

Abstract
Mutations within the gene encoding the DNA helicase RECQL4 underlie the autosomal recessive cancer-predisposition disorder Rothmund-Thomson syndrome, though it is unclear how these mutations lead to disease. Here, we demonstrated that somatic deletion of Recql4 causes a rapid bone marrow failure in mice that involves cells from across the myeloid, lymphoid, and, most profoundly, erythroid lineages. Apoptosis was markedly elevated in multipotent progenitors lacking RECQL4 compared with WT cells. While the stem cell compartment was relatively spared in RECQL4-deficent mice, HSCs from these animals were not transplantable and even selected against. The requirement for RECQL4 was intrinsic in hematopoietic cells, and loss of RECQL4 in these cells was associated with increased replicative DNA damage and failed cell-cycle progression. Concurrent deletion of p53, which rescues loss of function in animals lacking the related helicase BLM, did not rescue BM phenotypes in RECQL4-deficient animals. In contrast, hematopoietic defects in cells from Recql4Δ/Δ mice were fully rescued by a RECQL4 variant without RecQ helicase activity, demonstrating that RECQL4 maintains hematopoiesis independently of helicase activity. Together, our data indicate that RECQL4 participates in DNA replication rather than genome stability and identify RECQL4 as a regulator of hematopoiesis with a nonredundant role compared with other RecQ helicases.
Introduction
Germline mutations in RECQL4 are found in the majority of Rothmund-Thomson syndrome (RTS) patients (OMIM 268400) (1–5). RTS is a rare autosomal recessive disorder that presents with epithelial features (skin atrophy, hyper/hypopigmentation), congenital skeletal malformations (leading to short stature), premature aging, and an increased incidence of cancer, including osteosarcoma and hematological malignancy (4–6). RTS patients can present with multiple malignancies and are more susceptible to chemotherapy-induced malignancy (7, 8). Mutations in RECQL4 also associate with 2 additional syndromes, Rapadilino and Baller-Gerold syndrome (BGS), that share varying degrees of overlap in their clinical manifestation with RTS (6, 9). The in vivo functions of RECQL4 in mammalian systems have remained unclear. A better understanding of the functions of RECQL4 is likely to provide important insight into the diseases associated with RECQL4 mutation in humans.
RECQL4 is 1 of 5 human DNA helicases that have evolved from prokaryote recQ+ (9, 10). Similar to RECQL4, the related Werner (WRN) or Bloom (BLM) syndrome helicases are associated with familial cancer predisposition and aging syndromes. RECQL4 has been demonstrated to act as an ATP-dependent DNA helicase and to play a role in maintaining genome stability (11). In contrast to other RecQ helicases, RECQL4 displays additional homology in its N-terminal region to yeast Sld2 and may thereby participate in the initiation of DNA replication (12–18). The relative importance of the RecQ helicase function of RECQL4 compared with its role in DNA replication has not been ascertained. Mutations associated with RTS predominantly affect the helicase function and are largely absent from the N-terminal Sld2-like region of RECQL4 (1–5).
Murine models have revealed stark differences in survival depending on the region of Recql4 targeted. Missense mutations or hypomorphic Recql4 alleles are viable, whereas an N-terminal targeted allele was embryonic lethal very early (19–21). To bypass embryonic lethality, we generated the first conditional allele of Recql4 and investigated the role of RECQL4 upon widespread somatic deletion in the adult mouse. Recql4Δ/Δ mice developed acute BM failure, with progression to hematopoietic failure. There was evidence of highly elevated levels of cell death in vivo. The B cell and T cell lineage defects could be recapitulated in vitro, demonstrating a cell intrinsic requirement for RECQL4. Loss of RECQL4 caused an increase in DNA damage and an accumulation of cells in the S-phase of the cell cycle, most consistent with a DNA replication failure. Strikingly, and in agreement with this interpretation, hematopoietic colony formation and B and T cell differentiation could be fully rescued by a RecQ helicase inactive mutated RECQL4. Collectively, these data identify RECQL4 as a unique regulator of hematopoiesis that has functions distinct from other RecQ helicases in the regulation of hematopoiesis.
Results
Generation of Recql4fl/fl mice.
