

Abstract
Purpose.
Previous studies have demonstrated that peroxisome proliferator-activated receptor-alpha (PPARα) agonists have therapeutic effects in diabetic retinopathy, although the mechanism of action remains incompletely understood. The purpose of this study was to evaluate PPARα's protective effects in the ischemic retina, and to delineate its molecular mechanism of action.
Methods.
For the oxygen-induced retinopathy (OIR) model, wild-type (WT), and PPARα knockout (PPARα−/−) mice were exposed to 75% O2 from postnatal day 7 (P7) to P12 and treated with the PPARα agonist fenofibric acid (Feno-FA) from P12 to P16. At P17, the effects of Feno-FA on retinal glial fibrillary acidic protein (GFAP) expression, apoptotic DNA cleavage, and TUNEL labeling were analyzed. Cultured retinal cells were exposed to CoCl2 to induce hypoxia, and TUNEL staining and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein dye were used to measure apoptosis and reactive oxygen species (ROS) generation. Western blotting was used to measure GFAP levels and cell signaling.
Results.
Feno-FA decreased retinal apoptosis and oxidative stress in WT but not PPARα−/− OIR mice. Peroxisome proliferator-activated receptor-alpha knockout OIR mice showed increased retinal cell death and glial activation in comparison to WT OIR mice. Feno-FA treatment and PPARα overexpression protected cultured retinal cells from hypoxic cell death and decreased ROS levels. Nuclear hypoxia-inducible factor-α (HIF-1α) and nicotine adenine dinucleotide phosphate oxidase-4 (Nox 4) were increased in OIR retinas and downregulated by Feno-FA in WT but not in PPARα−/− mice.
Conclusions.
Peroxisome proliferator-activated receptor-alpha has a potent antiapoptotic effect in the ischemic retina. This protective effect may be mediated in part through downregulation of HIF-1α/Nox 4 and consequently alleviation of oxidative stress.
Keywords: apoptosis, diabetic retinopathy, HIF-1, hypoxia, ischemia, neurodegeneration, OIR
Activation and over-expression of PPARα protected retinal cells against ischemia-induced apoptosis. Further, PPARα ablation exacerbated ischemia-induced retinal glial activation and cell death. PPARα alleviated oxidative stress under ischemia via down-regulation of NADPH Oxidase 4 expression.
Introduction
Peroxisome proliferator-activated receptor-alpha (PPARα) is a ligand-activated transcription factor, and belongs to the nuclear receptor superfamily.1 It is highly expressed in the liver, and is also expressed in the microvascular, neuronal, and glial tissues of many organs, including the retina.1–4 Peroxisome proliferator-activated receptor-alpha is an attractive therapeutic target for many diseases and pathological conditions due in part to its antioxidant, antiinflammatory, and hypolipidemic effects.1,4–7 It has been shown to play a neuroprotective role in several diseases, and also to regulate vascular homeostasis.1,4,8–12 Recently, two perspective clinical studies identified that the PPARα agonist fenofibrate had a robust therapeutic effect in diabetic retinopathy (DR), although the molecular mechanism(s) of action remain poorly understood.13,14
Diabetic retinopathy is conventionally considered to be a microvascular complication, and indeed the microvascular lesions associated with DR are the primary diagnostic criteria and therapeutic target.15 However, retinal neurodegeneration has now been found to precede sight-threatening proliferative diabetic retinopathy (PDR) and contribute to its development and ultimately to vision loss.15–17
Ischemia/hypoxia contributes to DR development and pathophysiology, and ischemic retinopathy models exhibit phenotypes similar to that of DR, including an emerging role for neurodegeneration.18–22 Hypoxia's pathological effect in DR is complex, but is due in part to oxidative stress, namely increased reactive oxygen species (ROS) production.15,23,24
Many mechanisms contribute to hypoxic oxidative stress, including upregulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase family proteins.25 Nicotinamide adenine dinucleotide phosphate oxidases possess a catalytic NADPH oxidase (Nox) subunit, which generates ROS by catalyzing electron transport from NADPH to molecular oxygen, thus producing superoxide.25 There are seven Nox isoforms, including Nox 1 through 5 and dual oxidase (Duox) 1 and 2.25,26 Nox 2 and Nox 4 both contribute to neurodegeneration in multiple disease models, and inhibition or ablation of each isoform has been shown to have beneficial effects in diabetic and ischemic retinopathies, including neuroprotection.27,28 Regulation of Nox family members is complex, and is a topic of intense investigation, as Nox proteins represent a promising therapeutic target for ischemic disease.29
Hypoxia-inducible factor 1α (HIF-1α) is rapidly degraded in normoxic conditions, but is stabilized in hypoxia.30 Hypoxia-inducible factor 1α is protective in some contexts, but plays a pathophysiological role in many disease processes including diabetic and ischemic retinopathies.31 In ischemia, HIF-1α upregulates expression of Nox 4, which may contribute to hypoxic oxidative stress.32
In the present study, we investigated antiapoptotic and antioxidant effects of PPARα in the ischemic retina, and explored a novel mechanism by which PPARα suppresses retinal oxidative stress and neurodegeneration in an oxygen-induced retinopathy (OIR) model.
