<0.05 versus control podocytes; *p<0.01, **p<0.05 versus LPS-treated podocytes
To confirm these findings in vivo, we induced the over-expression of a constitutively active NFATc1 variant (NFATc1nuc) in podocytes of adult transgenic mice. In NFATc1nuc, the serine residues that are dephosphorylated by calcineurin are mutated to alanines, rendering this NFATc1 mutant form constitutively nuclear and active and insensitive to nuclear kinases [13]. Transgenic NFATc1nuc mice, which have been shown to develop massive proteinuria within 4 days of transgene induction [11], showed significantly elevated uPAR protein levels in isolated glomeruli when compared with control animals (Fig. 5m). This experiment indicates that NFAT activation in podocytes regulates glomerular uPAR expression in vivo.
ChIP assays followed by qPCR using the Plaur gene promoter-specific primer further confirmed the binding of NFATc1 to the promoter of Plaur gene (encoding uPAR) in podocytes (Fig. 5n). These results suggest that NFATc1 binds directly to the promoter of Plaur gene and upregulates uPAR in podocytes.
Because uPAR activates β3 integrin in podocytes [4, 6], we then analyzed the effect of 11R-VIVIT (a NFAT inhibitor) on the activation of podocyte β3 integrin. As shown in Fig. 6a, d, ionomycin (a NFAT activator) activated β3 integrin (p>0.01); in contrast, 11R-VIVIT (a NFAT inhibitor) inhibited the LPS-induced activation of β3 integrin in a dose-dependent manner. However, neither ionomycin (p> 0.05; Fig. 6b, c) nor 11R-VIVIT (p>0.05; Fig. 6e, f) had any effect on the expression of total β3 integrin protein. To show the effect of endogenous NFAT on the activation of β3 integrin, NFATc1-siRNA was used. NFATc1-siRNA-treated podocytes showed a significant reduction of β3 integrin activation compared to the control cells (p<0.01; Fig. 6g). However, NFATc1-siRNA had no effect on the expression of total β3 integrin (p>0.05; Fig. 6h, i). These results indicated that NFAT did not affect the expression of total β3 integrin, but affected its activation through the upregulation of podocyte uPAR.
Fig. 6.

NFATc1 affects β3 integrin activation, but not its surface expression. Ionomycin activation of NFAT was used as a positive control. Flow cytometry for ionomycin-treated podocytes showed that ionomycin (0.5–2 μM) activated β3 integrin (AP5 antibody labeling) in a dose-dependent manner (a) (p<0.01). However, ionomycin did not affect the expression of total β3 integrin mRNA (b) (p>0.05) or protein (c) (p>0.05). Same as ionomycin, LPS induced β3 integrin activation in podocytes (d) (p<0.01), but not the expression of total β3 integrin mRNA (e) (p>0.05) or protein (f) (p>0.05). Flow cytometry for 11R-VIVIT-treated podocytes showed that 11R-VIVIT (a NFAT inhibitor, 10–1,000 nM) inhibited the LPS-induced activation of β3 integrin in a dose-dependent manner (d) (p<0.01). However, 11R-VIVIT did not affect the expression of total β3 integrin mRNA (e) (p>0.05) or protein (f) (p>0.05). Same as 11R-VIVIT (a NFAT inhibitor), NFATc1-siRNA inhibited the LPS-induced activation of β3 integrin (g) (p<0.01), but not the expression of total β3 integrin mRNA (h) (p>0.05) or protein (i) (p>0.05). All values are expressed as the means±SD. §p<0.01 versus control podocytes; *p<0.01 versus LPS-treated podocytes
NFAT inhibition by 11R-VIVIT reduces proteinuria in LPS-induced proteinuric SCID mice
Given that NFAT mediates the regulation of uPAR-β3 integrin signaling leading to proteinuria, we then investigate whether NFAT inhibition using 11R-VIVIT has an anti-proteinuric effect in LPS-induced proteinuric SCID mice that are devoid of T and B cells [21]. As shown in Fig. 7a, mice receiving 11R-VIVIT showed an attenuated proteinuria in a dose-dependent manner (p<0.01).
Fig. 7.