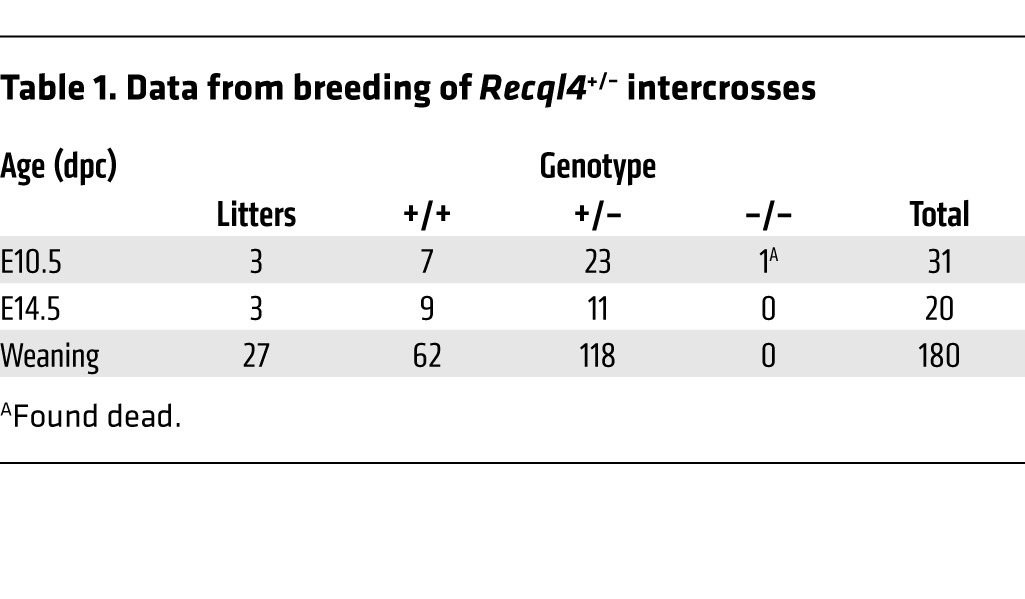
We generated a conditional allele of Recql4 by inserting loxP sites on either side of exons 9 and 10 (Recql4fl/fl) that encodes the start of the RecQ helicase domain, the location of the majority of mutations in RTS patients (refs. 5, 6, and Supplemental Figure 1, A–C; supplemental material available online with this article; doi:10.1172/JCI75334DS1). Homozygous Recql4fl/fl mice were viable and fertile and displayed no phenotype. This allele was efficiently excised on Cre expression, resulting in a loss of detectable protein (Supplemental Figure 1, D and E). Recql4+/– mice were normal and fertile, with no apparent basal phenotype or aging-induced phenotypes. Intercrosses of Recql4+/– did not yield any viable Recql4–/– pups at weaning. Analysis of embryos as early as E10.5 did not recover any viable Recql4–/– embryos. This early embryonic lethality of germline loss of RECQL4 is consistent with the previously reported germline N-terminal targeted allele (Table 1).
Table 1.
Data from breeding of Recql4+/– intercrosses
Widespread somatic deletion of Recql4 results in BM failure.
We crossed the Recql4fl/fl allele to the Rosa26-CreERT2 (R26-CreER; ref. 22) line to allow tamoxifen-induced somatic deletion across a broad range of tissues. At 7 weeks of age, cohorts of R26-CreERki/+Recql4+/+, R26-CreERki/+Recql4fl/+, and R26-CreERki/+Recql4fl/fl were fed tamoxifen containing chow for up to 30 days. This resulted in efficient recombination of the genomic locus in the BM (Figure 1A). Within approximately 3 weeks, R26-CreERki/+Recql4fl/fl (Recql4Δ/Δ) began to display signs of illness, most notably weight loss and pale tail tips and feet (Figure 1A and data not shown). Within 4 weeks, all Recql4Δ/Δ displayed this phenotype, with no effect on the R26-CreERki/+Recql4+/+ and R26-CreERki/+Recql4fl/+ controls.
Figure 1. Somatic deletion of Recql4 causes anemia and leucopenia.

(A) Schematic for the deletion strategy used in 7-week-old mice using R26-CreERT2 and genomic PCR demonstrating deletion of Recql4 in BM. (B) Leukocyte counts in the PB of control (+/+) and Recql4 deficient (Δ/Δ) mice. (C) Absolute numbers of leukocyte subsets in PB. (D) Platelet count. (E) rbc count. (F) Hemoglobin. (G) Hematocrit. (H) images of BM flushes from the indicated genotypes. (I) Representative histology of the tibia from epiphysis (region 1); trabecular region (arrowheads indicated adipocytes; region 2); and cortical region (region 3) of the bone. Scale bars: 50 μm; 20 μm (higher power image, region 3). Data expressed as mean ± SEM, Student’s t test. *P < 0.05; **P < 0.01. n ≥ 5 per genotype. Experiments were independently performed on at least 3 separate cohorts, with results pooled for presentation.