Materials and Methods
Chemicals
Fenofibric acid (Feno-FA) was purchased from AK Scientific (Union City, CA, USA). Dimethyl sulfoxide (DMSO) and GW6471 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cobalt chloride (CoCl2) was purchased from Thermo Fisher (Waltham, MA, USA).
Experimental Animals
Both wild-type (WT) and PPARα-knockout (PPARα−/−) mice in the C57-BL/6J background were bred in-house. Care, use, and treatment of all animals in this study were in strict agreement with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research and guidelines in the Care and Use of Laboratory Animals set forth by the University of Oklahoma (Oklahoma City, OK, USA).
OIR Model
Oxygen-induced retinopathy was induced as described previously.33,34 Briefly, newborn WT or PPARα−/− mice were exposed to hyperoxia (75% O2) from postnatal day 7 (P7) to P12 and returned to room air. Feno-FA suspended in DMSO was injected intraperitoneally (10 mg/kg/d) from P12 to P16, with the same volume of vehicle as a control. Normal control animals maintained in constant room air were injected with Feno-FA and vehicle from P12 to P16. For retinal homogenate and eyecup isolation, animals were euthanized, and perfused with prewarmed (37°C) PBS. Retinas were homogenized in Holt's Lysis buffer for Western blotting, and eyecups were fixed and sectioned as described below. All animal studies were representative of at least five animals per group in a minimum of two independent experiments.
Cell Culture
R28 retinal precursor cells were a kind gift of Gail Seigel.35 Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 1 mM D-glucose, 10% heat-inactivated fetal bovine serum (FBS; Cellgro, Manassas, VA, USA), and 1% antibiotic-antimycotic solution (Cellgro). All cells used for experiments were between passages 10 and 15 from the original stock.
Cell Death ELISA
Deoxyribonucleic acid cleavage was measured using an ELISA-based kit (Cell Death Detection ELISA; Roche, Indianapolis, IN, USA) as described previously with a few minor modifications.18 Sample preparation and the ELISA assay were performed as by Zheng et al.,18 and relative fragmentation was expressed as OD (405/490 nm) normalized by RC/DC (Bio-Rad; Hercules, CA, USA) OD (570 nm).
In Vivo TUNEL Staining and Immunohistochemistry
Eyes were enucleated and fixed in Prefer fixative (Anatech, Battle Creek, MI, USA) for 1 hour. Eyecups were cryoprotected in 30% sucrose overnight before being embedded in optimal cutting temperature (OCT) media (Tissue Tek; Sakura Finetek USA, Torrence, CA, USA), snap frozen, and sectioned at 10 μm.
A TMR Red TUNEL labeling kit (Roche) was used for TUNEL staining according to manufacturer's instructions and then mounted in Vectashield medium with diamidino-2-phenylindole (DAPI; Vector Laboratories; Burlingame, CA, USA) prior to imaging with a fluorescence microscope (Leica; Wetzlar, Germany). A minimum of six cross sections taken from six different animals were used in TUNEL analyses.
For colabeling studies, eyecups were TUNEL-labeled as described above and subsequently immunostained with antibodies for CD31 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or protein kinase Cα (PKCα Abcam, Cambridge, MA, USA) prior to being mounted in Vectashield and imaged. All colabeling studies were conducted on a minimum of three cross sections taken from three different animals.
Western Blot Analysis
Retinal homogenates were resolved by SDS-PAGE and blotted with specific antibodies. Antibodies for Nox 4 PPARα and β-actin were purchased from Santa Cruz Biotechnology, Inc. glial fibrillary acidic protein (GFAP) antibody was purchased from Abcam.
Nuclear Extraction and Western Blotting
An EpiSeeker nuclear extraction kit (Abcam) was used to generate retinal nuclear extracts according to manufacturer's instructions. Nuclear lysates were resolved by SDS-PAGE and blotted with anti–HIF-1α antibody (Novus Biologicals; Littleton, CO, USA) and TATA-Binding Protein (TBP) antibody (Santa Cruz Biotechnology, Inc.).
Adenovirus Preparation
Peroxisome proliferator-activated receptor-α–expressing adenovirus was generated previously.4 Preparation, amplification, and titration of the recombinant adenovirus were performed as described previously.36,37 Ad-β-galactosidase prepared previously was used as a control.31,32
Fluorescent Detection of Intracellular ROS
An ROS-sensitive, probe-based method was used as described previously.38 Prior to the assay, 75% confluent adherent R28 cells were treated with one or more of the following for 24 hours: 200 μM CoCl2, 25 μM Feno-FA, vehicle (DMSO), 5 μM GW6471 (a PPARα antagonist) and/or infected with Ad-PPARα or Ad-β-galactosidase (multiplicity of infection = 20). Western blotting was used to verify PPARα overexpression.
In Vitro TUNEL Assay
R28 cells were plated at 102 cells/mL on chamber slides and allowed to adhere overnight before treatment with 200 μM CoCl2 and 25 μM Feno-FA, or vehicle (DMSO). After 48 hours, the Roche TMR Red TUNEL Kit (Roche) was used according to manufacturer's instructions to stain cells. Cells were subsequently mounted in Vectashield media (Vector Laboratories) before being imaged using a fluorescence microscope (Leica). The total number of TUNEL-positive cells per chamber was counted, and data were expressed as the relative percentage of TUNEL-positive cells in comparison to control.
