

Abstract
Toll-like receptor (TLR) stimulation has been implicated as a major contributor to chronic inflammation. Among these receptors, TLR4 has been described as a key regulator of endogenous inflammation and has been proposed as a therapeutic target. Previously, we discovered by high-throughput screening a group of substituted pyrimido[5,4-b]indoles that activated a nuclear factor-κB reporter in THP-1 human monocytic cells. A biologically active hit compound was resynthesized, and derivatives were prepared to assess structure–activity relationships. The derived compounds activated cells in a TLR4/myeloid differentiation protein 2 (MD2)–dependent and CD14-independent manner, using the myeloid differentiation primary response 88 and Toll/IL-1 receptor domain–containing adapter-inducing interferon-β pathways. Two lead compounds, 1Z105 and 1Z88, were selected for further analysis based on favorable biologic properties and lack of toxicity. In vivo pharmacokinetics indicated that 1Z105 was orally bioavailable, whereas 1Z88 was not. Oral or parenteral doses of 1Z105 and 1Z88 induced undetectable or negligible levels of circulating cytokines and did not induce hepatotoxicity when administered to galactosamine-conditioned mice, indicating good safety profiles. Both compounds were very effective in preventing lethal liver damage in lipopolysaccharide treated galatosamine-conditioned mice. Orally administered 1Z105 and parenteral 1Z88 prevented arthritis in an autoantibody-driven murine model. Hence, these low molecular weight molecules that target TLR4/MD2 were well tolerated and effective in reducing target organ damage in two different mouse models of sterile inflammation.
Introduction
Persistent inflammation has been implicated in the pathogenesis of diverse chronic diseases such as rheumatoid arthritis, atherosclerosis, chronic hepatitis, and diabetes. The Toll-like receptor (TLR) family members are key contributors to these proinflammatory conditions. These pattern recognition receptors respond to molecular patterns in components of bacteria and viruses. In addition to their role in detecting microbial associated molecular patterns, the TLRs can also sense endogenous ligands and have been implicated in perpetuating inflammatory cascades in the absence of invading microbes or other pathogens. Identifying agents capable of safely modulating individual TLR activity is an area of intense investigation.
Among TLRs, TLR4 was discovered as a sensing receptor for bacterial lipopolysaccharide (LPS) (Beutler, 2000). Membrane-bound TLR4 recognizes LPS and signals with enhanced efficiency after forming a receptor complex with accessory proteins including myeloid differentiation protein 2 (MD2), LPS binding protein, and CD14 (Jiang et al., 2005; Kobayashi et al., 2006; Park et al., 2009). Docking the LPS-CD14 complex onto the TLR4/MD2 complex initiates signaling through both the myeloid differentiation primary response 88 (MyD88) and Toll/IL-1 receptor-domain–containing adapter-inducing interferon-β (TRIF) pathways (Kobayashi et al., 2006). MyD88-dependent signaling activates nuclear factor-κB (NF-κB), which leads to the production of proinflammatory cytokines, including IL-6, tumor necrosis factor α (TNFα), and IL-12. Alternatively, TLR4 signaling can use the TRIF pathway, which acts through interferon (IFN) regulatory factor 3, to promote the production of type I IFN (IFN α/β), IFN-inducible gene products, and an immune regulatory response (Yamamoto et al., 2004; Kobayashi et al., 2006).
In light of their capability to activate innate immune responses, TLR ligands have been implicated as potential immune modulating agents. These ligands can be used as immune activators such as vaccine adjuvants and can also be used as inflammatory suppressants when delivered as functional receptor antagonists. Natural or semisynthetic TLR4 ligands, including LPS and monophosphoryl lipid A (MPLA), exhibit adjuvant activity when used with vaccine antigens (Mata-Haro et al., 2007; McAleer and Vella, 2010) that confer protection against viruses. Eritoran (also known as E5564; Eisai Co., Ltd., Tokyo, Japan), a synthetic TLR4 antagonist, did not improve survival in sepsis patients in a phase III clinical trial (Barochia et al., 2011; Opal et al., 2013) but reduced influenza-induced lethality in mice (Shirey et al., 2013). Alternatively TLR ligands administered repeatedly can prevent or attenuate autoimmune disease in various mouse models (Hayashi et al., 2009). However, the clinical development of TLR4 ligands as immunotherapeutic agents has been impeded by their inflammatory side effects, which have been difficult to control using LPS and its semisynthetic derivatives.
A synthetic low molecular weight ligand for TLR4 would be more amenable to structural modification and formulation to attain the desired immunosuppressant, or immunostimulatory profile with limited toxicity, and oral bioavailability could be obtained. Hence, our laboratory conducted a cell-based high-throughput screening (HTS) campaign using a library with over 170,000 compounds to identify molecules that activated the human monocyte cell line, THP-1, stably integrated with a β-lactamase reporter gene under the control of a NF-κB response element. We identified a scaffold (here designated 1Z65) that was biologically active and targeted the TLR4/MD2 complex in both mouse and human cells. For structure–activity relationship (SAR) analysis, we prepared 42 derivatives and evaluated their receptor specificities and immunostimulatory profiles. Promising candidates, based on their chemical properties and immunologic profiles, were tested for bioavailability, safety, and efficacy in mouse models of sterile liver and joint inflammation.
Material and Methods
Animals.
Seven- to nine-week-old C57BL/6 (wild-type; WT) and Cd14−/−(C57BL/6 background) mice were purchased from the The Jackson Laboratories (Bar Harbor, ME). TrifLps2/Lps2 mice were kindly provided by Dr. Bruce Beutler (University of Texas Southwestern Medical Center, Dallas, TX). Tlr4−/− and Myd88−/− mice were a gift from Dr. Shizuo Akira (Osaka University, Osaka, Japan). These strains were backcrossed for 10 generations onto the C57BL/6 background at the University of California, San Diego (La Jolla, CA). Md2−/− mice (C57BL/6 background) were originally generated by Dr. K. Miyake (University of Tokyo, Tokyo, Japan) and were kindly provided by Dr. Lynn Hajjar (University of Washington, Seattle, WA) (Hajjar et al., 2012). KRN T-cell receptor transgenic mice were a gift from Drs. D. Mathis and C. Benoist (Harvard Medical School, Boston, MA) (Kouskoff et al., 1996; Mangialaio et al., 1999). All animal experiments were approved by the University of California, San Diego, Institutional Animal Care and Use Committee.
Reagents.
Compounds 247543 and 247514 were purchased from Life Chemicals (Burlington, ON, Canada). 1Z65 and derivatives were synthesized in our laboratory (Chan et al., 2013) as previously described and dissolved in dimethylsulfoxide as a 20–100 mM stock solution and kept in −20°C until use. Endotoxin levels of these drugs were determined by Endosafe (Charles River Laboratory, Wilmington, MA) and were less than 10 EU/µmol. The stock solutions and vehicle were diluted in the medium or normal saline with a final dimethylsulfoxide concentration of 0.5% (in vitro) or 10% (in vivo), which was also used as the vehicle control. Lipopolysaccharide (LPS; Escherichia coli 026;B6) and LPS-RS (LPS from Rhodococcus sphaeroides) were purchased from Sigma-Aldrich [St. Louis, MO]) and Invivogen (San Diego, CA), respectively. RPMI 1640 medium (Invitrogen, Carlsbad, CA) and Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) were supplemented with 10% fetal bovine serum (Omega Scientific, Inc. Tarzana, CA) and penicillin/streptomycin (Life Technologies, Grand Island, NY)
In Vitro Cytokine Induction in Bone Marrow–Derived Dendritic Cells, Bone Marrow–Derived Macrophages.