Analysis of the peripheral blood (PB) revealed severe multilineage cytopenias in Recql4Δ/Δ (Figure 1, B–G). Effects were observed across all lineages with myeloid, B lymphoid, and T lymphoid cell numbers severely reduced. Surprisingly, we observed no change in platelet numbers in the PB (Figure 1D). Accompanying the PB cytopenias, Recql4Δ/Δ mice displayed profound red cell aplasia in the setting of a moderately hypocellular BM (Figure 1, H and I). Consistent with the normal platelet count in the PB, megakaryocyte numbers were not reduced in the BM. The BM architecture was grossly disorganized, and there were distinctive changes in the BM microenvironment. There was an accumulation of adipocytes, known to impair hematopoietic support and often accompanying BM failure (Figure 1I and ref. 23). Autopsy did not reveal any gross pathology of other organs. All together, the Recql4Δ/Δ mice had BM failure with a relative sparing of the megakaryocyte lineage.
Closer examination of the BM of Recql4Δ/Δ animals revealed an approximately 70% decrease in BM cellularity (Figure 2A). Myeloid output was reduced and there were defects in B lymphoid development, predominantly at the transition from pre-proB to proB stages (Figure 2, B and C). BM erythropoiesis was reduced, with the most marked reductions occurring in the earliest nucleated erythroid populations as assessed by either CD71/Ter119 or Ter119/CD44/FSC phenotyping (Figure 2, D and E, and refs. 24, 25). When the expression pattern of Recql4 transcript was assessed in the Gene Expression Commons database, the most severely impacted lineages in Recql4Δ/Δ displayed a striking and specific overlap with the points of highest transcript expression (Supplemental Figure 2 and ref. 26). Splenic erythropoiesis was significantly decreased (Supplemental Figure 3). Thymic lymphopoiesis was reduced, and T cell progenitors were severely impaired. All of the CD4+ and CD8+ cell populations in the BM of Recql4Δ/Δ were TCRβ positive, indicating recirculating mature cells (Supplemental Figure 3). Due to the rapid BM failure, we assessed whether there were changes in apoptosis. Immunohistochemical analysis revealed an approximately 2.5-fold increase in cleaved caspase-3–positive cells in the BM of the Recql4Δ/Δ, suggesting that the rapid BM failure may be due to elevated apoptosis (Figure 2F).
Figure 2. BM failure following deletion of Recql4.

(A) BM cellularity per femur.(B) Numbers of granulocytes and macrophages per femur. (C) B lymphocyte number per femur and the indicated B cell subset. (D) Erythroid fractions based on CD71/Ter119 staining and representative FACS plots. (E) Erythroid fractions based on Ter119/CD44/FSC phenotype. (F) Analysis of cleaved caspase-3 staining in the BM. Scale bars: 100 μm (low power image); 50 μm (higher power image). Quantitation of percentage cleaved caspase-3 positive cells in each genotype from 3 hpf per sample, n = 3 per genotype. Data expressed as mean ± SEM, Student’s t test. *P < 0.05; **P < 0.001. n > 5 per genotype (A–E). Experiments were independently performed on at least 3 separate cohorts, with results pooled for presentation.
To determine whether BM failure was due to stem cell and/or progenitor depletion, we assessed these populations using multiple phenotypic approaches (27–30). Surprisingly, the phenotypic long- and short-term HSC numbers per femur were relatively unaffected by loss of Recql4 (Figure 3, A and C). However, there were significant changes in the myelo-erythroid progenitor populations. Granulocyte-macrophage progenitors (GMPs) and common myeloid progenitors (CMPs) were maintained at the expense of the megakaryocyte-erythroid progenitor (MEP) (Figure 3B). A higher fidelity analysis of the erythroid progenitors demonstrated substantially compromised pools of pre-MegE and CFU-Es (Figure 3, D and E). Colony forming cell assays demonstrated approximately 60% reduction in progenitors in Recql4Δ/Δ BM (Supplemental Figure 4). These data demonstrate that loss of RECQL4 leads to a rapid-onset, multilineage BM failure, demonstrating an essential requirement for RECQL4 in the maintenance of hematopoiesis.
Figure 3. Preservation of HSCs but depletion of erythroid progenitors following Recql4 loss.

(A) HSC and primitive progenitors based on analysis of LKS CD34/Flt3 staining profiles. (B) Myeloid progenitor frequency and representative FACS profiles. (C) HSC and primitive progenitors based on analysis of LKS CD150/CD105 staining profiles. (D) Myeloid progenitors per femur. (E) Erythroid and megakaryocyte progenitor frequency in the BM and representative FACS plot. Data expressed as mean ± SEM, Student’s t test. n > 5 per genotype. Experiments were independently performed on at least 3 separate cohorts, with results pooled for presentation.
Increased cell death of early progenitors and mature cells.