Statistical Analysis
Prism 6 (GraphPad, San Diego, CA, USA) was used to calculate a multivariable ordinary one-way ANOVA for all statistical analyses. P less than or equal to 0.05 was considered to be statistically significant.
Results
Protective Effect of PPARα Activation in Oxygen-Induced Retinopathy
The protective effect of PPARα was evaluated by quantifying retinal glial activation and retinal cell apoptosis in OIR mice. To evaluate the effect of Feno-FA on ischemia-induced retinal glial cell activation, GFAP levels were measured using Western blotting. Retinal GFAP expression was significantly increased in OIR mice in comparison to room air control mice (P ≤ 0.05), and decreased by Feno-FA treatment (P ≤ 0.05), indicating that ischemia-induced glial activation was alleviated by PPARα activation (Fig. 1A). A cell death ELISA was used to quantify total retinal cell apoptosis, and identified that retinal apoptosis was increased in OIR mice in comparison to room air control mice (P ≤ 0.0001) and decreased by Feno-FA treatment in OIR mice (P ≤ 0.001), suggesting that PPARα activation has an antiapoptotic effect in ischemia (Fig. 1B). Similarly, TUNEL-positive cells were also increased in the outer nuclear layer (ONL) and inner nuclear layer (INL) of OIR mice (P ≤ 0.001) and decreased by Feno-FA (P ≤ 0.05), further suggesting a protective effect (Fig. 1C). Finally, double staining demonstrated that TUNEL-positive cells did not colocalize with CD31, suggesting that apoptotic cells were nonvascular, and were likely retinal neurons and/or glial cells (Fig. 1D).
Figure 1.

Cytoprotective effect of Feno-FA (FA) in the retinas of WT OIR mice. (A) At P17, retinal GFAP was measured by Western blotting in room air (RA) and OIR mice treated with Vehicle control (Veh) or FA, semiquantified by densitometry, normalized by β-actin levels, and expressed as the ratio to that in the RA control (n = 6). (B) Retinal DNA fragmentation in P17 mouse retinas treated as above was quantified using a cell death ELISA and normalized to total protein concentration (n ≥ 5). (C) Apoptotic cells in retinal sections from mice treated as above were labeled with TUNEL staining (red), and the nuclei counterstained with DAPI (blue). TUNEL-positive cells were counted in retinal sections (n ≥ 6); scale bars: 10 μm. (D) Retinal sections were double stained with anti-CD31 antibody (green) and TUNEL (red), and the nuclei were counterstained with DAPI (blue) (n ≥ 3). Scale bars: 10 μm. All values are mean ± SEM; *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001. OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
To confirm that TUNEL-positive cells were retinal neurons, retinal sections were colabeled with TUNEL and PKCα antibody. Some of the TUNEL-positive cells colocalized with PKCα, suggesting that some apoptotic cells were amacrine or bipolar cells (Supplementary Fig. S1A). Further, many TUNEL-positive cells in OIR mice were localized to the ONL (Fig. 1C), suggesting that apoptotic photoreceptors were also present.
Dependence of Feno-FA's Effect Upon PPARα
To determine whether Feno-FA's protective effects in retinal ischemia were dependent upon PPARα, OIR was induced in PPARα−/− mice, which were then treated with Feno-FA at the same dose as in WT OIR mice, and retinal glial activation and apoptosis were quantified. Retinal GFAP levels were significantly increased in PPARα−/− OIR mice (P ≤ 0.05), but were not decreased by Feno-FA, unlike WT OIR mice (Fig. 2A). A cell death ELISA demonstrated that retinal apoptosis was increased in PPARα−/− OIR mice (P ≤ 0.0001) but was not affected by Feno-FA treatment (Fig. 2B). Similarly, TUNEL-positive cells were increased in the retinas of PPARα−/− OIR mice (P ≤ 0.0001), but were unchanged by Feno-FA treatment (Fig. 2C). Together, these data suggested that Feno-FA's protective effect was PPARα-dependent. Finally, TUNEL-positive cells did not colocalize with CD31 in PPARα−/− OIR mice (Fig. 2D), suggesting that apoptotic cells were nonvascular, as in WT animals.
Figure 2.

Lack of cytoprotective effect of FA in PPARα−/− OIR mice. (A) Retinal GFAP expression in P17 RA and OIR mice treated with Veh or FA was measured by Western blot analysis, semiquantified by densitometry, normalized by β-actin levels, and expressed as the ratio to that in the RA control (n = 6). (B) Deoxyribonucleic acid fragmentation in mouse retains treated as above was measured by cell death ELISA in retinal homogenates at P17 (n ≥ 5). (C) Apoptotic cells in retinal sections from mice treated as above were labeled by TUNEL (red), and the nuclei counterstained with DAPI (blue). TUNEL-positive retinal cells were counted in retinal sections (n ≥ 6); scale bars: 10 μm. (D) Retinal sections were double stained with the anti-CD31 antibody (green) and TUNEL (red), and the nuclei were counterstained with DAPI (blue; n ≥ 3). Scale bars: 10 μm. All values are mean ± SEM. *P ≤ 0.05, ****P ≤ 0.0001.
