

Abstract
Sigma-1 receptor (σ1R), an endoplasmic reticulum–chaperone protein, can modulate painful response after peripheral nerve injury. We have demonstrated that voltage-gated calcium current is inhibited in axotomized sensory neurons. We examined whether σ1R contributes to the sensory dysfunction of voltage-gated calcium channel (VGCC) after peripheral nerve injury through electrophysiological approach in dissociated rat dorsal root ganglion (DRG) neurons. Animals received either skin incision (Control) or spinal nerve ligation (SNL). Both σ1R agonists, (+)pentazocine (PTZ) and DTG [1,3-di-(2-tolyl)guanidine], dose dependently inhibited calcium current (ICa) with Ba2+ as charge carrier in control sensory neurons. The inhibitory effect of σ1R agonists on ICa was blocked by σ1R antagonist, BD1063 (1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride) or BD1047 (N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine dihydrobromide). PTZ and DTG showed similar effect on ICa in axotomized fifth DRG neurons (SNL L5). Both PTZ and DTG shifted the voltage-dependent activation and steady-state inactivation of VGCC to the left and accelerated VGCC inactivation rate in both Control and axotomized L5 SNL DRG neurons. The σ1R antagonist, BD1063 (10 μM), increases ICa in SNL L5 neurons but had no effect on Control and noninjured fourth lumbar neurons in SNL rats. Together, the findings suggest that activation of σR1 decreases ICa in sensory neurons and may play a pivotal role in pain generation.
Introduction
Once identified as an opioid receptor subtype, sigma-1 receptor (σ1R) is now recognized as an intracellular ligand-regulated chaperone protein that resides at the mitochondria-associated endoplasmic reticulum membrane (MAM) (Hayashi and Su, 2007; Su et al., 2010). After activation, σ1R traffics to various subcellular compartments, including the plasmalemma, and regulates diverse functions (Su et al., 2010). The σ1R is found in a range of tissues, including the central nervous system (Alonso et al., 2000; Matsumoto, 2007), where σ1R modulation of neuronal ion channels and activity is associated with neurologic and psychiatric conditions (Kourrich et al., 2012). We have recently demonstrated that σ1R is also present in the peripheral nervous system, in both sensory neurons and satellite glial cells of the dorsal root ganglion (DRG) (Bangaru et al., 2013). The σ1R has been shown to modulate opioid activity, by which σ1R activation reduces opioid analgesia (Chien and Pasternak, 1994), whereas σ1R blockade potentiates opioid analgesia (Chien and Pasternak, 1995; Mei and Pasternak, 2002). A direct involvement of σ1R in pain processing is also evident by modulation of hyperalgesic responses after nerve injury. Specifically, intrathecal injection of σ1R antagonists, such as BD1047 (N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine dihydrobromide) or S1RA, reduces painful behavior after peripheral nerve injury (Roh et al., 2008b; Romero et al., 2012), whereas σ1R knockout mice reduce central sensitization and diminish hyperalgesic responses after injury (de la Puente et al., 2009). Additionally, blockade of σ1R can prevent chemotherapy-induced neuropathic pain (Nieto et al., 2012). Finally, formalin- and capsaicin-induced inflammation pain is attenuated by σ1R knockout and by σ1R antagonist administration (Cendan et al., 2005; Entrena et al., 2009). Together, these findings suggest that σ1R plays an important role in pain modulation.
Voltage-gated calcium channels (VGCCs) control critical neuronal functions, including development, excitability, and synaptic transmission (Catterall, 2011). The σ1R is a recognized component of homeostatic system regulating cytoplasmic Ca2+ concentration (Hayashi et al., 2000; Hayashi and Su, 2007; Kourrich et al., 2012). Specifically, σ1R agonists inhibit activity-induced Ca2+ influx through direct interactions with L-type VGCCs (Hayashi et al., 2000; Tchedre et al., 2008; Mueller et al., 2013). Our previous findings demonstrate that painful peripheral nerve injury decreases Ca2+ current (ICa) through sensory neuron VGCCs (Hogan et al., 2000; McCallum et al., 2006) and increases neuronal excitability (Lirk et al., 2008). Thus, it is possible that σ1R activation is a component of the processes leading to neuropathic pain. Because this hypothesis has not previously been tested, we examined the regulation of VGCC function by σ1R activation in sensory neurons using the spinal nerve ligation (SNL) model of neuropathic pain (Kim and Chung, 1992). This model results in two populations of neurons, specifically the fifth lumbar (L5) DRG neurons that are directly axotomized by the ligation and the fourth lumbar (L4) neurons whose peripheral processes are exposed to the inflammatory effects of the degenerating L5 axonal fragments.
Materials and Methods
Animals.
All methods and use of animals were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (Taconic Farms, Hudson, NY) were housed individually in a room maintained at 22 ± 0.5°C and constant humidity (60 ± 15%) with an alternating 12-hour light/dark cycle. Food and water were available ad libitum throughout the experiments.
Nerve Injury.
Rats weighing 125–150 g were subjected to SNL modified from the original technique (Kim and Chung, 1992). Specifically, rats were anesthetized with 2% isoflurane in oxygen, and the right paravertebral region was exposed. The sixth lumbar transverse process was removed, after which the L5 and sixth lumbar spinal nerves were ligated with 6-0 silk suture and transected distal to the ligature. To minimize non-neural injury, no muscle was removed, muscles and intertransverse fascia were incised only at the site of the two ligations, and articular processes were not removed. The muscular fascia was closed with 4-0 resorbable polyglactin sutures, and the skin was closed with staples. Control animals received skin incision and closure only. After surgery, rats were returned to their cages and kept under normal housing conditions with access to pellet food and water ad lib. In all animals subjected to nerve injury, anatomically correct SNL was confirmed at the time of tissue harvest.
Sensory Testing.
We measured the incidence of a pattern of noxious stimulus-induced hyperalgesic behavior, which is a specific neuropathic-related pain behavior that we have previously documented to be associated with conditioned place avoidance (Hogan et al., 2004; Wu et al., 2010). Thus, noxious stimulation (22G spinal needle) was used as only inclusion criterion after peripheral nerve injury. On 3 different days between 12 and 17 days after surgery, right plantar skin was touched (10 stimuli/test) with a 22G spinal needle with adequate pressure to indent but not penetrate the skin. Whereas control animals respond with only a brief reflexive withdrawal, rats following SNL may display a complex hyperalgesia response that includes licking, chewing, grooming, and sustained elevation of the paw. The average frequency of hyperalgesia responses over the 3 testing days was tabulated for each rat. To assure high selectivity on effects associated with hyperalgesia (Hogan et al., 2004), only rats that displayed a hyperalgesia-type response after at least 20% of stimuli were used further after SNL in this study, which excludes approximately 35% of animal subjects. Because such nonresponder animals may result from various causes, including interanimal anatomic variation and sensory testing inconsistency, nonresponder animals were not studied further.