Bone marrow–derived dendritic cells (BMDC) and bone marrow–derived macrophages (BMDM) were prepared from C57BL/6 mice as described (Wu et al., 2007). BMDC (105 cells per well) or BMDM were plated in 96-well plates in 200 μl of complete RPMI 1640. The cells were incubated with graded concentrations of the compounds for 18 hour at 37°C, 5% CO2. After 18 hour incubation, the cell culture supernatants were collected. LPS (purified LPS, 10 ng/ml; Invivogen) or MPLA (1 μg/ml synthetic MPLA; Invivogen), Pam3Cys [(S)-(2,3-bis (palmitoyloxy)-(2RS)-propyl)-N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys4-OH, trihydrochloride; Invivogen], 1V136 (a TLR7 ligand), and oligonucleotide (ODN) 1826 (Invivogen) were used as stimulants. Levels of IL-6 and IFNγ-induced protein 10 (IP-10) in the culture supernatants were determined by enzyme-linked immunosorbent assay (BD Biosciences, La Jolla, CA) (Chan et al., 2009). The levels of type I IFN in the culture supernatants were determined by bioassay using L929 cells stably expressing an IFN-sensitive response element (ISRE) luciferase reporter provided by Dr. B. Beutler as previously described (Crozat et al., 2006). In brief, L929-ISRE cells were plated at 5 × 104 cells per well in complete DMEM medium in a 96-well white-walled clear-bottom plate. The L929-ISRE cells were then incubated in 50 μl of DMEM and 50 μl of mBMDC supernatant for 6 hours. The luciferase activities were measured using Steady-Glo or Bright Glo Luciferase Assay System (Promega, Madison, WI), employing mu-IFN Beta Standard (PBL Interferon Source, Piscataway, NJ) to generate a standard curve. To calculate the LogEC50 or the IC50, Y = 100/(1 + 10^[(LogEC50 − X)*HillSlope]) was used with Prism 6 (GraphPad Software, San Diego, CA).
In Vitro Assay Using Mouse TLR4 Reporter Cell Line.
Murine (m) TLR4 HEK Bluecells (2.5 × 104 cells per well of a 96-well plate; Invivogen) were incubated with 1Z105 in the presence and absence of LPS or LPS-RS. The culture supernatants were harvested after a 20- to 24-hour incubation period. Secreted alkaline phosphatase activity in the supernatants was determined by a colorimetric assay, using QuantiBlue (Invivogen) with absorbance read at 630 nm.
Cell Toxicity Assay.
Briefly, HepG2 cells were dispersed in 96-well plates (1 × 104/well) and treated with graded concentrations of each compound overnight. After 24 hours of drug treatment, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-dipheyl tetrazolium bromide)-based solution (0.5 mg/ml) was added to each well. Six or more hours later, the cells were lysed, and absorbance values at 570 and 650 nm were measured.
In Vivo Studies.
C57BL/6 mice were treated intraperitoneally with varying doses of compound or MPLA (from Salmonella minnesota; Sigma-Aldrich). Twenty-four hours after the treatment, mice were challenged with LPS (0.2 μg/animal from E. coli 026:B6; Sigma-Aldrich) and galactosamine (12 mg/animal; Sigma-Aldrich). To test the compounds for liver toxicity, varying doses of compound or MPLA were mixed with galactosamine (12 mg/animal; Sigma-Aldrich) and administered intraperitoneally to mice. Sera were collected 1.5 hours after LPS/galactosamine or compound/galactosamine challenge. In other experiments, treated mice were challenged with 20 μg i.p. LPS, and sera were collected 1.5 hours after injection.
Arthritic adult K/BxN mice were bled and their sera were pooled. To induce arthritis, recipient mice were injected with 150 μl i.p. of K/BxN serum, as described previously (Corr and Crain, 2002). Mice were treated every other day with vehicle intraperitoneally, 1Z88 (500 nmol), or 1Z105 (5 μmol) by gavage. Clinical and histologic arthritis scores were evaluated (Guma et al., 2010).
Pharmacokinetic Studies.
Mice were administered 1Z105 or 1Z88 intravenously (0.5 μmol) or p.o. by gavage (1 μmol). Sera were collected at 5, 30, 60, 90, 120, and 240 minutes after administration, centrifuged (14,000g for 10 minutes), and the resulting supernatant was mixed with an internal standard. After dilution with twice the volume of methanol, the mixtures were centrifuged (14,000g for 10 minutes) to remove precipitates and lyophilized. The dried samples were then reconstituted with a small volume of methanol and injected into an Agilent 1100 LC/MSD (Agilent Technologies, Santa Clara, CA) with a Supelco Discovery HS C18 column (5 µm, 50 × 2.1 mm i.d.) (Sigma-Aldrich). The samples were eluted with a linear gradient from 10:90 acetonitrile:water to 90:10 acetonitrile:water with 0.1% triflouroacetic acid at a flow rate of 0.4 ml/min for 8 minutes and monitored by tandem mass spectrometry. Compound concentrations in serum were calculated from peak area and extrapolated from prepared standards in serum with a lower limit of 10 nM. Area under curve values were calculated using Prism 6.0 and used to determine oral bioavailability (Kerns and Di, 2008).
Statistical Analysis.
The data are represented as mean ± S.E.M. Two-tailed Student’s t test or Mann–Whitney U test was used to compare two groups, and one-way analysis of variance with Dunnett’s post hoc test was used to compare multiple groups with the control. In the arthritis model, significance was determined by the Kruskal–Wallis method for area under the curve values for data points. Prism 6 statistical software was used to obtain P values for comparisons between groups (P<0.05 was considered significant).
Results
Cytokine Induction by Derivatives Is TLR4/MD2 Signaling–Dependent and CD14-Independent.
Through a cell-based HTS campaign we previously identified compounds that shared a drug-like pyrimido[5,4-b]indole scaffold (here designated 1Z65, 247543, and 247514), which targeted TLR4/MD2. We then synthesized 42 derivatives of a hit compound (1Z65) and performed a SAR using BMDC and a TLR4-NF-κB reporter cell line (Chan et al., 2013). The derivatives that retained the ability to stimulate NF-κB in the reporter cell line were synthesized in sufficient quantity for target confirmation and functional analysis (Table 1). First we examined the components of the TLR4 receptor complex as the molecular target(s) of the synthesized analogs.
TABLE 1.