Double immunostaining demonstrated that some TUNEL-positive cells colocalized with PKCα, suggesting that they were amacrine or bipolar cells (Supplementary Fig. S1B). Further, some TUNEL-positive cells in OIR mice were localized to the ONL (Fig. 2C), suggesting that apoptotic photoreceptors were also present.
Exacerbated Cell Death and Glial Activation in PPARα−/− OIR Animals
Oxygen-induced retinopathy–induced retinal glial activation and cell death were compared in WT versus PPARα−/− mice. Similar to Figure 1, retinal GFAP expression was increased in WT OIR mice in comparison to room air controls (P ≤ 0.05; Fig. 3A). Under identical OIR conditions, PPARα−/− mice showed a greater increase in retinal GFAP expression in comparison with WT mice (P ≤ 0.05; Fig. 3A). Further, a cell death ELISA demonstrated that PPARα−/− OIR mice had more prominent increases in retinal apoptosis in comparison to WT OIR animals (P ≤ 0.0001; Fig. 3B). Likewise, OIR induced a higher number of TUNEL-positive cells in PPARα−/− mice in comparison with WT mice (P ≤ 0.0001; Fig. 3C). However, P17 room air PPARα−/− mice showed no statistically significant increase in retinal glial activation or apoptosis (Figs. 3A–C) in comparison with WT room air mice, indicating that PPARα is critical to retinal cell survival under stress conditions.
Figure 3.

Increased OIR-induced retinal cell death and glial activation by PPARα ablation. (A) Retinal GFAP expression in P17 RA and OIR WT and PPARα−/− mice was measured by Western blot analysis, normalized by β-actin levels, and expressed as the ratio to that in WT RA control (n = 6). (B) Retinal DNA fragmentation in mice treated as above was measured by cell death ELISA and normalized by total retinal protein levels (n ≥ 5). (C) Apoptotic cells in retinal sections from mice treated as above were labeled with TUNEL staining (red), and nuclei were counterstained with DAPI (blue). TUNEL-positive cells were counted in retinal sections (n ≥ 6); scale bars: 10 μm. All values are mean ± SEM. *P ≤ 0.05, ****P ≤ 0.0001.
Protective Effect of Feno-FA In Vitro
Cultured R28 cells were treated with CoCl2 to induce hypoxia, and apoptosis and was quantified by TUNEL staining. The total number of TUNEL-positive cells per slide chamber was counted, and data were expressed as the percentage of TUNEL-positive cells in comparison with control. TUNEL-positive cells were increased by CoCl2 (P ≤ 0.0001) and decreased by Feno-FA (P ≤ 0.0001), suggesting that PPARα activation ahs a direct protective effect (Fig. 4A).
Figure 4.
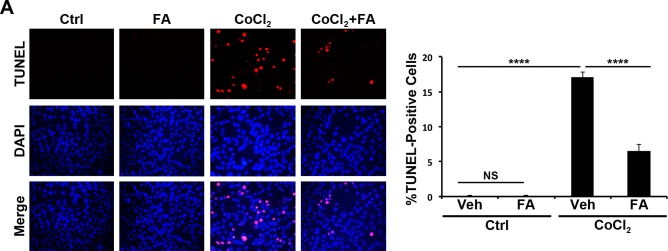
Protection of R28 cells by FA against hypoxia in vitro. (A) R28 cells were treated with CoCl2, Veh, or FA for 48 hours. Apoptotic cells were stained with TUNEL (red), and the nuclei counterstained with DAPI (blue). TUNEL-positive cells in each slide chamber were counted and expressed as percentage of total cells (mean ± SEM; n = 3). ***P ≤ 0.001.
Antioxidant Effects of Feno-FA and PPARα
R28 cells were exposed to CoCl2, and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein (Molecular Probes, Grand Island, NY, USA) was used to quantify intracellular ROS. Intracellular ROS generation was increased by CoCl2 (P ≤ 0.01) and decreased by Feno-FA (P ≤ 0.05), suggesting an antioxidant effect (Fig. 5A). This antioxidant effect was abolished by cotreatment with the PPARα antagonist GW6471, suggesting that Feno-FA's antioxidant effect was PPARα-dependent (Fig. 5A). Further, overexpression of PPARα with an adenoviral vector (Supplementary Fig. S2) was able to decrease CoCl2-induced ROS production (P ≤ 0.01; Fig. 5B).
Figure 5.

Antioxidant effects of FA and PPARα. (A) R28 cells were treated with CoCl2, FA, and GW6471 (PPARα antagonist) as indicated for 24 hours. (B) R28 cells were treated with CoCl2 and infected with Ad-PPARα or control virus Ad-β-galactosidase (β-gal) as indicated for 24 hours. Reactive oxygen species were then measured and expressed as percentage of normoxic control (mean ± SEM; n = 3). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
PPARα-Mediated Repression of Nox 4 in OIR
To delineate the mechanism by which PPARα activation suppresses ROS generation, retinal Nox 4 levels were measured using Western blotting in the retinae of OIR animals. In WT mice, retinal Nox 4 expression was increased in OIR (P ≤ 0.01) and decreased by Feno-FA treatment (P ≤ 0.01; Fig. 6A). Contrastingly, in PPARα−/− mice, OIR-induced Nox 4 upregulation (P ≤ 0.01) was unaffected by Feno-FA (Fig. 6B). Further, retinal Nox 4 upregulation induced by OIR was greater in PPARα−/− mice than in WT mice (P ≤ 0.05) under the same conditions, suggesting that PPARα functions as an endogenous inhibitor of Nox 4 expression in the ischemic retina (Fig. 6C).