Neuron Isolation and Plating.
Neurons were rapidly harvested from L4 and L5 DRGs during isoflurane anesthesia and decapitation 21–28 days after SNL or skin sham surgery. This interval was chosen because hyperalgesia is fully developed by this time (Hogan et al., 2004). Ganglia were incubated in 0.5 mg/ml Liberase TM (Roche, Indianapolis, IN) in Dulbecco’s modified Eagle’s medium/F12 with glutaMAX (Life Technologies, Grand Island, NY) for 30 minutes at 37°C, followed with 1 mg/ml trypsin (Sigma-Aldrich, St. Louis, MO) and 150 Kunitz units/ml DNase (Sigma-Aldrich) for another 10 minutes. After addition of 0.1% trypsin inhibitor (type II; Sigma-Aldrich), tissues were centrifuged, lightly triturated in neural basal media (1×; Life Technologies) containing 2% (v:v) B27 supplement (50×; Life Technologies), 0.5 mM glutamine (Sigma-Aldrich), 0.05 mg/ml gentamicin (Life Technologies), and 10 ng/ml nerve growth factor 7S (Alomone Laboratories, Jerusalem, Israel). Cells were then plated onto poly-L-lysine (70–150 kDa; Sigma-Aldrich)–coated glass coverslips (Deutsches Spiegelglas; Carolina Biologic Supply, Burlington, NC) and incubated at 37°C in humidified 95% air and 5% CO2 for at least 2 hours and were studied 3–8 hours after dissociation.
Solutions and Agents.
Unless otherwise specified, the bath contained Tyrode´s solution (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 10 glucose, 2 MgCl2, 10 HEPES, with an osmolarity of 297–300 mOsm and pH 7.40. The σ1R agonist, (+)pentazocine (PTZ) (Su et al., 2010), was obtained from the National Institute on Drug Abuse (Baltimore, MD). Other σ1R ligands, including DTG [1,3-di-(2-tolyl)guanidine], BD1047, and BD1063 (1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride) (Su et al., 2010), were purchased from Tocris (Ellisville, MO). Selective VGCC subtype antagonists used included nimodipine (Calbiochem, Billerica, MA) to block L-type current, with a dose (5 μM) that has been determined in previous studies (Oliveria et al., 2007; Wu et al., 2008; Duncan et al., 2013) and fully blocks L-type currents (Xu and Lipscombe, 2001). N-Type current was blocked with ω-conotoxin GVIA (GVIA, 200 nM; Tocris) (McCallum et al., 2011); ω-conotoxin MVIIC (MVIIC, 200 nM; Tocris) was used to block both N- and P/Q-type current (McCallum et al., 2011); and SNX-482 (200 nM; Tocris) was used to block R-type current (McCallum et al., 2011). Stock solutions of DTG, BD1063, and nimodipine were dissolved in dimethylsulfoxide, and subsequently diluted in the relevant bath solution such that final bath concentration of dimethylsulfoxide was 0.2% or less, which does not affect cytoplasmic Ca2+ concentration (Gemes et al., 2011). PTZ and other VGCC antagonists were dissolved in water to make stock solutions.
Whole-Cell Patch-Clamp Electrophysiological Recording.
Voltage and currents were recorded in small- to medium-size neurons (33.8 ± 0.2 µm, n = 222), using the whole-cell configuration of the patch-clamp technique. Patch pipettes (2–5 MΩ) were formed from borosilicate glass (Garner Glass, Claremont, CA) using a micropipette puller (P-97; Sutter Instruments, Novato, CA) and were then fire polished. Currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz through a 4-pole Bessel filter, and digitized at 10 kHz with a Digidata 1320 A/D interface and pClamp 9 software (Molecular Devices) for storage on a personal computer. After achieving giga-ohm seal and breakthrough, membrane capacitance was determined and access resistance was compensated (60–85%). Neurons with 10 MΩ access resistances after breakthrough were discarded. Experiments were performed 3–5 minutes after breakthrough, and at room temperature (25°C).
Seals were achieved in Tyrode’s solution. Voltage-induced currents flowing through VGCCs, referred to as ICa, although Ba2+ is the charge carrier, were recorded using an extracellular solution containing (in mM): 2 BaCl2, 4.8 CsCl, 2 MgCl2, 5 4-aminophyridine, 132 N-methyl-D-glucamine, 10 HEPES, and 5 D-glucose, pH 7.4, with an osmolarity of 300 mOsm. The internal pipette solution contained (in mM): 110 CsCl, 20 TEACl, 5 Mg-ATP, 0.4 Li4-GTP, 0.5 EGTA, 1 CaCl2, 1 MgCl2, 0.1 cAMP, and 10 HEPES, pH 7.2, with an osmolarity of 300 mOsm. To selectively record low voltage–activated T-type currents, a pipette solution that contained fluoride to facilitate high voltage–activated (HVA) ICa rundown was used (Fischer et al., 2014), which contained (in mM) 135 tetramethylammonium hydroxide, 10 EGTA, 40 HEPES, and 2 MgCl2, pH of 7.2, adjusted with hydrofluoric acid with an osmolarity of 300 mOsm. The free concentration for Ca2+ was calculated to be 211 nM using Maxchelator (http://maxchelator.stanford.edu). Voltage-clamp protocols were used to measure whole-cell ICa. Peak current amplitude was used in constructing current-voltage relationship, steady-state activation, and inactivation, as protocols are as described in relevant results and figure legends in the text. Current that remained after the application of Cd2+ (200 μM) at the end of each experiment was subtracted from measured currents to account for currents other than ICa, including leak currents. Currents are normalized against cell capacitance (pA/pF) to account for neuronal size. Data were included only from neurons with ICa rundown of less than 25% during the entire recording period. The 1000-µl recording chamber was superfused by gravity-driven flow at a rate of 3 ml/min.
Statistical Analysis.