Compound properties
| Group | Compound ID | Sructure | Molecular Mass | logP | PSA | H-Bond Donors | H-Bond Acceptors |
|---|---|---|---|---|---|---|---|
| g/mol | Å | n | |||||
| 1 | 1Z65 | 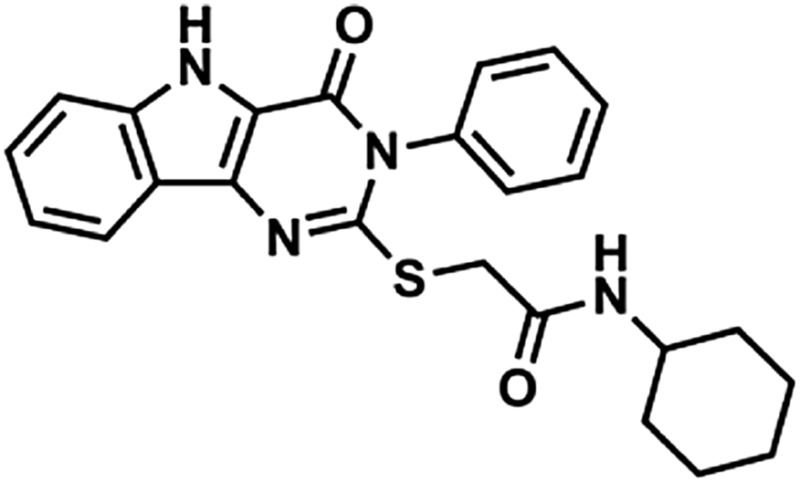 |
433 | 4.02 | 73.8 | 2 | 6 |
| 1 | 1Z103 | 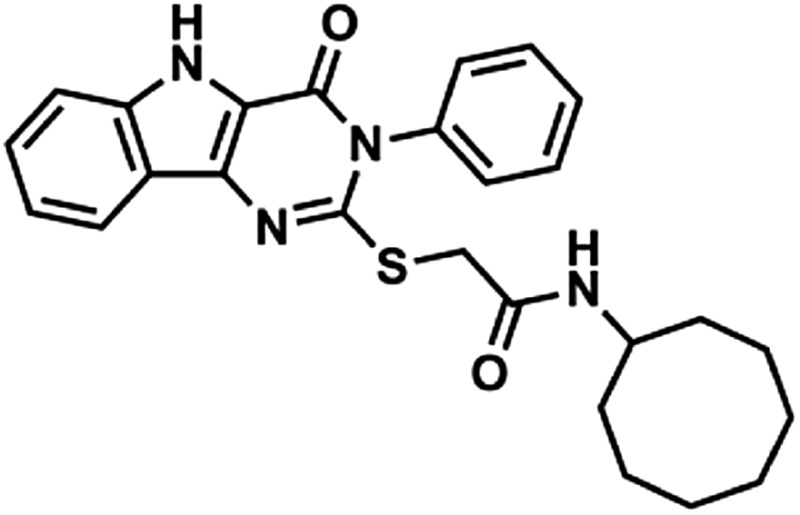 |
461 | 4.86 | 73.8 | 2 | 6 |
| 1 | 1Z82 | 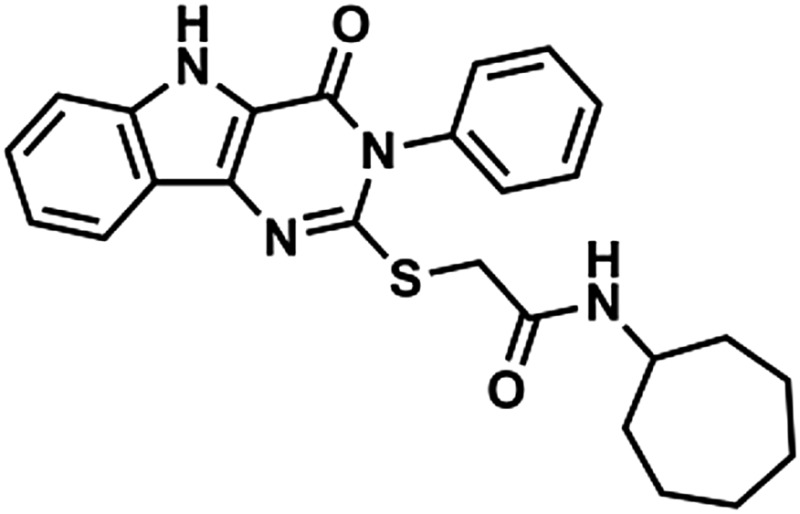 |
447 | 4.44 | 73.8 | 2 | 6 |
| 1 | 1Z105 | 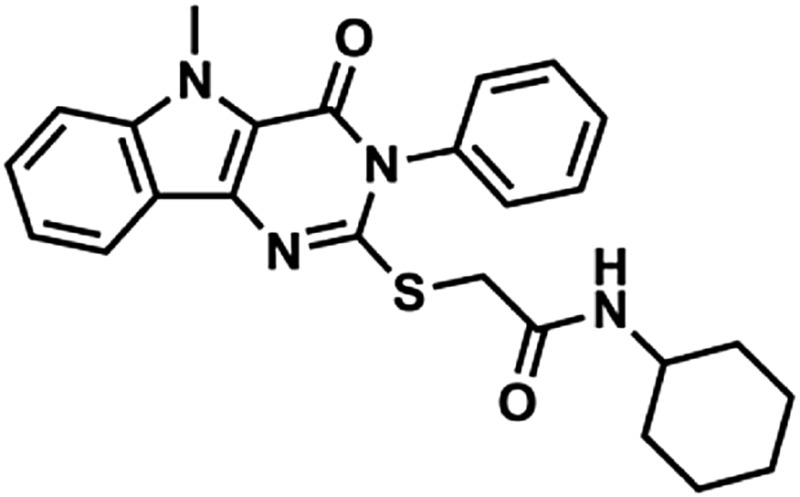 |
447 | 4.26 | 65.01 | 1 | 6 |
| 1 | 1Y82 | 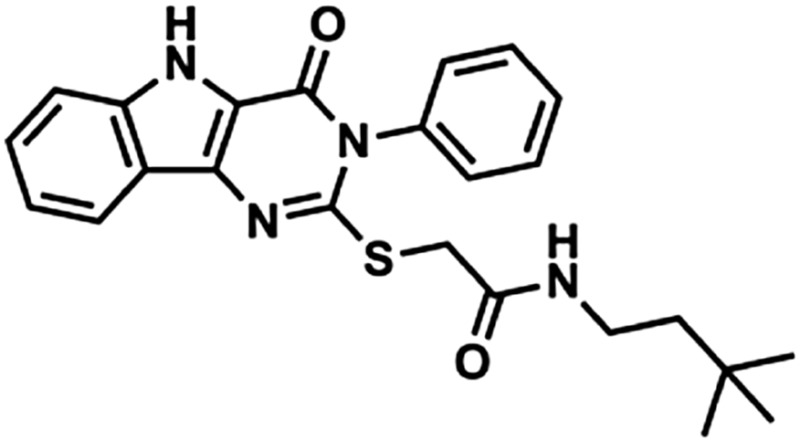 |
435 | 4.52 | 73.8 | 2 | 6 |
| 2 | 1Z88 | 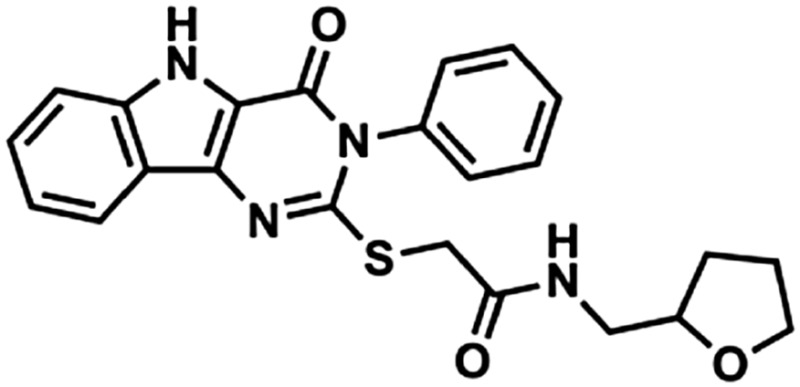 |
435 | 2.61 | 83.03 | 2 | 7 |
| 2 | 1Z104 | 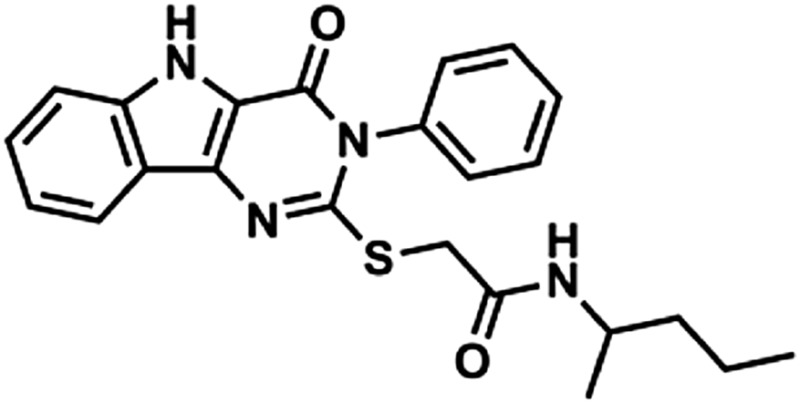 |
421 | 4.03 | 73.8 | 2 | 6 |
| 2 | 247514 | 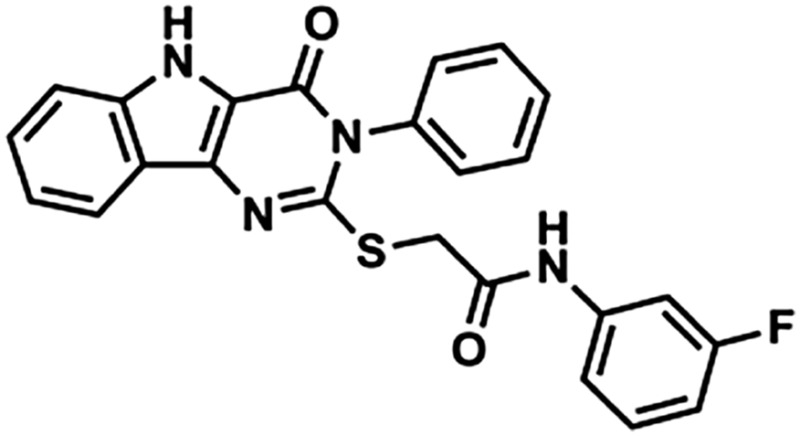 |
444 | 4.30 | 73.8 | 2 | 6 |
| 2 | 247543 |  |
440 | 4.62 | 73.8 | 2 | 6 |
Given that the two TLR4 signaling pathways, through MyD88 and TRIF, result in IL-6 or type I IFN secretion, respectively, we focused on the induction of IL-6 and IP-10, a surrogate marker of type I IFN production, by the compound. The ability to stimulate type I IFN was directly assayed vide infra and correlated with IP-10 (Fig. 2; Supplemental Fig. 1). The cytokine production profiles of WT stimulated BMDCs clustered into two distinct groups (Fig. 1, A and B). 1Z105, 1Z103, 1Z82, 1Y82, and the resynthesized hit compound 1Z65 induced high levels of IL-6, IP-10, and type 1 IFN (group 1), whereas 1Z88, 1Z104, and two compounds from the original library (247543 and 247524) induced only low levels of IL-6, but retained the ability to induce moderate levels of IP-10 and type I IFN (group 2).
Fig. 2.

Dose dependent effects on cytokine production and cell proliferation. (A) WT C57BL/6 BMDC were plated at 1 ×105 cells/well and were incubated with graded concentrations of each compound overnight. IL-6 levels were measured by enzyme-linked immunosorbent assay and type I IFN levels were measured by the reporter assay. Group 1 compounds (1Z65, 1Z82, 1Z103, 1Z105, and 1Y82) induced relatively high levels of both IL-6 and type 1 IFN. Group 2 compounds (1Z88, 1Z104, 247543, and 247524) induced low levels of IL-6 compared with type 1 IFN. (B) HepG2 cells in 96-well plates were exposed each compound for 24 hours, and viable cell numbers were measured by a tetrazolium (MTT) reduction assay. The Å570–650 was 1.14 ± 0.01 for the vehicle control-treated wells. Shown are the mean percentage Å570–650 values compared with the control for cells exposed to 10 µM ± S.E.M. of triplicates and are representative of two independent experiments.
Fig. 1.

Requirement for TLR4 and MD2, but not CD14, for cytokine production. BMDC prepared from WT (A and B), Tlr4−/−(C and D), Md2−/− (E and F), Cd14−/− (G and H), Myd88−/− (I and J), and TrifLps2/Lps2 (K and L) mice and were incubated with 5 μM of each compound overnight. Control wells were treated with vehicle (veh), LPS (10 ng/ml), MPLA (1 µg/ml), or a TLR7 ligand 1V136 (1 µM). IL-6 and IP-10 in the culture supernatants were determined by enzyme-linked immunosorbent assay. Data shown are means ± S.E.M. and are representative of two independent experiments.