Figure 6.

Inhibition of Nox 4 expression by PPARα in OIR. (A) At P17, retinal Nox 4 was measured by Western blotting in WT RA and OIR mice treated with Veh or FA, semiquantified by densitometry, normalized by β-actin levels, and expressed as the ratio to that in the RA control (n = 6). (B) At P17, retinal Nox 4 was measured by Western blotting in PPARα−/− RA and OIR mice treated with Veh or FA and quantified as above (n = 6). (C) In P17 WT and PPARα−/− RA and OIR mice, retinal Nox 4 expression was measured as described above (n = 6). All values are mean ± SEM. *P < 0.05, **P < 0.01.
PPARα-Mediated Repression of HIF-1α Activation in OIR
To elucidate the mechanism by which PPARα regulates Nox 4, we measured nuclear HIF-1α levels in the retinas of OIR mice. Oxygen-induced retinopathy increased nuclear levels of HIF-1α in the retinae of WT OIR mice (P ≤ 0.01), which was decreased by Feno-FA (P ≤ 0.01; Fig. 7A). Nuclear HIF-1α levels were increased in PPARα−/− OIR mice (P ≤ 0.01), but were unaffected by Feno-FA, suggesting that Feno-FA's effect was PPARα-dependent (Fig. 7B). Further, nuclear HIF-1α levels were higher in PPARα−/− OIR retinas than in WT OIR retinas (P ≤ 0.01), suggesting that PPARα plays an endogenous role in suppressing HIF-1α activation in hypoxia (Fig. 7C).
Figure 7.

Inihibition of HIf-1α by PPARα in OIR. (A) In P17 WT RA and OIR mice, retinal levels of nuclear HIF-1α were measured in Veh and FA-treated mice by Western blot analysis, normalized by TBP levels and expressed as the ratio to vehicle-treated RA control (N ≥ 5). (B) In P17 PPARα−/− RA and OIR mice, retinal nuclear HIF-1α levels were measured as above (N ≥ 5). (C) In WT and PPARα−/− RA and OIR mice, retinal nuclear HIF-1α levels were measured as described above and normalized to RA WT mice (N ≥ 5). All values are mean ± SEM. *P ≤ 0.05; **P ≤ 0.001.
Discussion
Here, we have identified for the first time that PPARα has a potent protective effect in retinal ischemia. Our results demonstrated that PPARα activation decreases retinal glial activation and apoptosis under ischemic stress, while PPARα ablation exacerbates retinal glial activation and retinal cell apoptosis in the same model. Further, we also identified that PPARα is able to repress the production of ROS and attenuate upregulation of the pro-oxidant gene Nox 4 by suppressing HIF-1α pathway under ischemia/hypoxia, establishing a molecular mechanism of action.
The retina consists of neuronal cells, glial cells, and vascular cells. To exclude the possibility that PPARα was inhibiting cell death in endothelial cells (EC) rather than neuronal/glial cells, we double-labeled retinal cross sections with TUNEL and CD31, an EC marker. Our results demonstrate that in P17 OIR animals, TUNEL-positive cells do not colocalize with CD31. Further, many TUNEL-positive cells were identified in the ONL, which consists of only photoreceptor cells. Colabeling of TUNEL-positive cells with PKCα in the INL provided further evidence that some of the apoptotic cells are amacrine or bipolar cells. Taken together, these results suggest that apoptotic cells at this time point are nonvascular, but are instead neuronal or glial cells. This indicates that PPARα improves neuroglial survival and integrity under ischemia.
Our prior studies have demonstrated that PPARα colocalizes with GFAP in the retina, indicating that it is expressed in Müller glia.4 Glial dysfunction is associated with retinal neurodegeneration,39 and it is possible that PPARα exerts its neuroprotective effect in part by modulating glial over-activation and dysfunction. However, we also demonstrated direct cytoprotective and antioxidant effects in cultured retinal neuronal cells under hypoxia. These results suggest that PPARα's beneficial effects may also be through a direct effect on retinal neurons in addition to improved glial function.
Clinical studies identified that the PPARα agonist fenofibrate decreased progression of DR in human patients,13,14 but provided little insight into which pathophysiological parameters PPARα was able to improve, or how it was able to do so on a molecular level. Diabetic retinopathy is traditionally considered a microvascular complication of diabetes.15,40 However, it has recently been discovered that neuronal cell death and glial activation precede clinically apparent vascular pathologies,15,16,41 and it is believed that these events affect the subsequent vascular dysfunction and pathological angiogenesis associated with PDR.15,16,34 Neurodegeneration is also thought to play a role in the vascular lesions associated with ischemic retinopathies, and is an emerging area of investigation and therapeutic target in many of these retinal disease models.18,20,27
Our group previously demonstrated that PPARα activation had a therapeutic effect in DR animal models, and that PPARα played an important role in vascular homeostasis, decreasing leukostasis, vascular leakage, and neovascularization in diabetic and OIR animal models.4,42 However, PPARα's effect on retinal neurodegeneration was previously unknown. We also identified in past studies that PPARα is downregulated in the retinas of human DR patients and in three DR animal models,4 and postulated that this decrease in retinal PPARα levels may contribute to the neurodegenerative events associated with early DR.