To characterize whole-cell current and voltage dependence of ICa, each cell was fit to the following Boltzmann equations: I = Gmax (V − VR)/1 + exp(V − V1/2/K) for voltage-dependent activation; I/Imax = 1/1 + exp(V − V1/2/K) for steady-state inactivation, where I is the current, Gmax is the maximum conductance, Imax is the maximum current, V1/2 is the voltage at which current is half-maximal, K is a slope factor describing voltage dependence of conductance, VR is the reversal potential for current, and V is the membrane potential. Steady-state activation and inactivation data were normalized to Gmax and Imax, respectively. Data from whole-cell patch-clamp recordings were analyzed with Axograph X (version 1.4.4; Axograph Scientifics, New Zealand). Prism (version 6.1; GraphPad Software, San Diego, CA) was used to perform paired or unpaired Student’s t test. Nonlinear regression was used to fit the dose-response curve to calculate the EC50. Unless specified, data were derived from at least three DRGs for every group. Data are reported as mean ± S.E.M. A P value less than 0.05 was considered significant.
Results
A total of 41 rats was used for the study, of which 26 were control animals and 15 were SNL. The frequency of hyperalgesic responses after noxious punctate mechanical stimulation was 38 ± 5% in SNL rats compared with 0% in Control rats (P < 0.001). Within a single testing session, we saw no pattern of accumulating sensitivity or accommodation to the stimuli. The accuracy of the SNL surgery was confirmed at the time of tissue harvest in all SNL animals. Whereas the SNL model alters pain behavior within a few days (Kim and Chung, 1992; Hogan et al., 2004), we have chosen to focus on the chronic phase (21 days) of neuropathic pain. This time point also allowed us to correlate our findings with previous study that had used that time point (Bangaru et al., 2013).
σ1R Activation Diminishes ICa through VGCC.
We have shown that σ1R is present throughout the DRG, including sensory neurons and satellite glial cells (Bangaru et al., 2013). To explore whether ICa in sensory neurons is modulated by σ1R, we first used a step voltage protocol. In Control DRG neurons, we found that both the PTZ and DTG reduced ICa within a dose-dependent fashion (Fig. 1). The EC50 for ICa blockade was 153.5 ± 0.0 for PTZ and 237.7 ± 0.1 μM DTG, with maximal observed blockade of approximately 74 and 70% (Fig. 1, C and D). The average time to reach peak effect was 109 ± 14 seconds for PTZ and 192 ± 22 seconds for DTG. Both agonists were reversible with 97.6 ± 1.1% recovery of ICa in 85 ± 5 seconds during washout of PTZ, and 94.8 ± 1.6% in 128 ± 15 seconds for DTG. The rapid onset and incomplete inhibition of ICa are consistent with previous reports of σ1R agonist actions in other neuronal tissues (Zhang and Cuevas, 2002; Tchedre et al., 2008). Overall, these results reveal σ1R regulation of ICa in DRG neurons.
Fig. 1.
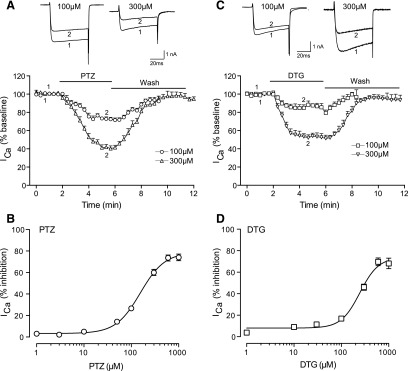
σ1R agonists inhibit voltage-gated calcium current in control sensory neurons. After whole-cell configuration, the ICa was measured with a square wave voltage command (holding potential at −90 mV and step to 0 mV for 100 milliseconds) using Ba2+ as charge carrier. PTZ (A) reduced ICa (sample traces, top panel; averaged time data, bottom panel) in a dose-related fashion (B), as did DTG (C and D). N = 4–10 for each concentration.
For further experiments, we chose to use low- to medium-range doses of PTZ (50 and 100 µM) and DTG (100 µM) to replicate the approximately 30% decrease of ICa in sensory neuron somata after axotomy in the SNL model (McCallum et al., 2006), and to avoid possible nonspecific effects of high-dose exposure to σ1R agonists, such as DTG (Matsumoto et al., 1995). To validate that the observed action of PTZ and DTG on ICa was via activation of the σ1R, we tested whether their effects were sensitive to blockade-selective σ1R antagonists (BD1047 and BD1063) (Matsumoto et al., 1995; Su et al., 2010).
To avoid possible nonselective effects of higher doses of BD1047 and BD1063, a low dose of antagonist, that is, 10 µM (Mueller et al., 2013), was used, which itself has no effect on ICa per se. We observed that DTG modulation of ICa was prevented by prior incubation with BD1063 (10 µM) (Fig. 2A). Although PTZ modulation of ICa was incompletely blocked by BD1063, we confirmed the specificity of PTZ by blockade using a related σ1R antagonist BD1047 (10 µM) (Fig. 2B). Both antagonists had minimal direct effect on ICa (BD1047, 3.8 ± 2.6% inhibition, n = 5; BD1063, 3.2 ± 2.1% inhibition, n = 5). These data indicate that PTZ and DTG modulate ICa through the activation of σ1R.
Fig. 2.

Effect of PTZ and DTG on ICa is mediated by σ1R. After whole-cell configuration, the ICa was measured with a square wave voltage command (holding potential at −100 mV and step to 0 mV for 50 milliseconds). Cells were preincubated with BD1047 or BD1063 for 15 minutes before PTZ and DTG administration. (A) Sample traces from control animal showed that DTG (10 μM) inhibited ICa and the inhibition was prevented by BD1063 (10 μM). (B) Sample traces from control animal showed that PTZ (100 μM) inhibited ICa and the inhibition was prevented by BD1047 (10 μM). Summary data of ICa inhibition were derived from the current density, which was normalized to its baseline current density. Mean ± S.E.M.; number in bars represents the sample size; ***P < 0.001. BL, baseline.
Mechanism of the σ1R Activation on ICa Kinetics.
From the current-voltage relationship, whole-cell ICa diminished significantly after PTZ (50 μM) and DTG (100 μM) bath perfusion (Fig. 3A), with peak current density diminished from −132.1 ± 5.7 at baseline compared with −95.1 ± 6.6 pA/pF (P < 0.05) after PTZ treatment, and from −145.6 ± 18.8 at baseline compared with −101.3 ± 7.8 pA/pF (P < 0.05) after DTG treatment. Boltzmann analysis revealed a reduction of Gmax of approximately 25% by PTZ and approximately 31% by DTG (Fig. 3A). We further characterized their effect on voltage-dependent activation, which showed a hyperpolarized shift of activation (Fig. 3B), in which V1/2 was reduced from −10.8 ± 1.7 mV at baseline to −14.4 ± 1.7 mV after PTZ (P < 0.01) and from −10.9 ± 2.4 mV to −14.7 ± 2.0 mV after DTG compared with P < 0.05. There was no effect of either agent on the Boltzmann slope factor K (baseline 3.52 ± 0.16 versus 3.47 ± 0.29 for PTZ; baseline 2.88 ± 0.26 versus 2.82 ± 0.23 for DTG).