Although TLR4 transmits an intracellular signal, there is a variable requirement by individual ligands for the MD2 and CD14 accessory molecules to fully engage an activation signal (Lu et al., 2008). MD2 is required for activation of NF-κB by LPS or MPLA (Kim et al., 2007; Ohto et al., 2007; Casella and Mitchell, 2013). CD14 is expressed on the surfaces of macrophages, dendritic cells, and neutrophils as a glycosylphosphatidylinositol-anchored membrane protein that cannot transmit a signal itself without a coreceptor. CD14 is also present in serum as a soluble isoform, which can also play a role in LPS recognition by TLR4/MD2 by recruiting ligands into the complex (Ulevitch and Tobias, 1995).
Hence, we tested the requirements for TLR4, MD2, and CD14 in stimulating cytokine secretion by the SAR derivatives using BMDC from TLR4, MD2, or CD14 null mice, compared with WT mice. The induction of IL-6 and IP-10 by all SAR derivatives was abrogated in TLR4 and MD2 null BMDCs (Fig. 1, C–F). However, the SAR derivatives induced similar levels of IL-6 and IP-10 from the CD14 null BMDCs compared with WT BMDCs (Fig. 1, G and H), whereas LPS was not able to stimulate CD14 null cells. In control experiments, a TLR7 ligand, 1V136, was able to stimulate TLR4, MD2, and CD14 null BMDCs to produce both cytokines at levels similar to WT cells.
TLR4/MD2 uses both the MyD88 and the TRIF signaling pathways. To test whether the compounds were selective for one of these pathways, we stimulated BMDCs that were deficient in the respective adaptor molecules. None of the compounds elicited IL-6 in MyD88 null cells (Fig. 1I); however group 1 compounds were able to stimulate IP-10 production in these cells (Fig. 1J). In the TRIF mutant cells there was little if any cytokine production by the compounds (Fig. 1, K and L). These data suggest that group 1 compounds require MyD88 for IL-6 production and TRIF for IP-10 production, and group 2 compounds are dependent on the presence of both adaptors to induce cytokine production.
Lead Candidate Selection: 1Z105 and 1Z88.
Since there were two distinct immunologic profiles observed when cells were tested with a single dose of SAR compounds, we examined whether these profiles were dose dependent and evaluated type 1 IFN production directly. BMDC were treated with graded doses of the compounds (Fig. 2A; Supplemental Fig. 1), and IL-6 levels and type I IFN levels were measured in the supernatants. The cytokine production profiles again clustered into two distinct groups (Fig. 2A). 1Z105, 1Z103, 1Z82, 1Y82, and the resynthesized hit compound 1Z65 induced high levels of both IL-6 and type 1 IFN (group 1), whereas 1Z88, 1Z104, and two compounds from the original library (247543 and 247524) induced low levels of IL-6 but retained the ability to induce moderate levels of type I IFN (group 2). The group 1 derivatives stimulated robust levels of IL-6 with EC50 values of 0.16 and 0.27 µM, respectively, for 1Y82 and 1Z105. Of the group 1 derivatives, 1Y82 and 1Z105 were the most potent in inducing type I IFN with EC50 values of 1.4 and 1.9 µM, respectively. Group 2 compounds stimulated little if any IL-6 but retained the ability to stimulate type I IFN to reach a level of approximately 60% of the group 1 compounds. The EC50 values for stimulating type I interferon were 4.9 and 5.5 µM for 1Z88 and 1Z104, respectively. Control wells included LPS (10 ng/ml) or MPLA (1 µg/ml), which stimulated IL-6 (17.3 ± 2.4 and 3.8 ± 0.2 ng/ml, respectively). The type 1 IFN level induced by MPLA (1 µg/ml) was 53 ± 1.5 arbitrary units.