In the OIR model, newborn rodents are exposed to hyperoxia (75% O2) to induce vasoobliteration. Subsequent return to room air results in retinal ischemia and consequently neurodegeneration, oxidative stress, vascular leakage, and pathological neovascularization.20,34 Although OIR is not a diabetic animal model, the OIR model is physiologically and molecularly similar to DR, as it induces retinal inflammation, NV, and retinal neurodegeneration, similar to PDR.27,43 Further, ischemia/hypoxia contributes to the development and progression in DR, and plays a role in retinal apoptosis.15 Therefore, in order to delineate PPARα's protective effects in retinal ischemia, we used the OIR model, and have now demonstrated for the first time that PPARα has a potent neuroprotective effect in the ischemic retina.
Because the retina is the most metabolically demanding tissue in the body, it is highly susceptible to ischemic stress.23 Ischemia thus plays a pathological role in many eye diseases including ROP, glaucoma, central vein occlusion, retinal detachment, and AMD.23,44–46 Further, hypoxic oxidative stress contributes to neurodegeneration in many of these models.23 Our present findings suggest that PPARα also has significant therapeutic potential in these ischemic retinal diseases, much as in DR.
Neurodegeneration occurs in early DR, prior to the onset of overt microvascular dysfunction.47 Once considered a prognostic indicator for DR, neurodegeneration is now known to contribute to the vascular abnormalities characteristic of advanced PDR. Neural activity in the retina regulates retinal blood flow through neurovascular coupling, wherein neural simulation releases vasodilating factors.48,49 Neurodegeneration impairs the neurovascular coupling response, precipitating endothelial dysfunction and vasoconstriction.48,49 Further, DR-associated glial activation and excitotoxicity upregulate vascular growth factors, leading to increased vascular permeability and subsequent breakdown of the blood–retinal barrier.50 It is now thought that inhibiting this early neurodegenerative event may prevent development of downstream microvascular abnormalities, although no clinically available treatments target this aspect of the disease.
Our prior work demonstrated that PPARα alleviates retinal leukostasis, vascular leakage, and neovascularization in a DR model, and thus indicated that it has beneficial effects on diabetic vascular homeostasis.42 The present study has now demonstrated that PPARα has a neuroprotective effect in retinal ischemia, and is likely to exert a similar effect in DR. However, whether PPARα indeed plays a protective role against DR-associated retinal neurodegeneration is a topic of future investigation. Nonetheless, this dual effect of PPARα in both neuroprotection and vascular homeostasis makes it a highly desirable therapeutic target for diabetic and other retinopathies, and may account for fenofibrate's unprecedented efficacy in clinical trial.
Oxidative stress plays a major causative role in the development and progression of most diabetic complications, including DR.51 Oxidative stress in the diabetic retina can be induced by many mechanisms, including ischemia/hypoxia resulting from impaired vascular function.51,52 Peroxisome proliferator-activated receptor-alpha's antioxidant and neuroprotective properties in other ischemic diseases are well documented,1 and we hypothesized that PPARα may potentially decrease ischemic oxidative stress to attenuate retinal apoptosis. Our results confirm that PPARα activation and overexpression indeed reduces ROS generation in retinal neuronal cells.
The NADPH oxidases are a relatively newly recognized protein family, and since their discovery 30 years ago, they have been found to play diverse roles in many physiological and pathophysiological processes.53 Nox proteins play pathological roles in many diseases, contributing to oxidative stress and subsequent neuronal apoptosis, inflammation, and vascular dysfunction.54–56 Nox 4 has been shown to play a pathological role in DR, while Nox 2 ablation was reported to have a neuroprotective effect in ischemic retinopathy.27,28 These reports suggest that Nox 2 and Nox 4 are both implicated in ischemic neuronal injury, and that inhibition or ablation of these proteins may have a protective, antioxidant effect in ischemia/hypoxia.56,57 Here, we demonstrate for the first time that PPARα negatively regulates Nox 4 expression in ischemia. This has broad-reaching implications for not only PPARα's therapeutic effects in diabetic and ischemic retinopathies, but also contributes to the understanding of PPARα's antioxidant properties and molecular mechanisms of action in other contexts.
We and others have previously demonstrated that PPARα suppresses HIF-1α, potentially by enhancing its interaction with von Hippel-Lindau tumor suppressor (pVHL), which mediates HIF-1α degradation.42,58 In turn, HIF-1α enhances transcription of Nox 4.32 Our results in the OIR retina are consistent with previous findings, suggesting that PPARα likely decreases hypoxia-induced Nox 4 upregulation by accelerating HIF-1α degradation.
In summary, our studies identified novel antiapoptotic and antioxidant activities of PPARα in retinal ischemia, and established that it negatively regulates HIF-1α/Nox 4 signaling, which is likely responsible in part for its antioxidant effect. Therefore, PPARα activation has therapeutic potential for retinal oxidative and inflammatory disorders such as DR and other ischemic retinal diseases.