Fig. 3.

σ1R modulates kinetic properties of VGCC in control animals. (A) ICa was measured with a square wave voltage command (200 milliseconds; 10 mV increment from −100 to 50 mV; holding potential at −65 mV). Average inward current density (pA/pF) against current-voltage relationship (I-V) elicited by PTZ (50 μM) and DTG (100 μM) was fit to a single Boltzmann function (see Materials and Methods). (B) Voltage-dependent activation of VGCC was derived from the I-V curve by Boltzmann analysis. (C) The steady-state inactivation of ICa was measured during a 4-second square wave depolarization (−100 to 30 mV in 10-mV increments) with ICa determined by a subsequent test pulse (20 milliseconds to 0 mV). Results are normalized to maximal peak current (I/Imax). (D) A simple step protocol (−100 to 0 mV for 2 seconds) was applied to measure the inactivation kinetics by a two-exponential function (τ1 and τ2). Inset figures revealed summary data of maximal conductance (Gmax; A), voltage at which current is half-maximal (V1/2; B and C), and inactivation constant (τ; D). Mean ± S.E.M.; number in bars represents the sample size; *P < 0.05; **P < 0.01; ***P < 0.001. BL, baseline.
To investigate the effect of σ1R ligands on VGCC kinetics, we further characterized the effects of PTZ and DTG on steady-state inactivation, which showed a shift in a hyperpolarizing direction (V1/2 at baseline of −28.4 ± 2.0 mV versus −38.0 ± 2.8 mV after PTZ, P < 0.01; baseline −26.4 ± 1.5 mV versus −33.7 ± 2.5 mV for DTG, P < 0.01) (Fig. 3C), whereas the Boltzmann slope factor K did not change (baseline −15.09 ± 0.80 versus −14.96 ± 0.47 for PTZ; baseline −15.85 ± 0.99 versus −16.13 ± 1.37 for DTG).
The kinetics of inactivation was next studied during a simple step voltage command and was fit with a two-exponential function (τ1 and τ2). Although no significant change was noted in the initial fast phase (baseline 0.17 ± 0.02 seconds versus 0.15 ± 0.02 seconds after PTZ; baseline 0.14 ± 0.01 seconds versus 0.13 ± 0.01 seconds after DTG), both PTZ and DTG decreased the slow phase constant (τ2) (Fig. 3D).
Together, these findings indicate that σ1R activation in sensory neurons reduces Gmax, reduces the current available at each Vm, and accelerates the rate of ICa inactivation, which will have the effect of decreasing ICa during neuronal function. However, σ1R ligands also result in VGCC activation at less depolarized Vm, which would lead to greater current generation with neuronal firing.
Selectivity of the σ1R Effect on VGCC Subtypes.
Sensory neurons express a range of HVA channel subtypes, including a predominance of N- and L-type, and a smaller representation of P/Q- and R-type currents, as well as low voltage–activated T-type currents (McCallum et al., 2011). To identify the VGCC subtypes that contribute to the changes noted above, we employed step voltage commands and recorded the effects of PTZ (100 μM) on ICa while selectively blocking channel subtypes. In neurons from Control animals, sensitivity to PTZ persisted during blockade of N-type currents (application of GVIA, 200 nM; Fig. 4A) and during blockade of L-type currents (nimodipine, 5 μM; Fig. 4B), demonstrating nonselectivity of σ1R regulation across VGCC subtypes and inhibition of the major HVA current subtypes in sensory neurons (McCallum et al., 2011). T-Type ICa, isolated by blockade of HVA VGCCs and depolarizations to −30 mV, also showed sensitivity to PTZ (Fig. 4, C and D).
Fig. 4.
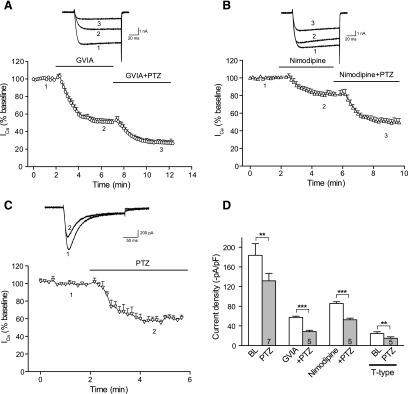
Inhibition of σ1R agonists on ICa encompasses multiple ICa subtypes in control sensory neurons. (A) In whole-cell configuration, ICa was measured with a square wave voltage command (holding potential at −90 mV and step to 0 mV for 150 milliseconds). Sample current traces (top) and averaged data (bottom panel) show response to σ1R activation by PTZ (100 µM) after blockade of N-type channels with ω-conotoxin GVIA (GVIA, 200 nM). (B) Similarly, sensitivity to PTZ is demonstrated after blockade of L-type channels with nimodipine (5 µM). (C) T-type ICa was triggered by depolarizations from a holding potential at −100 mV and step to −30 mV after incubation (20 minutes) with R-type current blocker SNX-482 (200 nM) and P/Q-type current blocker ω-conotoxin MVIIC (200 nM), and addition of L-type current blocker nimodipine (5 µM) and GVIA (200 nM) to the recording bath. These currents also showed sensitivity to PTZ. (D) Summary data. Mean ± S.E.M.; number in bars represents the sample size; **P < 0.01; ***P < 0.001. BL, baseline.
Effect of Sensory Neuron Axotomy on σ1R Regulation of ICa.