To better understand the mechanism for the BM failure, we analyzed tamoxifen-treated R26-CreERki/+Recql4+/+, R26-CreERki/+Recql4fl/+, and R26-CreERki/+Recql4fl/fl cohorts at earlier time points, before they displayed any clinical disease (14 days; Figure 4A). The PB and BM phenotypes were similar to those seen after 30 days of tamoxifen, albeit less severe (Table 2 and Figure 4, B–G). Using annexinV/7AAD staining, we found increased proportions of dead cells (annexinV+/–7AAD+) in the Recql4Δ/Δ BM, most pronounced in the multipotent progenitor (MPP) (Figure 4H), committed progenitor fractions (Figure 4, I and J), and erythroid populations (Figure 4K and Supplemental Table 1). Thus, the selective death of progenitor populations leads to the observed BM failure. There were significantly elevated levels of DNA damage in the Recql4Δ/Δ BM and thymus as assessed by γH2A.X staining, a marker of DNA breaks (Figure 5, A–C). In addition, increased intracellular cleaved caspase-3 was apparent in thymocytes in vivo (Figure 5C), correlating with the decreased thymus cellularity. These results demonstrate that deletion of Recql4 causes the elimination of proliferative progenitor cells through increased DNA damage and ultimately apoptosis.
Figure 4. Loss of RECQL4 compromises survival of the progenitor compartment.

(A) Schematic for the deletion strategy used in 7-week-old mice and genomic PCR in BM after 14 days of tamoxifen. (B) Femur cellularity. (C) B lymphocyte number per femur and the indicated subsets. (D) Erythroid fractions based on Ter119/CD44/FSC phenotype. (E) HSC and primitive progenitors based on analysis of LKS CD150/CD105. (F) HSC and primitive progenitors based on analysis of LKS CD34/Flt3. (G) Myeloid progenitor frequency and representative FACS profile. (H) Percentage dead (7AAD+AnnexinV+/–) of the LKS CD150/CD105 populations. (I) Percentage dead among erythroid progenitors in the BM. (J) Increased death of the proB and preB cell populations in the BM. (K) Analysis of death in the committed erythroid cells using Ter119/CD44/FSC fractionation. Fold difference appears below indicated P values. Data expressed as mean ± SEM, Student’s t test (Supplemental Table 1). *P < 0.05, **P < 0.01 or as indicated. n ≥ 3 per genotype.
Table 2.
PB parameters after 14 days of tamoxifen food
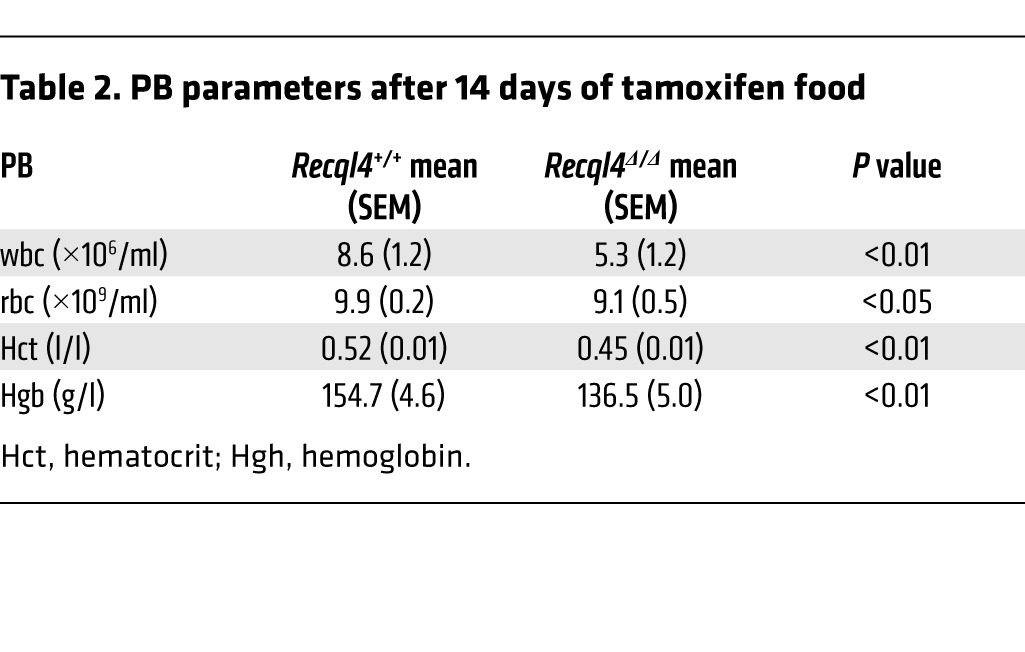
Figure 5. Increased DNA damage and apoptosis of lymphoid cells following loss of Recql4.