Acknowledgments
Supported by National Institutes of Health (Bethesda, MD, USA) Grants EY012231, EY018659, EY019309, and GM104934, American Heart Association (Dallas, TX, USA) Grant 12PRE12030078, and the Oklahoma Center for the Advancement of Science & Technology (OCAST; Oklahoma City, OK, USA).
Disclosure: E. Moran, None; L. Ding, None; Z. Wang, None; R. Cheng, None; Q. Chen, None; R. Moore, None; Y. Takahashi, None; J.-X. Ma, None
References
- 1. Bordet R, Ouk T, Petrault O, et al. PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans. 2006; 34: 1341–1346 [DOI] [PubMed] [Google Scholar]
- 2. Hiukka A, Maranghi M, Matikainen N, Taskinen MR. PPARalpha: an emerging therapeutic target in diabetic microvascular damage. Nat Rev Endocrinol. 2010; 6: 454–463 [DOI] [PubMed] [Google Scholar]
- 3. IJ A, Tan NS, Gelman L, et al. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004; 23: 2083–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu YC, Chen Y, Ding L, et al. Pathogenic role of diabetes-induced PPAR-alpha down-regulation in microvascular dysfunction. Proc Natl Acad Sci U S A. 2013; 110: 15401–15406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sugden MC, Warlow MP, Holness MJ. The involvement of PPARs in the causes, consequences and mechanisms for correction of cardiac lipotoxicity and oxidative stress. Curr Mol Pharmacol. 2012; 5: 224–240 [PubMed] [Google Scholar]
- 6. Gervois P, Mansouri RM. PPARalpha as a therapeutic target in inflammation-associated diseases. Expert Opin Ther Targets. 2012; 16: 1113–1125 [DOI] [PubMed] [Google Scholar]
- 7. Tenenbaum A, Fisman EZ. Fibrates are an essential part of modern anti-dyslipidemic arsenal: spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc Diabetol. 2012; 11: 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sehgal N, Kumawat KL, Basu A, Ravindranath V. Fenofibrate reduces mortality and precludes neurological deficits in survivors in murine model of Japanese encephalitis viral infection. PLoS One. 2012; 7: e35427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhateja DK, Dhull DK, Gill A, et al. Peroxisome proliferator-activated receptor-alpha activation attenuates 3-nitropropionic acid induced behavioral and biochemical alterations in rats: possible neuroprotective mechanisms. Eur J Pharmacol. 2012; 674: 33–43 [DOI] [PubMed] [Google Scholar]
- 10. Mysiorek C, Culot M, Dehouck L, et al. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr Neurovasc Res. 2009; 6: 181–193 [DOI] [PubMed] [Google Scholar]
- 11. Deplanque D, Gele P, Petrault O, et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003; 23: 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006; 116: 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keech AC, Mitchell P, Summanen PA, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007; 370: 1687–1697 [DOI] [PubMed] [Google Scholar]
- 14. Group AS, Group AES, Chew EY, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010; 363: 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012; 366: 1227–1239 [DOI] [PubMed] [Google Scholar]
- 16. Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011; 52: 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gastinger MJ, Singh RS, Barber AJ. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci. 2006; 47: 3143–3150 [DOI] [PubMed] [Google Scholar]
- 18. Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007; 48: 361–367 [DOI] [PubMed] [Google Scholar]
- 19. Heidary G, Vanderveen D, Smith LE. Retinopathy of prematurity: current concepts in molecular pathogenesis. Semin Ophthalmol. 2009; 24: 77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Narayanan SP, Suwanpradid J, Saul A, et al. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS One. 2011; 6: e22460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mowat FM, Gonzalez F, Luhmann UF, et al. Endogenous erythropoietin protects neuroretinal function in ischemic retinopathy. Am J Pathol. 2012; 180: 1726–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dorfman AL, Cuenca N, Pinilla I, Chemtob S, Lachapelle P. Immunohistochemical evidence of synaptic retraction, cytoarchitectural remodeling, and cell death in the inner retina of the rat model of oxygen-induced retinopathy (OIR). Invest Ophthalmol Vis Sci. 2011; 52: 1693–1708 [DOI] [PubMed] [Google Scholar]
- 23. Li SY, Fu ZJ, Lo AC. Hypoxia-induced oxidative stress in ischemic retinopathy. Oxid Med Cell Longev. 2012; 2012: 426769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kermorvant-Duchemin E, Sapieha P, Sirinyan M, et al. Understanding ischemic retinopathies: emerging concepts from oxygen-induced retinopathy. Doc Ophthalmol. 2010; 120: 51–60 [DOI] [PubMed] [Google Scholar]
- 25. Kleikers PW, Wingler K, Hermans JJ, et al. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl). 2012; 90: 1391–1406 [DOI] [PubMed] [Google Scholar]
- 26. Kahles T, Brandes RP. NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci. 2012; 69: 2345–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yokota H, Narayanan SP, Zhang W, et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest Ophthalmol Vis Sci. 2011; 52: 8123–8131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes. 2010; 59: 1528–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spychalowicz A, Wilk G, Sliwa T, Ludew D, Guzik TJ. Novel therapeutic approaches in limiting oxidative stress and inflammation. Curr Pharm Biotechnol. 2012; 13: 2456–2466 [PubMed] [Google Scholar]