To further support the selection of lead candidate compounds, the cytotoxicities of these compounds were evaluated by MTT assay using HepG2 cells (Supplemental Fig. 1, G and H). The viability of cells incubated with the group 1 compounds: 1Z65, 1Z82, 1Z103, 1Z105, and 1Y82 at 10 μM were 38, 59, 93, 99, and 93%, respectively, compared with the vehicle control (Fig. 2B). Among the compounds in group 2, 1Z104 was slightly cytotoxic at 10 μM (89% of control viability), whereas the other compounds (1Z88, 247543, and 247514) did not show cytotoxicity in HepG2 cells (98–100%) (Fig. 2B). Hence 1Z65, 1Z103, 1Z82, 1Y82, and 1Z104 exhibited cytoxicity in this assay and were thus excluded from in vivo studies.
In the hit-to-lead phase of drug discovery, it is important to consider structural properties including molecular weight, lipophilicity (as measured by logP), polar surface area (PSA), and number of hydrogen bonds (both donors and acceptors) (Kerns and Di, 2008). Retrospective computational studies of successful lead compounds have suggested the following criteria for inclusion in the lead selection process: molecular weight < 500, logP < 4.2, PSA < 100, H-bond donors < 5, H-bond acceptors < 9 (Kerns and Di, 2008). With these criteria in mind, we compared the structural properties of the active compounds in groups 1 and 2 and observed that 1Z105 and 1Z88 were among the best candidates in their respective groups (Table 1). Whereas essentially all of the compounds met the requirements of PSA < 100 and molecular weight < 460, 1Z105 had fewer H-bonds and was less toxic than other members in its group, and 1Z88 had a lower logP than other members in its group.
On the basis of chemical properties, relative potency and lack of cytotoxicity, 1Z105 (group 1) and 1Z88 (group 2) were proposed as candidates for further study. The potential for toxicity of these compounds was retested on primary BMDC using MTT. The viability for BMDC treated with 10 µM 1Z65, 1Z105, and 1Z88 were 47, 92, and 122%, respectively (Supplemental Fig. 2).
Exposure to 1Z88 and 1Z105 Desensitizes Cells to Subsequent TLR Activation.
As the lead compounds targeted the TLR4/MD2-MyD88 pathway, we tested their ability to interfere with subsequent TLR stimulation. Prior exposure of innate immune cells like monocytes/macrophages to LPS causes them to become refractory or hyposensitive to subsequent LPS challenge, a phenomenon called “LPS (endotoxin) tolerance.” Pretreatment with a low dose of LPS can allow sustained SHIP phosphorylation and increase the expression of the IL-1 receptor–associated kinase (IRAK) decoy-IRAK isform M (IRAKM). These two mechanisms then limit the ability of TLR ligands to transmit a signal through the MyD88-NF-κB pathway (Medvedev et al., 2006; Bao and Liu, 2013).
We thus assessed whether 1Z105 and 1Z88 could functionally inhibit TLR4 activation at low drug concentrations. BMDM initially exposed to varying concentrations of 1Z105 or 1Z88 were restimulated with LPS and the IL-6 release (Fig. 3A) or IP-10 (Supplemental Fig. 3) into the culture supernatant was measured. Pretreatment with as low as 0.63 µM 1Z105 significantly reduced the IL-6 secretion by subsequent challenge with LPS (Fig. 3A). A similar inhibition was observed when 1Z88 was used, although 1Z88 required higher doses to desensitize BMDM to rechallenge. However, simultaneous delivery of LPS and 1Z105 or 1Z88 did not diminish NF-κB activation in a reporter cell line (Supplemental Fig. 4).
Fig. 3.
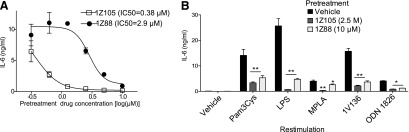
1Z105 and 1Z88 desensitize cells to TLR ligand stimulation. (A) Desensitization to LPS challenge was tested by pretreating C57BL/6 BMDM plated at 5 × 104 cells/well with vehicle or graded concentrations of 1Z105 or 1Z88 overnight. The cells were then challenged with LPS (10 ng/ml), and cytokine release in the culture supernatants after 18 hours was assessed by enzyme-linked immunosorbent assay (ELISA). Data shown are mean ± S.E.M. (B) 1Z105 and 1Z88 inhibits activity of other TLRs. BMDM were pretreated with vehicle, 2.5 μM 1Z105, or 10 μM 1Z88 overnight. The cells were then challenged with the TLR activators: Pam3Cys (2.5 µg/ml, TLR2), pure LPS (10 ng/ml, TLR4), MPLA (1 µg/ml, TLR4), 1V136 (1 µM, TLR7), and ODN1826 (0.1 µM, TLR9) overnight, and IL-6 levels in the culture supernatants were determined by ELISA. Data shown are mean ± S.E.M. (*P < 0.05; **P < 0.001 by analysis of variance and Dunnett's post hoc test).
Considering that 1Z105 and 1Z88 were able to desensitize cells to a TLR4 rechallenge, we also tested for the “cross tolerance” phenomenon in which pretreatment with a TLR ligand can limit signal transmission through MyD88- NF-κB by other TLRs. We chose doses of 1Z105 and 1Z88 that were able to fully suppress IL-6 (Fig. 3A) and IP10 (Supplemental Fig. 3) production by LPS stimulated BMDC. Hence, BMDCs were pretreated with vehicle, 2.5 μM 1Z105 or 10 μM 1Z88 overnight. These cells were then challenged with ligands to TLR4 and other TLRs that converge on the MyD88 signaling pathway. 1Z105 and 1Z88 pretreatment reduced the level of IL-6 released after challenge with Pam3Cys (TLR2), pure LPS (TLR4), MPLA (TLR4), 1V136 (TLR7), and ODN1826 (TLR9), indicating that these compounds not only reduce subsequent TLR4 stimulation, but also attenuate signaling by other MyD88-dependent TLRs (Fig. 3B).
1Z105 Is Orally Bioavailable, Nontoxic, and Retains TLR Tolerizing Activity.
We further evaluated the pharmacokinetics of 1Z105 and 1Z88. The serum concentrations of 1Z105 and 1Z88 after intravenous administration peaked right after injection (5 minutes) and declined within 60 minutes (Fig. 4, A and B). Compound 1Z105, but not 1Z88, was also detectable in the serum after oral administration. The peak concentration was observed at 60 minutes (Fig. 4A). The area under the curve after intravenous and oral administration was 374 ± 111 and 84 ± 7 (mean ± S.E.M.). The calculated oral bioavailability of 1Z105 was 11%. After oral administration, the concentration of 1Z105 in sera increased in a dose-dependent manner and the maximal sera concentration was detected at a 5 μmol dose (Fig. 4C).
Fig. 4.