(A) Intracellular cleaved caspase-3 and γH2AX costaining B cell fractions of the BM. (B) Quantitation of cleaved caspase-3 and γH2AX in B cells as either the percentage or absolute number of positive cells per femur. (C) Total BM and thymus cleaved caspase-3 and γH2AX. Data expressed as mean ± SEM, Student’s t test. *P < 0.05. n ≥ 3 per genotype.
RECQL4 is essential for HSCs to repopulate hematopoiesis.
R26-CreER mediates deletion of Recql4 in many cell types, including hematopoietic cells. To ascertain whether the BM failure was reflective of a hematopoietic cell–intrinsic role for RECQL4, we performed a series of BM transplants and employed an additional HSC-specific Cre strain (31–34). Competitive transplantation of BM derived from tamoxifen-treated R26-CreERki/+Recql4+/+, R26-CreERki/+Recql4fl/+, and R26-CreERki/+Recql4fl/fl donors was performed (data not shown). This approach assesses the potential of the HSCs remaining after Recql4 deletion, but does not exclude a role for RECQL4 in regulating the supportive potential of the BM microenvironment. Surprisingly, PB chimerism was comparable among all genotypes (Supplemental Figure 5). Isolation of the donor-derived cells revealed a profound selection against cells deleted for Recql4, with retention of an unexcised allele resulting in Recql4Δ/fl cells (Supplemental Figure 5). Similar negative selection phenotypes have been reported with floxed alleles for other genes with essential hematopoietic functions, such as Tel, Mll, and Adar1 (35–37).
To directly address the hematopoietic-intrinsic role of RECQL4, we transplanted nondeleted fetal liver or adult BM from R26-CreERki/+Recql4+/+, R26-CreERki/+Recql4fl/+, and R26-CreERki/+Recql4fl/fl into congenic recipients. After 4 to 6 weeks of recovery, chimerism was assessed, and then the recipients were placed on tamoxifen-containing chow. The only cells expressing Cre under these conditions are the hematopoietic cells, directly addressing the hematopoietic-intrinsic requirement. In both cases, long-term chimerism was comparable between groups, and isolation of donor cells from the BM and genomic PCR revealed homozygous deletion of Recql4 could not be maintained and was selected against, resulting in reconstitution by cells that had retained the floxed allele (Figure 6A).
Figure 6. Profound selection against Recql4 deletion in HSCs.

(A) Fetal liver or whole BM was isolated from donors of the indicated genotypes and transplanted into irradiated CD45.1 recipients. Hematopoiesis was allowed to establish for 5 weeks; then chimerism was assessed (4-week time point). Recipients were then placed on tamoxifen food for 30 days. PB was serially monitored at the indicated times, and total chimerism (CD45.2+ve) and myeloid chimerism (Mac-1+ve) are shown from each independent experiment. At end point, CD45.2 cells were isolated from the BM and genomic PCR performed to determine RECQL4 status;data shown are representative genotyping results from 1 of the experiments; n = 5 recipients/genotype/experiment. (B) hScl-CreERT R26eYFP Recql4 mice of the indicated genotypes were fed tamoxifen diet for 30 days. One cohort was left for long-term observation (upper panels). At end point, eYFP+ve cells were isolated from the BM and genomic PCR performed to assess Recql4 status; the percentage of eYFP was assessed in the PB and BM. A second cohort was analyzed at the end of the 30-day feeding of tamoxifen. Femur cellularity, contribution of eYFP+ve cells to each phenotypic population, and genomic PCR were performed. n > 3 per genotype per experiment. Data expressed as mean ± SEM, Student’s t test. *P < 0.05.
To corroborate the hematopoietic intrinsic function of Recql4 we employed the hScl-CreERT strain, which allows specific gene deletion in the HSCs and primitive progenitors without the need for prior HSC transplant (37–39). hScl-CreERT R26-eYFPki/kiRecql4+/+, R26-eYFPki/kiRecql4fl/+, and R26-eYFPki/kiRecql4fl/fl were fed tamoxifen chow for 30 days to induce efficient gene deletion in HSCs. hScl-CreERT R26-eYFPki/kiRecql4fl/fl did not present with a phenotype. At more than 5 months after tamoxifen cessation, eYFP+ cells from the BM were isolated and gene deletion assessed. Consistent with previous results, we only detected stable deletion of a single floxed allele in eYFP-positive BM cells of the Recql4fl/fl mice (Figure 6B). Mice analyzed immediately following cessation of tamoxifen did not have any changes in contribution to hematopoiesis of the eYFP-positive cells (Figure 6B). Transplant studies demonstrate that RECQL4 is essential for multilineage hematopoiesis and reconstitution by HSCs. Furthermore, using 3 distinct HSC analysis methods, we demonstrate very strong selection against deletion of Recql4.
Specific intrinsic requirements for Recql4 in B and T cell development.