σ1R antagonism attenuates behavioral manifestations of pain in experimental traumatic neuropathy (Roh et al., 2008b; Romero et al., 2012). Because painful nerve injury is accompanied by reduction of ICa in axotomized sensory neurons (Hogan et al., 2000; McCallum et al., 2006), which in turn results in elevated sensory neuron excitability (Lirk et al., 2008), our new finding of σ1R regulation of ICa raises the possibility that σ1R activation contributes to pain after nerve injury by elevated sensitivity of injured neurons to σ1R agonists. To test the hypotheses, we determined whether sensitivity of ICa to σ1R agonists is increased in injured neurons. Consistent with our previous findings (Hogan et al., 2000; McCallum et al., 2006), baseline ICa was depressed in axotomized L5 sensory neurons after SNL. Treatment of these neurons with PTZ (50 μM) and DTG (100 μM) further diminished ICa and Gmax (Fig. 5A). Voltage-dependence of activation was shifted in hyperpolarized direction by PTZ, but not by DTG, whereas steady-state inactivation was shifted left for both agonists (Fig. 5, B and C). Slope factor K was unchanged except for a small increase for activation during DTG (baseline 3.14 ± 0.35 versus 3.80 ± 0.41 for DTG; P < 0.05). Kinetics of inactivation showed decreased τ2 after PTZ and DTG administration, as well as decreased τ1 after DTG (Fig. 5D). Thus, the ability of σ1R agonists to modulate ICa persists after neuronal injury. We further analyzed whether the size of these effects of σ1R activation is altered after injury. Although most effects in injured neurons were comparable in scale to control neurons, σ1R activation by DTG produced an enhanced shift of the V1/2 of steady-state inactivation and accelerated the slow phase of ICa inactivation (reduced τ2) after axotomy (Table 1). This heightened action of σ1R would have the effect of reducing ICa further in injured neurons.
Fig. 5.

σ1R modulates kinetics of VGCC in axotomized animals. After whole-cell configuration, the current-voltage relationship (I-V), voltage-dependent activation, and steady-state inactivation of the ICa were measured as described in Fig. 4 using Ba2+ as charge carrier in SNL L5 DRG sensory neurons. (A) Average inward current density (pA/pF) against I-V relationship elicited by PTZ (50 μM) and DTG (100 μM) was fit to a single Boltzmann function (see Materials and Methods). (B) Voltage-dependent activation of VGCC was derived from the I-V curve. (C) In steady-state inactivation, V1/2 was shifted toward a hyperpolarization direction. (D) Sample traces showed that both PTZ and DTG administration decreased slow inactivation constant (τ2). Inset figures revealed summary data of maximal conductance (Gmax; A), voltage at which current is half-maximal (V1/2; B and C), and inactivation constant (τ; D). Mean ± S.E.M.; number in bars represents the sample size; *P < 0.05; **P < 0.01; ***P < 0.001. BL, baseline.
TABLE 1.
Injury effect of σ1R agonists on ICa kinetics
Sample size: n = 6–8 for Gmax, activation and inactivation, n = 10–13 for inactivation kinetics; mean ± S.E.M.
| PTZ |
DTG |
|||
|---|---|---|---|---|
| Control | SNL | Control | SNL | |
| Gmax (change, %) | −25.8 ± 11.3 | −30.7 ± 5.3 | −30.5 ± 4.3 | −31.9 ± 6.9 |
| Activation V1/2 (ΔmV) | −3.6 ± 0.6 | −4.3 ± 1.4 | −3.8 ± 1.2 | −2.2 ± 1.1 |
| Activation slope (Δ%) | −0.3 ± 9.7 | −4.3 ± 14.5 | −4.6 ± 15.6 | −24.7 ± 10.8 |
| S-S inactivation V1/2 (ΔmV) | −9.6 ± 1.6 | −8.6 ± 2.9 | −7.3 ± 1.6 | −11.6 ± 1.7* |
| S-S inactivation slope (Δ%) | 0.2 ± 3.0 | 1.0 ± 4.0 | −0.8 ± 6.4 | 6.1 ± 5.6 |
| Inactivation τ1 (Δs) | −0.02 ± 0.02 | 0.01 ± 0.01 | −0.01 ± 0.01 | −0.03 ± 0.01 |
| Inactivation τ2 (Δs) | −0.92 ± 0.22 | −0.92 ± 0.19 | −0.57 ± 0.10 | −1.09 ± 0.17* |
S-S, steady-state; V1/2, voltage at which current is half-maximal; τ, inactivation kinetic constant; Δ, difference between baseline and agonist treatment.
P < 0.05.
Whereas the σ1R antagonist BD1063 did not affect peak ICa in control and noninjured L4 neurons after SNL (Fig. 6, A and B), we found increased peak ICa in SNL L5 neurons following σ1R antagonism (Fig. 6C). This supports the view that ongoing σ1R activation contributes to suppression of ICa in sensory neurons after injury.
Fig. 6.
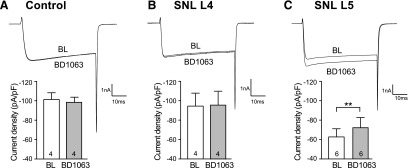
σ1R antagonist, BD1063 (10 μM), increased ICa in axotomized fifth (SNL L5) DRG sensory neurons. After whole-cell configuration, the ICa was measured with a square wave voltage command (holding potential at −100 mV and step to 0 mV for 20 milliseconds) using Ba2+ as charge carrier. Sample current traces (A and B) showed that BD1063 (10 μM) did not affect ICa in control and noninjured neighboring fourth (SNL L4) DRG neurons. (C) BD1063 administration increased the ICa in SNL L5 neurons. Summary data demonstrated that application of BD1063 (10 μM) was able to increase in SNL L5 but not control and SNL L4 neurons. Mean ± S.E.M.; number in bars represents the sample size; **P < 0.01. BL, baseline.
Discussion
Our interest in identifying the functional effects of σ1R activation stems from our recognition of their expression in DRG sensory neurons (Bangaru et al., 2013), combined with behavioral studies demonstrating analgesia of σ1R blockade following nerve injury (Roh et al., 2008b; Romero et al., 2012). Several key findings emerge from our examination of σ1R effects in control and injured sensory neurons. We show that σ1R activation inhibits ICa through actions on multiple VGCC subtypes. Furthermore, nerve injury is accompanied by amplified σ1R suppression of VGCCs, based on the finding that σ1R antagonist augments ICa in SNL L5, but not in control neurons. We have noted prolonged hyperalgesia after SNL (at least 10 weeks; see Kim and Chung, 1992), but have not yet extended our σ1R analysis to this time frame. Together, these observations suggest that σ1R regulates Ca2+ influx during sensory neuron activity and participates in generating neuropathic pain.