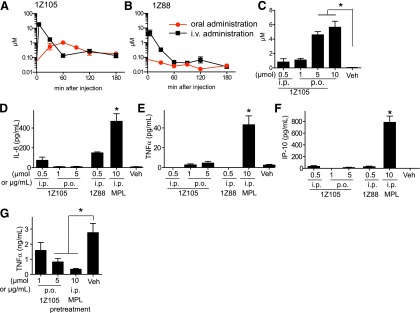
Pharmacokinetics of 1Z105 and 1Z88 after intravenous and oral administration. Groups of C57BL/6 mice (n = 3) were injected intravenously with 0.5 μmol 1Z105 (A) or 0.5 µmol 1Z88 (B) in 10% dimethylsulfoxide (DMSO) or gavaged with 1 μmol 1Z105 (A) or 1Z88 (B) in 10% DMSO. Sera were collected 5, 30, 60, 120, and 180 minutes postadministration. Compound concentrations in serum were measured by liquid chromatography–tandem mass spectrometry and were calculated from peak area and extrapolated from internal standards. (C) Groups of C57BL/6 mice (n = 3) were gavaged with the indicated doses of 1Z105 and bled after 1 hour. Sera concentrations of 1Z105 were determined as above. *P < 0.05 by one way analysis of variance (ANOVA) with Dunnett’s post hoc testing. (D–F) C57BL/6 mice were treated with an intraperitoneal injection of vehicle, 10 µg MPLA, 0.5 µmol 1Z105, or 0.5 µmol 1Z88, or by gavage with 1 or 5 µmol 1Z105. The sera were collected 2 hours after drug administration and tested by enzyme-linked immunosorbent assay for IL-6 (D), TNFα (Ε), and IP-10 (F) were measured by Luminex multiplex bead assay. *P < 0.05 by one-way ANOVA with Dunnett’s post hoc testing compared with vehicle. (G) C57BL/6 mice (n = 3/group) were gavaged with vehicle or 1 or 5 μmol 1Z105 or injected intraperitoneally with 10 µg MPLA. Twenty-four hours later, mice were injected intraperitoneally with 20 μg LPS. TNFα levels in the sera 2 hours after injection were determined by Luminex multiplex assay. Data shown are mean ± S.E.M. of pooled two independent experiments. *P < 0.05 by one way ANOVA with Dunnett’s post hoc testing.
The clinical development of TLR active agents as therapeutics has been hindered by their inflammatory side effects due to release of TNF and other cytokines. Systemic cytokine induction narrows the therapeutic window needed to achieve an effective treatment dose (Engelich et al., 2001). To study whether 1Z105 and 1Z88 stimulated in vivo proinflammatory cytokine release at the doses needed to suppress LPS stimulation, 1Z105 and 1Z88 were given intraperitoneally, and 1Z105 was also tested by oral administration. In prior studies peak levels of cytokine release in the sera of mice occurred about 1 to 2 hours after intraperitoneal injection. Sera at 2 hours after injection had very low or undetectable levels of TNFα, IL-6, or IP-10 (Fig. 4, D–F) after administration of both agents. In addition, no behavioral changes were observed in the treated animals.
These results showed that in vivo administration of low doses of 1Z88 or 1Z105 did not result in systemic proinflammatory cytokine release. However, the results could also indicate a lack of activity in vivo. To further assess the in vivo effects of oral 1Z105 we again used the TLR tolerance model. In this model pretreatment with a TLR4 ligand can attenuate subsequent challenge by LPS as demonstrated in vitro in Fig. 3, A and B. Here WT mice were orally pretreated with 1Z105 (1 and 5 μmol) and then were intraperitoneally injected with LPS. The oral administration of 5 μmol 1Z105 significantly reduced release of TNF by subsequent challenge with LPS (Fig. 4G).
Beneficial Effects of 1Z105 and 1Z88 on Galactosamine-Induced Liver Toxicity.
The lack of a proinflammatory response to systemic 1Z105 or 1Z88 administration suggested that these compounds might be useful in attenuating inflammation. To test this possibility, the endotoxin-induced liver injury model was used (Fig. 5, A and B). In this model, mice that are primed with galactosamine succumb to acute hepatic inflammation and failure after injection with LPS, a TLR4 ligand. To assess if the TLR4/MD2 ligands could prevent toxicity in this model, WT mice were intraperitoneally treated with 1Z105, 1Z88, or MPLA and challenged with LPS plus galactosamine. More than 80% of mice treated with 1Z105, 1Z88, and MPLA survived, whereas only 22% of vehicle-treated mice survived 24 hours after LPS/galactosamine challenge (Fig. 5A). This finding was mirrored by the levels of TNF release in the sera (Fig. 5B).
Fig. 5.

Lack of systemic cytokine induction attenuates mortality from galactosamine hepatotoxicity. (A) Pretreatment reduces LPS/galactosamine induced mortality. Groups of C57BL/6 mice were pretreated intraperitoneally with vehicle, 10 µg MPLA, 0.5 µmol 1Z105, or 0.5 µmol 1Z88. Twenty-four hours later, mice were injected intraperitoneally with LPS (0.2 μg/mouse) and galactosamine (12 mg/mouse). Survival was monitored over the next 24 hours. (B) Levels of TNFα in sera 2 hours after LPS injection were measured by enzyme-linked immunosorbent assay. Data shown are mean ± S.E.M. of two independent experiments. *P < 0.05 by one-way analysis of variance with Dunnett’s post hoc testing. (C) C57BL/6 mice were administered with indicated doses of 1Z105, LPS, or MPLA mixed with galactosamine (1 mg per animal). The mortality was monitored for 24 hours. DMSO, dimethylsulfoxide.
1Z105 and 1Z88 were shown to be safe by lack of nonspecific immune activation (Fig. 4, D and F). In a model of chemically induced liver failure, galactosamine is coadministered with LPS, and TLR4 ligation is required for lethality. 1Z105 was tested as a surrogate for LPS in this model. When 1Z105 was delivered with galactosamine at doses that were protective, no mortality of mice after administration with galactosamine was detected (Fig. 5C).
1Z105 and 1Z88 Prevent Autoantibody-Induced Arthritis.
In rheumatoid arthritis, the production of cytokines and chemokines has been partially attributed to innate immune mechanisms. TLR4 has been identified as a prominent candidate, because several endogenous synovial ligands bind to it and may thereby promote and sustain joint inflammation (Ditzel, 2004). Hence, we tested intraperitoneal treatment with 1Z88 or oral treatment with 1Z105 by gavage in a model of arthritis, induced by transfer of K/BxN sera, that is autoantibody-mediated and dependent on the innate immune system. Mice that were treated with the drugs had significantly reduced paw swelling (Fig. 6, A and B). The 1Z105 treatment significantly decreased histologic inflammation, bone erosion, and cartilage damage (Fig. 6, C–I).
Fig. 6.
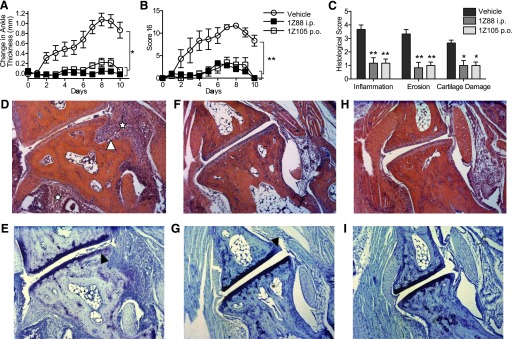
1Z88 and 1Z105 prevent the onset of innate inflammatory arthritis. Groups (n = 6) of mice were injected with pooled arthritogenic KRN sera intraperitoneally. Mice received every other day treatment with vehicle, 0.50 µmol i.p. 1Z88. An additional group received 5 µmol 1Z105 by gavage. Paw swelling (A) was measured and the arthritis was clinically (B) scored daily. Data shown are mean ± S.E.M. of pooled two independent experiments. *P < 0.001; **P < 0.01 were determined by the Kruskal–Wallis method for area under the curve values for individual mice. (C) On day 10 the mice were killed, one hind paw per mouse was prepared for histology, and sections were stained with H&E [for inflammation (☆) and erosion (▵)] and toluidine blue (for cartilage damage, ▴). The inflammation, erosion, and cartilage scores were statistically lower in the 1Z88 and the 1Z105 treated mice. *P < 0.05; **P < 0.01 by one-way analysis of variance with Dunnett’s post hoc testing. Shown are examples (50× magnification) of H&E and toluidine blue stained talonavicular joints of the vehicle- (D and E), 1Z88- (F and G), and 1Z105- (H and I) treated animals.