To dissect B and T cell development in greater detail, we utilized an in vitro culture system. Lineage-negative, Sca-1–positive, c-Kit–positive (LKS+) cells were isolated from untreated R26-CreERki/+Recql4+/+ and R26-CreERki/+Recql4fl/fl BM and placed into culture with tamoxifen for 5 days (Figure 7A). Cells were then transferred to differentiation cultures: an OP9 stromal cell layer for B cell development or OP9 cells expressing the Notch ligand delta-like 1 (OP9-DL1) for T cell development (40–42). Recql4Δ/Δ cells cultured under B cell conditions displayed a mild proliferative disadvantage (data not shown) and recapitulated the impaired differentiation detected in vivo with impaired transition from pre–proB to proB cells (Figure 7B and Supplemental Figure 6). On OP9-DL1, Recql4 deficiency caused impairment at multiple stages, with total T cell output dramatically reduced. DN2b and DN3a subsets, phases where proliferation slows and stalls before β-selection, accumulated with a loss of more mature and proliferative T cell subsets. (Figure 7, C–E).
Figure 7. RECQL4 is intrinsically required for B and T cell development.

(A) Outline of culture system to assess the requirement for RECQL4 in B and T cell development. (B) B cell development from the indicated genotype at 14 days after addition to the OP9 cultures. (C) Representative FACS plots from DN2 and DN3/4 T cell development at 14 days after addition to OP9-DL1 cells. (D) Quantitation of T cell development from DN1-DN4. (E) Analysis of DN2-4 subset development following loss of Recql4. (F) Representative FACS plots of intracellular DNA damage (γH2AX) and DNA content. (G) Quantitation of cell-cycle distribution and DNA damage in T cell cultures at day 14. (H) Cell-cycle distribution and DNA damage in B cell cultures at day 14. Data expressed as mean ± SEM, Student’s t test. *P < 0.05; **P < 0.01. n = 4 per genotype. Experiments were independently performed on 2 separate occasions, with results pooled for presentation.
The differentiation stages affected by RECQL4 deficiency overlap closely with the peak expression of Recql4 transcript in B and T cells and temporally precede B and T cell receptor rearrangement, suggesting that the RECQL4 requirement is not linked to V(D)J recombination and DNA repair (Figure 7C). There were significantly increased levels in γH2A.X staining in Recql4Δ/Δ cultures during S-phase specifically and an accumulation of cells in S-phase (Figure 7, F–H, and Supplemental Figure 6C). Our data demonstrate that loss of RECQL4 results in increased replicative DNA damage, reminiscent of recent studies linking RECQL4 orthologues in other organisms to essential roles in DNA replication (12, 15, 16).
RECQL4 is dispensable for terminal erythroid differentiation.
Deletion of Recql4 using the R26-CreER model revealed a severe impact on erythroid progenitors that was not seen in the closely related myeloid progenitors (CMP and GMP, Figure 2). To determine whether there was an erythroid intrinsic requirement of RECQL4, independent of the role in lineage commitment, we selectively deleted Recql4 in erythroid progenitors using Epor-Cre (43, 44). Surprisingly we did not detect a difference in PB erythroid indices (Figure 8A). The deletion efficiency was comparable between genotypes and the erythroid progenitor populations were similar (Figure 8, B–D). BM erythropoiesis was largely normal, with the exception of a mild reduction at the latest stages of differentiation (Figure 8, E and F). A modest, but not statistically significant, increase in the CD71+Ter119+ erythroblasts in the spleen was seen. In contrast to the negative selection using Rosa-Cre-ERT2 and Scl-CreERT (Figure 6), isolated eYFP-positive cells from the BM confirmed efficient Epor-Cre–mediated recombination of both alleles of Recql4 (Figure 8H). Therefore, RECQL4 is dispensable for the differentiation of committed erythroid cells.
Figure 8. RECQL4 is dispensable for terminal erythroid differentiation.

(A) PB parameters of erythroid-specific Epor-CreR26eYFPki/kiRecql4fl/+ and Epor-CreR26eYFPki/kiRecql4fl/fl mutants. (B) Percentage eYFP-positive cells in each population for the indicated genotypes. (C) Numbers of erythroid progenitors per femur. (D) Numbers of myelo-erythroid progenitors per femur. (E) Erythroid fractions based on Ter119/CD44/FSC phenotype. (F) Erythroid fractions based on Ter119/CD71 phenotype. (G) Erythroid populations in the spleen. (H) Genomic PCR from eYFP+ve BM cells demonstrating efficient deletion of Recql4 using Epor-Cre. Data expressed as mean ± SEM, Student’s t test. n ≥ 4 per genotype.
Concurrent loss of p53 does not rescue RECQL4 deficient hematopoiesis.