NFAT inhibitor (11R-VIVIT) reduces proteinuria in LPS-induced proteinuric SCID mice. a Compared with untreated LPS mice, 11R-VIVIT-treated LPS mice showed a significant reduction in proteinuria in a dose-dependent manner with a maximum response at a dose of 15 mg kg−1day−1. b Double immunofluorescence staining for uPAR (red) and synaptopodin (synpo, green), a podocyte marker, in glomeruli from LPS mice. 11R-VIVIT inhibits uPAR expression. c Quantitative real-time RT-PCR performed on kidney glomerulus isolated from LPS mice shows that 11R-VIVIT inhibits Plaur mRNA (encoding uPAR) expression. d Double immunofluorescence staining for activeβ3 integrin (detected by AP5 antibody, red) and synaptopodin (synpo, green) in glomeruli. 11R-VIVIT reduces AP5 labeling. e, f Neither 11R-VIVIT nor LPS fails to affect the expression of total β3 integrin protein (red) or ITGB3 mRNA (encoding β3 integrin). All values are expressed as the means±SD. *p<0.01 versus untreated LPS mice
Next, we examined the effect of 11R-VIVIT on uPAR expression in vivo. Similar as in vitro, 11R-VIVIT significantly reduced uPAR expression compared to the control mice that had received only LPS (p<0.01; Fig. 7b, c). Finally, we examined the effect of 11R-VIVIT on β3 integrin activation in vivo. 11R-VIVIT largely prevented β3 integrin activation (Fig. 7d). However, the expression of total β3 integrin in podocytes was unchanged in LPS mice with or without 11R-VIVIT administration (p>0.05; Fig. 7e, f). These data showed that 11R-VIVIT inhibited uPAR expression and, with that, suppressed β3 integrin activation, but not its expression. Further supporting the pathogenic role of an increased β3 integrin activation in podocytes, it was found that 11R-VIVIT prevented the loss of podocyte slit diaphragm-associated proteins, nephrin and CD2AP, in LPS-treated mice (ESM Supplementary figures A, B, C).
Discussion
The present study shows that the inhibition of the calcineurin–NFAT pathway in podocytes activates the NFAT-mediated transcription of the podocyte urokinase receptor, resulting in podocyte β3 integrin activation and subsequent proteinuria. Using CsA or the NFAT inhibitor 11R-VIVIT results in a decrease of uPAR expression and in a reduction of β3 integrin activation and proteinuria. Recent studies suggested a novel role for podocyte β3 integrin activation in podocyte injury and proteinuria through uPAR or its soluble form, suPAR [4, 6]. While suPAR is increased in the sera of the majority of FSGS patients to cause an outside-in activation of podocyte β3 integrin [6], the present study suggests that the activation of β3 integrin can additionally occur from the podocyte itself utilizing enhanced podocyte uPAR expression [4], which is a consequence of induced NFAT signaling. Thus, our present findings extend the role of uPAR-β3 integrin signaling suggesting that calcineurin/NFAT activation is upstream of podocyte uPAR, contributing to podocyte injury and proteinuria [10, 11] (Fig. 8).
Fig. 8.

Involvement of uPAR-β3 integrin signaling in calcineurin/NFAT activation-induced podocyte injury and proteinuria. Our in vivo results show that calcineurin-NFAT inhibition reduced proteinuria and inhibited uPAR expression and β3 integrin activation in NTX rats and LPS mice. Given recent evidence of the involvement of uPAR-β3 integrin signaling in podocyte dysfunction and proteinuria [4, 6], it is possible that the anti-proteinuric effect of CsA may be attributed to its inhibition of uPAR signaling in podocytes. The results from NFATc1nuc transgene mice, along with those from Wang et al. and Nijenhuis et al. [10, 11], suggested a NFAT activation-induced podoycte injury and proteinuria in mice. Importantly, the present results show that NFAT is able to regulate uPAR expression. All together, these findings suggest the involvement of uPAR signaling in calcineurin/NFAT activation-induced podocyte injury and proteinuria
We show a marked anti-proteinuric effect of CsA in two proteinuria models including 5/6 nephrectomy rats (NTX rats) [19, 20] and LPS-induced proteinuric SCID mice (LPS mice) [12]. The latter model suggests that proteinuria through NFAT/uPAR is independent on T cells, which is in line with previous studies [9, 22, 23]. The anti-proteinuric effect of CsA in the LPS model may be attributed to calcineurin inhibition in podocytes instead of T cells, affecting the stability of synaptopodin [9] and reducing uPAR expression. Furthermore, both models express high podocyte uPAR [4, 7], yet improved in the presence of CsA, suggesting similar injury pathways.
It is interesting that uPAR is identified as a NFAT target gene in podocytes as NFAT activation is relevant to several proteinuric diseases that affect podocytes. Clearly, in FSGS, there is a strong increase in circulating suPAR that might act in concert with podocyte uPAR, whereas in other glomerular disorders short of elevated suPAR, podocyte uPAR might be sufficient to drive podocyte injury. Future studies will clarify the combination effect of suPAR and uPAR on podocyte β3 integrin signaling.