- 30. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007; 2007: [DOI] [PubMed] [Google Scholar]
- 31. Vadlapatla RK, Vadlapudi AD, Mitra AK. Hypoxia-inducible factor-1 (HIF-1): a potential target for intervention in ocular neovascular diseases. Curr Drug Targets. 2013; 14: 919–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diebold I, Petry A, Hess J, Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010; 21: 2087–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin M, Chen Y, Jin J, et al. Ischaemia-induced retinal neovascularisation and diabetic retinopathy in mice with conditional knockout of hypoxia-inducible factor-1 in retinal Muller cells. Diabetologia. 2011; 54: 1554–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994; 35: 101–111 [PubMed] [Google Scholar]
- 35. Seigel GM. Review: R28 retinal precursor cells: the first 20 years. Mol Vis. 2014; 20: 301–306 [PMC free article] [PubMed] [Google Scholar]
- 36. Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005; 102: 12413–12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takahashi Y, Moiseyev G, Chen Y, Ma JX. Identification of conserved histidines and glutamic acid as key residues for isomerohydrolase activity of RPE65, an enzyme of the visual cycle in the retinal pigment epithelium. FEBS Letters. 2005; 579: 5414–5418 [DOI] [PubMed] [Google Scholar]
- 38. Zhang B, Ma JX. SERPINA3K prevents oxidative stress induced necrotic cell death by inhibiting calcium overload. PLoS One. 2008; 3: e4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieth E, Gardner TW, Barber AJ, Antonetti DA; for the Penn State Retina Research Group. Retinal neurodegeneration: early pathology in diabetes. Clin Exp Ophthalmol. 2000; 28: 3–8 [DOI] [PubMed] [Google Scholar]
- 40. Durham JT, Herman IM. Microvascular modifications in diabetic retinopathy. Curr Diab Rep. 2011; 11: 253–264 [DOI] [PubMed] [Google Scholar]
- 41. Imai H, Singh RS, Fort PE, Gardner TW. Neuroprotection for diabetic retinopathy. Dev Ophthalmol. 2009; 44: 56–68 [DOI] [PubMed] [Google Scholar]
- 42. Chen Y, Hu Y, Lin M, et al. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes. 2013; 62: 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu X, Zhang B, McBride JD, et al. Anti-angiogenic and anti-neuroinflammatory effects of kallistatin through interactions with the canonical Wnt pathway. Diabetes. 2013; 62: 4228–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cherecheanu AP, Garhofer G, Schmidl D, Werkmeister R, Schmetterer L. Ocular perfusion pressure and ocular blood flow in glaucoma. Curr Opin Pharmacol. 2013; 13: 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McAllister IL. Central retinal vein occlusion: a review. Clin Exp Ophthalmol. 2012; 40: 48–58 [DOI] [PubMed] [Google Scholar]
- 46. Zarbin MA. Age-related macular degeneration: review of pathogenesis. Eur J Ophthalmol. 1998; 8: 199–206 [DOI] [PubMed] [Google Scholar]
- 47. Simo R, Hernandez C; for the European Consortium for the Early Treatment of Diabetic Retinopathy. Neurodegeneration is an early event in diabetic retinopathy: therapeutic implications. Br J Ophthalmol. 2012; 96: 1285–1290 [DOI] [PubMed] [Google Scholar]
- 48. Metea MR, Newman EA. Signalling within the neurovascular unit in the mammalian retina. Exp Physiol. 2007; 92: 635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Riva CE, Logean E, Falsini B. Visually evoked hemodynamical response and assessment of neurovascular coupling in the optic nerve and retina. Prog Retin Eye Res. 2005; 24: 183–215 [DOI] [PubMed] [Google Scholar]
- 50. Cervantes-Villagrana AR, Garcia-Roman J, Gonzalez-Espinosa C, Lamas M. Pharmacological inhibition of N-methyl d-aspartate receptor promotes secretion of vascular endothelial growth factor in Müller cells: effects of hyperglycemia and hypoxia. Curr Eye Res. 2010; 35: 733–741 [DOI] [PubMed] [Google Scholar]
- 51. Madsen-Bouterse SA, Kowluru RA. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord. 2008; 9: 315–327 [DOI] [PubMed] [Google Scholar]
- 52. Arden GB, Sivaprasad S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev. 2011; 7: 291–304 [DOI] [PubMed] [Google Scholar]
- 53. Krause KH, Lambeth D, Kronke M. NOX enzymes as drug targets. Cell Mol Life Sci. 2012; 69: 2279–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao HM, Zhou H, Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci. 2012; 33: 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Radermacher KA, Wingler K, Langhauser F, et al. Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid Redox Signal. 2013; 18: 1418–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kleinschnitz C, Grund H, Wingler K, et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010; 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011; 11: 27–35 [DOI] [PubMed] [Google Scholar]
- 58. Zhou J, Zhang S, Xue J, et al. Activation of peroxisome proliferator-activated receptor alpha (PPARalpha) suppresses hypoxia-inducible factor-1alpha (HIF-1alpha) signaling in cancer cells. J Biol Chem. 2012; 287: 35161–35169 [DOI] [PMC free article] [PubMed] [Google Scholar]