Given the extensive apoptosis and DNA damage observed in Recql4Δ/Δ BM, we tested whether concurrent p53 deletion would protect from these effects. p53 plays a critical role in cell cycle control and apoptosis phenotypes of both the Blm–/– and Wrn–/– mouse (45–47). In Fanconi anemia (FA), in which BLM helicase is implicated, knockdown of p53 can rescue many hematopoietic phenotypes (48, 49). We generated R26-CreERki/+Recql4fl/+, R26-CreERki/+Recql4fl/fl, R26-CreERki/+p53fl/fl, and R26-CreERki/+Recql4fl/flp53fl/fl (dKO) cohorts and placed all on tamoxifen chow for 30 days. Concurrent deletion of p53 was not able to rescue the hematopoietic phenotypes seen in Recql4Δ/Δ (Figure 9, A–H, and Supplemental Table 2). Loss of p53 does not modify the Recql4Δ/Δ BM failure, differentiating Recql4 from the closely related Blm in the regulation of hematopoiesis.
Figure 9. Concurrent p53 deletion does not modify the RECQL4 deficient phenotype.

(A) Schematic of analysis of Recql4 p53 double mutants. (B) PB leukocyte and rbc counts. (C) BM cellularity. (D) HSC and MPP number per femur. (E) BM B cell populations. (F) BM erythroid populations based on CD71/Ter119 staining. (G) Thymus cellularity. (H) Thymic CD4/CD8 populations. There are no statistical differences between the R26-CreER Recql4Δ/Δ and R26-CreER Recql4Δ/Δp53Δ/Δ (dKO) for any of the parameters assessed at day 30 of analysis. Data expressed as mean ± SEM, ordinary 1-way ANOVA with multiple comparisons test. Statistical comparison only shown for Recql4Δ/Δ versus dKO. Full statistical analysis of all comparisons in Supplemental Table 2. n ≥ 4 per genotype.
RecQ helicase–independent role for Recql4 in hematopoiesis.
To determine to what extend loss of RECQL4 RecQ helicase activity or another function (such as Sld2-like replicative functions) is responsible for the hematopoietic defects, we tested the ability of GFP-fused human WT RECQL4 (RECQL4WT) or a RecQ helicase–dead variant (RECQL4K508A) to rescue Recql4Δ/Δ cells (11, 17). RECQL4K508A has an alanine substitution of the conserved lysine in the Walker A motif of the RecQ helicase domain that is essential for ATP hydrolysis (50–52). LKS+ cells were isolated from R26-CreERki/+Recql4+/+ and R26-CreERki/+Recql4fl/fl and either placed directly into colony assays with 4-OHT or cultured and infected with retrovirus expressing WT or mutant GFP-RECQL4 (Figure 10A).
Figure 10. The RecQ DNA helicase function of RECQL4 is not required for hematopoiesis.

(A) Outline of experimental design and constructs tested for rescue. (B) Genomic PCR from cells of each population treated for 5 days with tamoxifen, demonstrating efficient gene deletion, and CFU assays counted at day 7 from each population with tamoxifen added to the methylcellulose. (C) Analysis of the rescue of colony formation in cells of the indicated genotypes infected either with WT or K508A helicase defective RECQL4; GFP–ve cells were included as a control. (D) Fold change in GFP levels and representative FACS plots relative to initial transduction levels over 21 days of culture for cells of the indicated genotypes. (E) B cell development is normalized by the K508A mutant. (F) Rescue of T cell development by helicase defective RECQL4. Data expressed as mean ± SEM, Student’s t test. n = 4 per genotype. Experiments were independently performed on 2 separate occasions with results pooled for presentation.
Deletion of Recql4 reduced colony formation from the LKS+ cells by approximately 70% compared with controls (Figure 10B). There was complete rescue of colony formation by LKS+ cells following the overexpression of RECQL4WT or RECQL4K508A and deletion of the endogenous Recql4 alleles (Figure 10C). Serving as an internal control, GFP–ve cells from the R26-CreERki/+Recql4fl/fl cultures were not rescued. We also performed B and T cell cultures and observed an outgrowth of GFP+ve cells from the Recql4Δ/Δ cultures with overexpression of either RECQL4WT or RECQL4K508A (Figure 10D). Over 21 days of culture, the Recql4Δ/Δ cultures became almost completely composed of GFP+ve cells. When B and T cell differentiation was assessed, rescue was found with both RECQL4WT and RECQL4K508A (Figure 10, E and F, and Supplemental Figure 7). Collectively, these studies demonstrate that the requirement for RECQL4 in hematopoiesis was RecQ helicase independent.