In sensory neurons, L- and N-type currents contribute to the majority of ICa (McCallum et al., 2011). A direct interaction between σ1R and L-type VGCC accounts for ICa in hippocampal and retinal ganglion neurons (Sabeti et al., 2007; Tchedre et al., 2008), whereas other observations show σ1R regulation of N- and P/Q-type as well in cortical neurons (Lu et al., 2012). We demonstrate that σ1R inhibits multiple VGCC subtypes in sensory neurons based on the findings that sensitivity of HVA to σ1R agonist remains during nimodipine and GIVA blockade and isolated T-type current can be inhibited by σ1R agonist. Activation of σ1R inhibits VGCCs, but also alters their kinetics. Unlike other VGCC inhibitors (Catterall, 2011), σ1R agonists decrease channel Gmax but also shift the activation and inactivation curves in a hyperpolarized direction, as has also been observed in sympathetic and parasympathetic peripheral neurons (Zhang and Cuevas, 2002). The leftward shift of the voltage dependence of activation would generally result in larger currents during neuronal depolarization, although the size of this shift is small. In contrast, the decreased Gmax, hyperpolarized V1/2 of inactivation, and accelerated ICa inactivation following σ1R activation may combine to substantially depress ICa during neuronal function. Together, it is likely that the action of σ1R on VGCCs results in an overall decrease of Ca2+ influx.
Why high doses (≥100 μM) of σ1R agonists are required on voltage-gated ion channel (Church and Fletcher, 1995; Lupardus et al., 2000; Zhang and Cuevas, 2002) compared with its high receptor affinity (McCann et al., 1994; Matsumoto, 2007; Su et al., 2010) is not clearly understood. This could be due to the influence of the cellular environment, such as σ1R-binding partners, post-translational modifications, isoform variations in σ1R (Shioda et al., 2012), or direct interaction with the VGCC independent of the σ1R high-affinity binding site (Church and Fletcher, 1995; Tchedre et al., 2008). Although nonspecific effects by high-concentration σ1R agonist cannot be ruled out, blockade of σ1R agonist effects on ICa supports our view that agonist action on ICa represents a relevant specific interaction between σ1R and ICa.
The means by which σ1R controls Ca2+ channel function is unknown. Upon activation, σ1R relocates from MAM to plasmalemma (Su et al., 2010). This process requires approximately 10 minutes for initiation of σ1R-dependent secondary signaling (Brent et al., 1997; Hayashi et al., 2000). Our observation of a much quicker onset time (2–3 minutes) for σ1R activity suggests an effect at the level of the plasmalemma directly on VGCCs, as has also been shown in other tissues for VGCCs (Zhang and Cuevas, 2002; Tchedre et al., 2008) and voltage-gated K+ channels (Wilke et al., 1999). There are two possible anatomic explanations for the rapid onset of σ1R agonist effects. Although σ1Rs are enriched in microsomal fractions, σ1Rs are also found in the plasmalemma, even in the absence of the known triggers of translocation (Alonso et al., 2000). Alternatively, σ1R located in MAM may be positioned adequately close to the plasmalemma for rapid ligand activation. In addition to the rapid σ1R action, further evidence for a direct interaction between σ1R and VGCCs is the lack of an effect on the slope of voltage-dependent activation and steady-state inactivation of VGCCs, which would be expected if σ1R acted through signaling pathways, such as G protein and protein phosphorylation (Mavlyutov et al., 2010). Our data thus support a close proximity interaction between the σ1R and VGCCs.
A distinct feature of sensory neurons is that lowered inward ICa has the dominant, overriding effect of decreasing outward current through Ca2+-activated K+ channels, thus reducing afterhyperpolarization and thereby increasing excitability (Malmberg and Yaksh, 1994). Following nerve trauma, ICa is reduced in DRG neurons (Hogan et al., 2000; McCallum et al., 2006), which leads to decreased activation of Ca2+-activated K+ currents. The resulting loss in afterhyperpolarization and reduced membrane input resistance lead to hyperexcitability of primary afferent fibers and enhanced pain (Sapunar et al., 2005; Lirk et al., 2008). The analgesic agents gabapentin and pregabalin, commonly used for neuropathic pain, also inhibit ICa (Hendrich et al., 2008; Patel et al., 2013), resulting in an apparent paradox. However, the roles of ICa are not uniform throughout the sensory neuron. At the dorsal horn (DH), ICa drives neurotransmission (Auer and Ibanez-Tallon, 2010), including that for pain pathways (Chen et al., 2009). Contrasting effects are found elsewhere (impulse generation and propagation), in which repetitive firing is controlled by K+ channels activated by ICa (Berkefeld et al., 2010). Further complexity is introduced into the system because ICa controls neurotransmission in both the excitatory (excitatory amino acid, neuropeptide) and inhibitory (GABA, glycine) pathways in the DH (Bourinet et al., 2014), which in parallel compete for influence on the aggregate output of DH projection neurons. Thus, the final outcome of ICa loss cannot be easily anticipated from current understanding of integrated pain mechanisms.
Can the actions of σ1R on VGCCs account for the loss of ICa following nerve injury? Although expression of σ1R protein is decreased in axotomized sensory neurons (Bangaru et al., 2013), we show that σ1R agonists influence the function of ICa in injured neurons at a level equal to or greater than in control neurons (Table 1). From this, we infer that the per-receptor efficacy of σ1R signaling is amplified following injury, and that the intrinsic activity of σ1R antagonist is increased after axotomy. In addition, the sensitivity of ICa to σ1R blockade after injury (Fig. 6) reveals the possible persisting activity of an endogenous σ1R ligand, whereas control neurons are unresponsive to σ1R blockade. The residual effects of an endogenous σ1R ligand after dissociation on ICa may result from persistence of the ligand bound to the σ1R, but also may be the consequence of persisting effects downstream from σ1R activation, such as elevated channel expression in the membrane or σ1R-triggered channel modification, for example, G protein signaling (Tokuyama et al., 1997; Kim et al., 2010; Brimson et al., 2011). Neurosteroids such as pregnanolone are endogenous σ1R agonists (Maurice, 2004) and are tonic release after injury (Mensah-Nyagan et al., 2008), so these ligands may account for this observation that σ1R activation persists selectively in injured neurons, and may also contribute to sensory neuronal hyperexcitability (Sapunar et al., 2005). Supporting this view, allopregnanolone, a neurosteroid σ1R antagonist (Maurice, 2004), has been shown to have analgesic effect on neuropathic pain (Patte-Mensah et al., 2014). Additional supportive data show that intrathecal σ1R agonists elicit hypersensitivity (Roh et al., 2008a), whereas antagonist and σ1R knockout reduce injury-induced hypersensitivity (Roh et al., 2008b; Romero et al., 2012).
Both systemic and central administration of σ1R antagonists are effective in attenuating nociception after peripheral nerve injury (Roh et al., 2008b; Entrena et al., 2009; Romero et al., 2012; Bura et al., 2013). Although systemically delivered σ1R can be expected to act on VGCCs throughout the body, and therefore have uncertain final consequences on pain due to the heterogeneous effects noted above, selective targeting of σ1R antagonism may allow specific actions. We predict that antagonism of sensory neuron σ1R at peripheral sites (including the DRG) may relieve pain by rescuing ICa. Because of the complexity of ICa signaling in the DH, results may be harder to anticipate.