Discussion
In a previous study, we identified by HTS small molecules that activated THP-1 NF-κB reporter cells. Further experiments with a hit compound (1Z65) identified TLR4/MD2 as the molecular target. In this study, we evaluated the immunologic and toxicity profiles of multiple derivatives of 1Z65 that retained activity in the NF-κB reporter cell line. All of the compounds required both TLR4 and MD2 but not CD14 for IL-6 and IP-10 induction. These cytokines have been reported to be dependent on the MyD88 and the TRIF pathways, respectively.
Activation of the TLR4 signaling pathway by bacterial LPS requires orchestrated action of LPS-binding protein, MD2, TLR4, and CD14 (Moore et al., 2000; Fitzgerald et al., 2004). CD14 is a glycosylphosphatidylinositol-anchored, membrane-bound, or secreted soluble protein that cannot independently signal. For lipid-based TLR ligands, CD14 is not required for TNF release but is required for MyD88-independent (TRIF) signaling (Jiang et al., 2005). The CD14 coreceptor aids the recruitment of various ligands, including LPS, lipoteichoic acid, ceramide, or poly(I:C)/double-stranded RNA to their respective TLRs, facilitating dimerization (Lee et al., 2006; Akashi-Takamura and Miyake, 2008; Baumann et al., 2010). In addition, CD14 also acts as a carrier protein to introduce LPS or other molecules into the intracellular endosomal components that trigger activation of the interferon regulatory factor 3 type I IFN pathway (Jiang et al., 2005; Baumann et al., 2010). However we observed CD14-independent induction of IP-10 (a surrogate for type I IFN production) by 1Z105 and 1Z88, suggesting that these small molecule TLR4 ligands can bypass the CD14 requirement to access the endosomal components of dendritic cells and macrophages and to stimulate IFN release.
The ability to limit TLR-triggered proinflammatory cytokine release has been demonstrated in preclinical models to be an effective strategy to prevent both innate and adaptive immune-mediated tissue injury. The paradoxical ability of TLR ligands to dampen the cellular response to other TLR activators was previously described (Fan and Cook, 2004; Hayashi et al., 2009). Various TLR active agents have been reported to be effective in preclinical models of inflammatory bowel disease, arthritis, multiple sclerosis, Alzheimer's disease, and asthma (Francis and Durham, 2004; Abdollahi-Roodsaz et al., 2007; Michaud et al., 2013). There are multiple complex mechanisms that influence the "TLR tolerance" phenomenon. Prominent among the feedback inhibitors of TLR activation is the IRAKM that reduces signaling through the MyD88 pathway. IRAKM has a variant in the terminal domain that renders it defective in transmitting a signal, such that it functions as a biologic decoy protein or an innate "dominant negative" protein (Hayashi et al., 2009). Other mechanisms for feedback inhibition of TLR-induced activation cascades include upregulation of the SHIP-1 phosphatase, miRNA-induced alterations in protein synthesis and degradation, and induction of suppressors of cytokine signaling proteins (Medvedev et al., 2006; Hayashi et al., 2009; Bao and Liu, 2013). The complexity of the cross-tolerance phenomenon has led to an intensive search for novel drugs that can restrain the proinflammatory effects of TLR stimulation without altering other cellular functions.
Here we demonstrated the ability of both 1Z88 and 1Z105 to reduce TLR4-mediated hepatic injury and cytokine storm in the LPS/galactosamine mouse model that requires TLR4. The K/BxN serum transfer arthritis model requires MyD88, but TLR4 is dispensable. The course of K/BxN arthritis in TLR4-deficient mice is the same as in WT mice (Christianson et al., 2011). However, treatment with either parenteral 1Z88 or oral 1Z105 minimized joint inflammation in this robust arthritis model. The therapeutic efficacy of the two agents may be attributable to inhibition of MyD88 signaling by negative regulators. Thus although 1Z88 and 1Z105 are specific for TLR4, they exert broad anti-inflammatory effects.
Several TLR4 ligands, like MPLA, aminoalkyl glucosaminide phosphates, glucopyranosyl lipid adjuvant, and E6020 were developed as semisynthetic vaccine adjuvants (Ismaili et al., 2002; Ishizaka and Hawkins, 2007; Johnson, 2008; Arias et al., 2012). These contain modified lipid A scaffolds that are not absorbed from the gastrointestinal tract unless the intestinal epithelial barrier is compromised. Our pharmacokinetics study showed that 1Z105 was orally bioavailable and nontoxic at doses that inhibited inflammation. Further formulation should improve the oral adsorption of this compound.
Both orally or parenterally administered 1Z105 reduced TNFα release in sera by high-dose LPS (20 μg/animal) alone or after LPS challenge in galactosamine preconditioned mice. The doses of 1Z105 that suppressed proinflammatory cytokine storm by subsequent LPS challenge did not cause proinflammatory cytokine release in sera. In preliminary experiments we have not yet achieved a toxic oral dose of 1Z105 in mice. Although MPLA is known as a detoxified form of LPS (Casella and Mitchell, 2013), it has a narrower therapeutic window than 1Z105. In contrast to MPLA, 1Z105 did not cause any mortality when injected into mice preconditioned with galactosamine (Fig. 5). These results document that 1Z105 is a safe and orally available TLR4 ligand that can restrain inflammatory processes that depend on MyD88 signaling.
The TLR4 antagonist Eritoran has been investigated for its ability to limit the cytokine storm produced by LPS release in Gram-negative bacterial infections (Kim et al., 2007; Barochia et al., 2011). In recent clinical trials this approach has not improved patient survival (Opal et al., 2013). However Eritoran does not inhibit MyD88 activation by TLR4 independent pathways as 1Z88 and 1Z105 do. Orally active and well tolerated synthetic TLR modulators, such as 1Z105, might be useful in many noninfectious inflammatory diseases. Their relatively short duration of pharmacokinetics would allow for treatment interruption in the setting of an acute infection.
In summary, we have described synthetic low molecular weight TLR4 ligands, 1Z105 and 1Z88, that target the TLR4/MD2 complex. Although these drugs can elicit proinflammatory cytokine release in vitro at high concentrations, when used in vivo they inhibit the production of these cytokines by diverse stimuli that stimulate the MyD88 pathway. One of these molecules, 1Z105, was modestly orally bioavailable and reduced joint inflammation in murine model of arthritis.
Supplementary Material
Acknowledgments
The authors thank Dr. Lynn Hajjar (University of Washington) for providing Tlr4−/−Md2−/− mice, Dr. Karen Messer (UCSD) for statistical consultation, and Richard Mathewson for technical and editorial assistance.
Abbreviations
- BMDC
bone marrow–derived dendritic cells
- BMDM
bone marrow–derived macrophages
- DMEM
Dulbecco’s modified Eagle’s medium
- HTS
high-throughput screen
- IFN
interferon
- IP-10
interferonγ-induced protein 10
- IRAK
IL-1 receptor–associated kinase
- ISRE
interferon-sensitive response element
- LPS
lipopolysaccharide
- MD2
myeloid differentiation protein 2
- MPLA
monophosphoryl lipid A
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-dipheyl tetrazolium bromide
- MyD88
myeloid differentiation primary response 88
- NF-κB
nuclear factor-κB
- Pam3Cys
(S)-(2,3-bis(palmitoyloxy)-(2RS)-propyl)-N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys4-OH, trihydrochloride
- PSA
polar surface area
- SAR
structure–activity relationship
- TLR
Toll-like receptor
- TNFα
tumor necrosis factor α
- TRIF
Toll/IL-1 receptor domain–containing adapter-inducing interferon-β
- WT
wild-type
Authorship Contributions
Participated in research design: Hayashi, Corr, Cottam, Carson.