The motility of cultured podocytes serves as a surrogate for foot process dysfunction in vivo and suggests that cytoskeletal dynamics are altered during injury. In keeping with this concept, CsA, 11R-VIVIT, or NFATc1-siRNA reduced LPS- or ionomycin-induced pathological podocyte motility and inhibited podocyte motility in a NFATc1-dependent manner, similar to CsA inhibiting cell motility and tissue infiltration of T cells [29] or NFAT blockade used to halt the invasion and motility of breast cancer cells [28].
Results from Wang et al. and Nijenhuis et al. [10, 11] suggested a novel NFAT activation-induced podoycte injury and proteinuria in mice. Our in vivo experiments show that the calcineurin–NFAT axis directly drives uPAR expression and that the inhibition of this pathway by CsA or 11R-VIVIT reduced uPAR expression and β3 integrin activation in NTX rats and LPS mice. Interestingly, when we analyzed uPAR expression in the inducible, podocyte-specific NFATc1nuc mouse model, we could detect elevated glomerular uPAR protein levels 4 days after transgene induction. Since in these mice transgene expression as well as proteinuria can also be detected in a significant manner starting at 4 days upon induction [11], it appeared likely that uPAR is a direct target gene of NFAT and that its expression is causative involved in the development of podocyte damage. ChIP assays followed by qPCR using the Plaur gene promoter-specific primer further confirmed the binding of NFATc1 to the promoter of the Plaur gene (encoding uPAR) in podocytes, suggesting that NFATc1 binds directly to the promoter of the Plaur gene and upregulates uPAR in podocytes. All together, these findings suggest that uPAR-β3 integrin signaling is involved in calcineurin/NFAT activation-induced podocyte injury and proteinuria. NFAT likely activates several pathways in podocytes that contribute to the induction and/or maintenance of proteinuric kidney disease, like the recently reported positive feedback loop involving NFAT-induced TRPC6 expression [11]. All together, this could explain the sustained activation of the uPAR-β3 integrin axis in progressive glomerular diseases, such as diabetic nephropathy where podocyte uPAR is induced [4].
Previous studies have shown that, instead of causing nonspecific transcriptional alterations, NFATc1 activation leads to distinctive changes in transcription and cellular behavior in different cell populations [30–32]. However, although our in vivo and in vitro results suggests a potential link between NFAT activation and increased uPAR-β3 integrin signaling, the molecular mechanism of NFAT and uPAR expression is not fully addressed. Although the downregulation of uPAR expression seems to be a specific response to NFATc1-siRNA manipulation, it might at least partially reflect a regulation loop encompassing calcineurin–NFAT, uPAR, and β3 integrin since the pleiotropic effects of the used activators LPS and ionomycin do not rule out the possibility of other relevant signaling pathways, e.g., MAPK/Erk cascade, which in turn could lead to increased AP1 known to regulate uPAR expression [33]. The understanding of the NFAT-uPAR-β3 integrin pathway will help develop novel anti-proteinuric strategies that likely have strong effects on podocyte dynamic behavior, resulting in improved kidney health.
Acknowledgments
This work was supported by grants from the Guangdong Science & Technology Department and in part by grants from the National Institute of Health (DK073495 and DK089394 to Dr. Reiser). We thank Dr. P Mundel (Massachusetts General Hospital, Boston, USA) for providing conditionally immortalized mouse podocytes.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00109-012-0960-6) contains supplementary material, which is available to authorized users.
Disclosures Jochen Reiser and Changli Wei are inventors on pending and issued patents on the development of novel therapeutics for proteinuric kidney diseases. They stand to gain royalties from their commercialization.
Contributor Information
Bin Zhang, Email: vistazhang66@yahoo.cn, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China. Southern Medical University, Guangzhou, China.
Wei Shi, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China.
Juan Ma, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China. Southern Medical University, Guangzhou, China.
Alexis Sloan, Department of Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Christian Faul, Department of Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Changli Wei, Department of Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Jochen Reiser, Department of Medicine, Rush University Medical Center, Chicago, IL 60612, USA.
Yun Yang, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China. Southern Medical University, Guangzhou, China.
Shuangxin Liu, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China.
Wenjian Wang, Department of Nephrology, Guangdong General Hospital, Guangdong Academy of Medical Sciences, 106 Zhongshan No. 2 Road, Guangzhou 510080, China.