Together, our observations suggest a mechanism by which the σ1R may mediate neuropathic pain. Elevated endogenous σ1R ligand levels and amplified σ1R signaling combine to suppress ICa in injured sensory neurons. Whereas decreased ICa might be expected to reduce neurotransmitter release in DH pain pathways and therefore result in analgesia, a different result is expected in the periphery, where ICa is required for natural suppression of repetitive firing via opening of Ca2+-activated K+ channels, and for filtering of high-frequency pulse trains at the sensory neuron T-junction in the DRG (Hogan et al., 2008; Lirk et al., 2008). We have identified loss of ICa in sensory neuron somata as a reliable consequence of painful nerve injury (Hogan et al., 2000; McCallum et al., 2006, 2011) that, like σ1R activation, acts through all VGCC subtypes. Thus, σ1R may provide a useful target for analgesia therapy in painful neuropathy.
Abbreviations
- BD1047
N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine dihydrobromide
- BD1063
1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride
- DH
dorsal horn
- DRG
dorsal root ganglion
- DTG
1,3-di-(2-tolyl)guanidine
- Gmax
maximum channel conductance
- HVA
high voltage–activated
- ICa
Ca2+ current
- Imax
maximal current
- L4
fourth lumbar
- L5
fifth lumbar
- MAM
mitochondria-associated endoplasmic reticulum membrane
- PTZ
(+)pentazocine
- σ1R
sigma-1 receptor
- SNL
spinal nerve ligation
- V1/2
voltage at which current is half-maximal
- VGCC
voltage-gated calcium channel
Authorship Contributions
Participated in research design: Pan, Kwok, Hogan, Wu.
Conducted experiments: Pan, Guo, Wu.
Performed data analysis: Pan, Wu.
Wrote or contributed to the writing of the manuscript: Kwok, Hogan, Wu.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant K01-DA024751] (to H.-e.W.); and the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R01-NS42150] (to Q.H.).
References
- Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, Maurice T. (2000) Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience 97:155–170 [DOI] [PubMed] [Google Scholar]
- Auer S, Ibañez-Tallon I. (2010) “The King is dead”: checkmating ion channels with tethered toxins. Toxicon 56:1293–1298 [DOI] [PubMed] [Google Scholar]
- Bangaru ML, Weihrauch D, Tang QB, Zoga V, Hogan Q, Wu HE. (2013) Sigma-1 receptor expression in sensory neurons and the effect of painful peripheral nerve injury. Mol Pain 9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B, Schulte U. (2010) Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 90:1437–1459 [DOI] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. (2014) Calcium-permeable ion channels in pain signaling. Physiol Rev 94:81–140 [DOI] [PubMed] [Google Scholar]
- Brent PJ, Herd L, Saunders H, Sim AT, Dunkley PR. (1997) Protein phosphorylation and calcium uptake into rat forebrain synaptosomes: modulation by the sigma ligand, 1,3-ditolylguanidine. J Neurochem 68:2201–2211 [DOI] [PubMed] [Google Scholar]
- Brimson JM, Brown CA, Safrany ST. (2011) Antagonists show GTP-sensitive high-affinity binding to the sigma-1 receptor. Br J Pharmacol 164:772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bura AS, Guegan T, Zamanillo D, Vela JM, Maldonado R. (2013) Operant self-administration of a sigma ligand improves nociceptive and emotional manifestations of neuropathic pain. Eur J Pain 17:832–843 [DOI] [PubMed] [Google Scholar]
- Catterall WA. (2011) Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3:a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendán CM, Pujalte JM, Portillo-Salido E, Montoliu L, Baeyens JM. (2005) Formalin-induced pain is reduced in sigma(1) receptor knockout mice. Eur J Pharmacol 511:73–74 [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramanyan S, Lai AY, Todd KG, Smith PA. (2009) Effects of sciatic nerve axotomy on excitatory synaptic transmission in rat substantia gelatinosa. J Neurophysiol 102:3203–3215 [DOI] [PubMed] [Google Scholar]
- Chien CC, Pasternak GW. (1994) Selective antagonism of opioid analgesia by a sigma system. J Pharmacol Exp Ther 271:1583–1590 [PubMed] [Google Scholar]
- Chien CC, Pasternak GW. (1995) Sigma antagonists potentiate opioid analgesia in rats. Neurosci Lett 190:137–139 [DOI] [PubMed] [Google Scholar]
- Church J, Fletcher EJ. (1995) Blockade by sigma site ligands of high voltage-activated Ca2+ channels in rat and mouse cultured hippocampal pyramidal neurones. Br J Pharmacol 116:2801–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Puente B, Nadal X, Portillo-Salido E, Sánchez-Arroyos R, Ovalle S, Palacios G, Muro A, Romero L, Entrena JM, Baeyens JM, et al. (2009) Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain 145:294–303 [DOI] [PubMed] [Google Scholar]
- Duncan C, Mueller S, Simon E, Renger JJ, Uebele VN, Hogan QH, Wu HE. (2013) Painful nerve injury decreases sarco-endoplasmic reticulum Ca²⁺-ATPase activity in axotomized sensory neurons. Neuroscience 231:247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entrena JM, Cobos EJ, Nieto FR, Cendán CM, Gris G, Del Pozo E, Zamanillo D, Baeyens JM. (2009) Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain 143:252–261 [DOI] [PubMed] [Google Scholar]
- Fischer G, Pan B, Vilceanu D, Hogan QH, Yu H. (2014) Sustained relief of neuropathic pain by AAV-targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther 21:44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. (2011) Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci 31:3536–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Maurice T, Su TP. (2000) Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J Pharmacol Exp Ther 293:788–798 [PubMed] [Google Scholar]
- Hayashi T, Su TP. (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610 [DOI] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. (2008) Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci USA 105:3628–3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan Q, Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Ljubkovic M, Gemes G, Sapunar D. (2008) Restoration of calcium influx corrects membrane hyperexcitability in injured rat dorsal root ganglion neurons. Anesth Analg 107:1045–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. (2004) Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology 101:476–487 [DOI] [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. (2000) Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain 86:43–53 [DOI] [PubMed] [Google Scholar]