Conducted experiments: Hayashi, Corr, Crain, Yao, Caneda, Chan.
Contributed new reagents or analytic tools: Cottam, Chan.
Performed data analysis: Hayashi, Corr, Crain, Yao, Cottam, Chan, Carson.
Wrote or contributed to the writing of the manuscript: Hayashi, Corr, Cottam, Carson.
Footnotes
This work was supported in part by a grant from the National Institutes of Health National Institute of Allergy and Infectious Diseases [Grant HHSN272200900034C] (D.A.C.); the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant R21-AR062236-02]; and an Innovative Research Grant from the Rheumatology Research Foundation (M.Co.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, van der Meer JW, Netea MG, van den Berg WB. (2007) Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum 56:2957–2967 [DOI] [PubMed] [Google Scholar]
- Akashi-Takamura S, Miyake K. (2008) TLR accessory molecules. Curr Opin Immunol 20:420–425 [DOI] [PubMed] [Google Scholar]
- Arias MA, Van Roey GA, Tregoning JS, Moutaftsi M, Coler RN, Windish HP, Reed SG, Carter D, Shattock RJ. (2012) Glucopyranosyl Lipid Adjuvant (GLA), a Synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS ONE 7:e41144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao M, Liu YJ. (2013) Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. Protein Cell 4:40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. (2011) Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin Drug Metab Toxicol 7:479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Blüml S, Grebien F, Bruckner M, Pasierbek P, Aumayr K, Planyavsky M, et al. (2010) CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med 207:2689–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B. (2000) Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12:20–26 [DOI] [PubMed] [Google Scholar]
- Casella CR, Mitchell TC. (2013) Inefficient TLR4/MD-2 heterotetramerization by monophosphoryl lipid A. PLoS ONE 8:e62622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M, Hayashi T, Kuy CS, Gray CS, Wu CC, Corr M, Wrasidlo W, Cottam HB, Carson DA. (2009) Synthesis and immunological characterization of toll-like receptor 7 agonistic conjugates. Bioconjug Chem 20:1194–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M, Hayashi T, Mathewson RD, Nour A, Hayashi Y, Yao S, Tawatao RI, Crain B, Tsigelny IF, Kouznetsova VL, et al. (2013) Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J Med Chem 56:4206–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, Yaksh TL. (2011) Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain 152:2881–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corr M, Crain B. (2002) The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J Immunol 169:6604–6609 [DOI] [PubMed] [Google Scholar]
- Crozat K, Georgel P, Rutschmann S, Mann N, Du X, Hoebe K, Beutler B. (2006) Analysis of the MCMV resistome by ENU mutagenesis. Mamm Genome 17:398–406 [DOI] [PubMed] [Google Scholar]
- Ditzel HJ. (2004) The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med 10:40–45 [DOI] [PubMed] [Google Scholar]
- Engelich G, White M, Hartshorn KL. (2001) Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol 69:50–56 [PubMed] [Google Scholar]
- Fan H, Cook JA. (2004) Molecular mechanisms of endotoxin tolerance. J Endotoxin Res 10:71–84 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Golenbock DT. (2004) Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect 6:1361–1367 [DOI] [PubMed] [Google Scholar]
- Francis JN, Durham SR. (2004) Adjuvants for allergen immunotherapy: experimental results and clinical perspectives. Curr Opin Allergy Clin Immunol 4:543–548 [DOI] [PubMed] [Google Scholar]
- Guma M, Kashiwakura J, Crain B, Kawakami Y, Beutler B, Firestein GS, Kawakami T, Karin M, Corr M. (2010) JNK1 controls mast cell degranulation and IL-1beta production in inflammatory arthritis. Proc Natl Acad Sci USA 107:22122–22127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar AM, Ernst RK, Fortuno ES, 3rd, Brasfield AS, Yam CS, Newlon LA, Kollmann TR, Miller SI, Wilson CB. (2012) Humanized TLR4/MD-2 mice reveal LPS recognition differentially impacts susceptibility to Yersinia pestis and Salmonella enterica. PLoS Pathog 8:e1002963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Gray CS, Chan M, Tawatao RI, Ronacher L, McGargill MA, Datta SK, Carson DA, Corr M. (2009) Prevention of autoimmune disease by induction of tolerance to Toll-like receptor 7. Proc Natl Acad Sci USA 106:2764–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaka ST, Hawkins LD. (2007) E6020: a synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev Vaccines 6:773–784 [DOI] [PubMed] [Google Scholar]
- Ismaili J, Rennesson J, Aksoy E, Vekemans J, Vincart B, Amraoui Z, Van Laethem F, Goldman M, Dubois PM. (2002) Monophosphoryl lipid A activates both human dendritic cells and T cells. J Immunol 168:926–932 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. (2005) CD14 is required for MyD88-independent LPS signaling. Nat Immunol 6:565–570 [DOI] [PubMed] [Google Scholar]
- Johnson DA. (2008) Synthetic TLR4-active glycolipids as vaccine adjuvants and stand-alone immunotherapeutics. Curr Top Med Chem 8:64–79 [DOI] [PubMed] [Google Scholar]
- Kerns EH, Di L. (2008) Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization, Academic Press, Amsterdam [Google Scholar]
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, et al. (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906–917 [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Saitoh S, Tanimura N, Takahashi K, Kawasaki K, Nishijima M, Fujimoto Y, Fukase K, Akashi-Takamura S, Miyake K. (2006) Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J Immunol 176:6211–6218 [DOI] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. (1996) Organ-specific disease provoked by systemic autoimmunity. Cell 87:811–822 [DOI] [PubMed] [Google Scholar]
- Lee HK, Dunzendorfer S, Soldau K, Tobias PS. (2006) Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity 24:153–163 [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. (2008) LPS/TLR4 signal transduction pathway. Cytokine 42:145–151 [DOI] [PubMed] [Google Scholar]
- Mangialaio S, Ji H, Korganow AS, Kouskoff V, Benoist C, Mathis D. (1999) The arthritogenic T cell receptor and its ligand in a model of spontaneous arthritis. Arthritis Rheum 42:2517–2523 [DOI] [PubMed] [Google Scholar]
- Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. (2007) The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316:1628–1632 [DOI] [PubMed] [Google Scholar]
- McAleer JP, Vella AT. (2010) Educating CD4 T cells with vaccine adjuvants: lessons from lipopolysaccharide. Trends Immunol 31:429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev AE, Sabroe I, Hasday JD, Vogel SN. (2006) Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res 12:133–150 [DOI] [PubMed] [Google Scholar]
- Michaud JP, Hallé M, Lampron A, Thériault P, Préfontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, et al. (2013) Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci USA 110:1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Andersson LP, Ingalls RR, Monks BG, Li R, Arnaout MA, Golenbock DT, Freeman MW. (2000) Divergent response to LPS and bacteria in CD14-deficient murine macrophages. J Immunol 165:4272–4280 [DOI] [PubMed] [Google Scholar]
- Ohto U, Fukase K, Miyake K, Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316:1632–1634 [DOI] [PubMed] [Google Scholar]
- Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, et al. ACCESS Study Group (2013) Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309:1154–1162 [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458:1191–1195 [DOI] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, et al. (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497:498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulevitch RJ, Tobias PS. (1995) Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol 13:437–457 [DOI] [PubMed] [Google Scholar]
- Wu CC, Hayashi T, Takabayashi K, Sabet M, Smee DF, Guiney DD, Cottam HB, Carson DA. (2007) Immunotherapeutic activity of a conjugate of a Toll-like receptor 7 ligand. Proc Natl Acad Sci USA 104:3990–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Takeda K, Akira S. (2004) TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol 40:861–868 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.