References
- 1.Zandi-Nejad K, Eddy AA, Glassock RJ, Brenner BM. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl. 2004;(92):S76–S89. doi: 10.1111/j.1523-1755.2004.09220.x. [DOI] [PubMed] [Google Scholar]
- 2.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 3.Reiser J, Oh J, Shirato I, Asanuma K, Hug A, Mundel TM, Honey K, Ishidoh K, Kominami E, Kreidberg JA, et al. Podocyte migration during nephritic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem. 2004;279:34827–34832. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 4.Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 5.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 6.Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap HK, Saleem M, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17:952–960. doi: 10.1038/nm.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B, Xie S, Shi W, Yang Y. Amiloride off-target effect inhibits podocyte urokinase receptor expression and reduces proteinuria. Nephrol Dial Transplant. 2012;27:1746–1755. doi: 10.1093/ndt/gfr612. [DOI] [PubMed] [Google Scholar]
- 8.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 9.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–1666. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, Bakker M, Bindels RJ, de Boer RA, Möller CC, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, et al. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med. 2004;10:305–309. doi: 10.1038/nm994. [DOI] [PubMed] [Google Scholar]
- 16.Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Tertilt C, Bopp T, Heib V, Becker M, Taube C, Schild H, et al. Specific and redundant roles for NFAT transcription factors in the expression of mast cell-derived cytokines. J Immunol. 2006;177:6667–6674. doi: 10.4049/jimmunol.177.10.6667. [DOI] [PubMed] [Google Scholar]
- 17.Høyer-Hansen G, Rønne E, Solberg H, Behrendt N, Ploug M, Lund LR, Ellis V, Danø K. Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain. J Biol Chem. 1992;267:18224–18229. [PubMed] [Google Scholar]
- 18.Pass J, Jögi A, Lund IK, Rønø B, Rasch MG, Gårdsvoll H, Lund LR, Ploug M, Rømer J, Danø K, et al. Murine monoclonal antibodies against murine uPA receptor produced in gene-deficient mice: inhibitory effects on receptor-mediated uPA activity in vitro and in vivo. Thromb Haemost. 2007;97:1013–1022. [PubMed] [Google Scholar]
- 19.Kuhlmann A, Haas CS, Gross ML, Reulbach U, Holzinger M, Schwarz U, Ritz E, Amann K. 1,25-Dihydroxyvitamin D3 decreases podocyte loss and podocyte hypertrophy in the subtotally nephrectomized rat. Am J Physiol Renal Physiol. 2004;286:F526–F533. doi: 10.1152/ajprenal.00316.2003. [DOI] [PubMed] [Google Scholar]
- 20.Amann K, Nichols C, Tornig J, Schwarz U, Zeier M, Mall G, Ritz E. Effect of ramipril, nifedipine, and moxonidine on glomerular morphology and podocyte structure in experimental renal failure. Nephrol Dial Transplant. 1996;11:1003–1011. [PubMed] [Google Scholar]
- 21.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 22.Sever S, Altintas MM, Nankoe SR, Möller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007;117:2095–2104. doi: 10.1172/JCI32022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comper WD. Is the LPS-mediated proteinuria mouse model relevant to human kidney disease? Nat Med. 2009;15:133–134. doi: 10.1038/nm0209-133a. [DOI] [PubMed] [Google Scholar]
- 24.Yaddanapudi S, Altintas MM, Kistler AD, Fernandez I, Möller CC, Wei C, Peev V, Flesche JB, Forst AL, Li J, et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest. 2011;121:3965–3980. doi: 10.1172/JCI58552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degryse B, Resnati M, Czekay RP, Loskutoff DJ, Blasi F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J Biol Chem. 2005;280:24792–24803. doi: 10.1074/jbc.M413954200. [DOI] [PubMed] [Google Scholar]
- 26.Pelletier AJ, Kunicki T, Quaranta V. Activation of the integrin alpha v beta 3 involves a discrete cation-binding site that regulates conformation. J Biol Chem. 1996;271:1364–1370. doi: 10.1074/jbc.271.3.1364. [DOI] [PubMed] [Google Scholar]
- 27.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 28.Jauliac S, López-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4:540–544. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 29.Datta A, David R, Glennie S, Scott D, Cernuda-Morollon E, Lechler RI, Ridley AJ, Marelli-Berg FM. Differential effects of immunosuppressive drugs on T-cell motility. Am J Transplant. 2006;6:2871–2883. doi: 10.1111/j.1600-6143.2006.01553.x. [DOI] [PubMed] [Google Scholar]
- 30.Winslow MM, Pan M, Starbuck M, Gallo EM, Deng L, Karsenty G, Crabtree GR. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Pan M, Winslow MM, Chen L, Kuo A, Felsher D, Crabtree GR. Enhanced NFATc1 nuclear occupancy causes T cell activation independent of CD28 costimulation. J Immunol. 2007;178:4315–4321. doi: 10.4049/jimmunol.178.7.4315. [DOI] [PubMed] [Google Scholar]
- 32.Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 33.Okan E, Drewett V, Shaw PE, Jones P. The small-GTPase RalA activates transcription of the urokinase plasminogen activator receptor (uPAR) gene via an AP1-dependent mechanism. Oncogene. 2001;20:1816–1824. doi: 10.1038/sj.onc.1204260. [DOI] [PubMed] [Google Scholar]