- Kim FJ, Kovalyshyn I, Burgman M, Neilan C, Chien CC, Pasternak GW. (2010) Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol 77:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363 [DOI] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, Bonci A. (2012) The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci 35:762–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Huang CY, Ljubkovic M, Sapunar D, Hogan Q. (2008) Modulators of calcium influx regulate membrane excitability in rat dorsal root ganglion neurons. Anesth Analg 107:673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CW, Lin TY, Wang CC, Wang SJ. (2012) σ-1 Receptor agonist SKF10047 inhibits glutamate release in rat cerebral cortex nerve endings. J Pharmacol Exp Ther 341:532–542 [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Wilke RA, Aydar E, Palmer CP, Chen Y, Ruoho AE, Jackson MB. (2000) Membrane-delimited coupling between sigma receptors and K+ channels in rat neurohypophysial terminals requires neither G-protein nor ATP. J Physiol 526:527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. (1994) Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. J Neurosci 14:4882–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto RR. (2007) Receptors: historical perspective and background, in Sigma Receptors (Matsumoto RR, Bowen WD, Su T-P, eds) pp 1–23, Springer, New York [Google Scholar]
- Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR. (1995) Characterization of two novel sigma receptor ligands: antidystonic effects in rats suggest sigma receptor antagonism. Eur J Pharmacol 280:301–310 [DOI] [PubMed] [Google Scholar]
- Maurice T. (2004) Neurosteroids and sigma1 receptors, biochemical and behavioral relevance. Pharmacopsychiatry 37(Suppl 3):S171–S182 [DOI] [PubMed] [Google Scholar]
- Mavlyutov TA, Epstein ML, Andersen KA, Ziskind-Conhaim L, Ruoho AE. (2010) The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons: an anatomical and behavioral study. Neuroscience 167:247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. (2006) Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology 105:160–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum JB, Wu HE, Tang Q, Kwok WM, Hogan QH. (2011) Subtype-specific reduction of voltage-gated calcium current in medium-sized dorsal root ganglion neurons after painful peripheral nerve injury. Neuroscience 179:244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann DJ, Weissman AD, Su TP. (1994) Sigma-1 and sigma-2 sites in rat brain: comparison of regional, ontogenetic, and subcellular patterns. Synapse 17:182–189 [DOI] [PubMed] [Google Scholar]
- Mei J, Pasternak GW. (2002) Sigma1 receptor modulation of opioid analgesia in the mouse. J Pharmacol Exp Ther 300:1070–1074 [DOI] [PubMed] [Google Scholar]
- Mensah-Nyagan AG, Kibaly C, Schaeffer V, Venard C, Meyer L, Patte-Mensah C. (2008) Endogenous steroid production in the spinal cord and potential involvement in neuropathic pain modulation. J Steroid Biochem Mol Biol 109:286–293 [DOI] [PubMed] [Google Scholar]
- Mueller BH, 2nd, Park Y, Daudt DR, 3rd, Ma HY, Akopova I, Stankowska DL, Clark AF, Yorio T. (2013) Sigma-1 receptor stimulation attenuates calcium influx through activated L-type voltage gated calcium channels in purified retinal ganglion cells. Exp Eye Res 107:21–31 [DOI] [PubMed] [Google Scholar]
- Nieto FR, Cendán CM, Sánchez-Fernández C, Cobos EJ, Entrena JM, Tejada MA, Zamanillo D, Vela JM, Baeyens JM. (2012) Role of sigma-1 receptors in paclitaxel-induced neuropathic pain in mice. J Pain 13:1107–1121 [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, Sather WA. (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto-Rostro M, Margas W, Ferron L, Chaggar K, Crews K, Ramirez JD, Bennett DL, Schwartz A, et al. (2013) α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci 33:16412–16426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG. (2014) Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain. Prog Neurobiol 113:70–78 [DOI] [PubMed] [Google Scholar]
- Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, Han HJ, Beitz AJ, Lee JH. (2008a) Intrathecal administration of sigma-1 receptor agonists facilitates nociception: involvement of a protein kinase C-dependent pathway. J Neurosci Res 86:3644–3654 [DOI] [PubMed] [Google Scholar]
- Roh DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, Han HJ, Beitz AJ, Na HS, Lee JH. (2008b) Intrathecal injection of the sigma(1) receptor antagonist BD1047 blocks both mechanical allodynia and increases in spinal NR1 expression during the induction phase of rodent neuropathic pain. Anesthesiology 109:879–889 [DOI] [PubMed] [Google Scholar]
- Romero L, Zamanillo D, Nadal X, Sánchez-Arroyos R, Rivera-Arconada I, Dordal A, Montero A, Muro A, Bura A, Segalés C, et al. (2012) Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br J Pharmacol 166:2289–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti J, Nelson TE, Purdy RH, Gruol DL. (2007) Steroid pregnenolone sulfate enhances NMDA-receptor-independent long-term potentiation at hippocampal CA1 synapses: role for L-type calcium channels and sigma-receptors. Hippocampus 17:349–369 [DOI] [PubMed] [Google Scholar]
- Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. (2005) Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology 103:360–376 [DOI] [PubMed] [Google Scholar]
- Shioda N, Ishikawa K, Tagashira H, Ishizuka T, Yawo H, Fukunaga K. (2012) Expression of a truncated form of the endoplasmic reticulum chaperone protein, σ1 receptor, promotes mitochondrial energy depletion and apoptosis. J Biol Chem 287:23318–23331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. (2010) The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci 31:557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T. (2008) Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci 49:4993–5002 [DOI] [PubMed] [Google Scholar]
- Tokuyama S, Hirata K, Ide A, Ueda H. (1997) Sigma ligands stimulate GTPase activity in mouse prefrontal membranes: evidence for the existence of metabotropic sigma receptor. Neurosci Lett 233:141–144 [DOI] [PubMed] [Google Scholar]
- Wilke RA, Lupardus PJ, Grandy DK, Rubinstein M, Low MJ, Jackson MB. (1999) K+ channel modulation in rodent neurohypophysial nerve terminals by sigma receptors and not by dopamine receptors. J Physiol 517:391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Gemes G, Zoga V, Kawano T, Hogan QH. (2010) Learned avoidance from noxious mechanical simulation but not threshold semmes weinstein filament stimulation after nerve injury in rats. J Pain 11:280–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. (2008) Distinct inhibition of voltage-activated Ca2+ channels by delta-opioid agonists in dorsal root ganglion neurons devoid of functional T-type Ca2+ currents. Neuroscience 153:1256–1267 [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. (2001) Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cuevas J. (2002) Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol 87:2867–2879 [DOI] [PubMed] [Google Scholar]