Discussion
The role of RECQL4 in vivo has been poorly characterized compared with related RecQ helicases. The relative importance of its contribution to genome stability via its RecQ helicase domain and to DNA replication via its Sld2 homology region have not been delineated. Our conditional Recql4 allele demonstrated an essential function for Recql4 in the maintenance of hematopoiesis. This phenotype was associated with widespread apoptosis of progenitor cells and increased replicative DNA damage. Recql4Δ/Δ HSCs were not transplantable, and there was profound selection for retention of 1 unexcised allele. The requirement for RECQL4 was hematopoietic intrinsic and can be observed across B cell, T cell, and myeloid progenitors. Using viral complementation of null cells, we established that the RecQ helicase function of RECQL4 is not required for its role in hematopoiesis. Therefore, the primary role of RECQL4 in hematopoiesis is as a participant in DNA replication rather than the regulation of genome stability. Consistent with this interpretation is the lack of mutations affecting the Sld2 homology regions in RTS patients, which would be expected to be nonviable based on our analysis (3, 6). Clarification of the function of RECQL4 is important for our understanding of RTS and the mutation spectrum that is observed in patients. This should improve interpretation and analysis of sequence variants in people with a suspected RTS diagnosis.
The analysis of the conditional allele clarifies the previous ambiguity related to the phenotype of Recql4 KO mice. Three germline Recql4 alleles have been described. Homozygous loss of exons 5–8 resulted in very early embryonic death (20), mirroring the early embryonic lethality of the germline deletion of exons 9–10 reported with our KO allele (Supplemental Figure 1). An in-frame deletion of exon 13 caused a deficiency in helicase activity and was associated with neo- and postnatal lethality and other defects (19). A third mouse lacking exons 9–13 showed palate and limb defects, aneuploidy, poikiloderma, and cancer predisposition (21). Interestingly, in the 2 lines where viable animals were recovered at birth, aberrantly truncated Recql4 transcripts can be found. Exon 13 nulls expressed a transcript lacking exon 13, and several shortened truncated transcripts encompassing exons 1–12 can be observed (19). Exon 9–13 KOs express 3′ truncated transcripts fusing exons 1–8 to the PGK cassette used to disrupt the gene (21). Thus, the viable hypomorphic alleles are able to generate an N-terminal fragment containing the Sld2 domain but lacking helicase activity, suggesting that the essential function of RECQL4 resides in its DNA replication function rather than its helicase activity. This is consistent with evidence from the analysis of cells engineered to express only the N-terminal region of RECQL4, which lacks the RecQ helicase domain. These cells had a normal basal proliferation rate but impaired DNA damage response. This demonstrated that expression of only the N-terminal, Sld2 homology region of RECQL4 protein is sufficient for normal proliferation (51).
Comparison of conditional KO of Recql4 to that of other RecQ helicases revealed some striking differences in hematopoietic activity. Wrn–/– are not reported to have a hematological phenotype (53, 54). Blm–/– died at approximately E14.5 and had a distinct defect in erythropoiesis, including inclusion bodies but no other described problems, and they survived much later in development than the Recql4–/– (55). Unlike Blm–/– animals, the concurrent loss of p53 had no effect on the outcome of deletion of Recql4 on the hematopoietic system (47). Recql5–/– are homozygous viable without reported hematologic phenotypes, but develop tumors at long latency (~50% of mice), with both hematologic and solid tumors represented (56). We therefore conclude that RECQL4 plays a nonredundant role with other RecQ helicases in hematopoieisis.
The requirement for RECQL4 in hematopoiesis is not uniform. The megakaryocyte lineage is preserved and platelet numbers in the PB remain unchanged. PB platelets have a normal morphology in RECQL4-deficient animals, and the numbers of megakaryocytes in the BM were comparable between controls and RECQL4-deficient animals (Recql4+/+ 6.2 ± 1.43 megakaryocytes per high-power field (hpf); Recql4Δ/Δ 6.23 ± 1.30 megakaryocytes/hpf; 10 hpf/tibia/animal assessed, n = 3 per genotype, data expressed as mean ± SEM). The other populations of cells that are spared consist of the HSCs and primitive progenitor populations, in contrast to the severely compromised committed progenitors. Recently, a population of phenotypic HSCs has been identified that possess a platelet-specific gene expression signature and display an ability at the single-cell level to directly differentiate into megakaryocytes and related myeloid lineages (57, 58). One explanation could be that differentiation of committed progenitors from HSCs is impaired except toward the megakaryocytic progenitors. Alternatively, megakaryocytes may be unique in their lack of requirement for RECQL4. During polyploidization, there is a reduction in the expression of genes involved in DNA replication and repair, including known binding partners of RECQL4 (59, 60). We assessed the expression of Recql4 in murine megakaryocytes via analysis of publicly available microarray data sets. In 2 independent data sets, Recql4 is below the detection threshold in the megakaryocyte populations (60). We therefore propose that the main determinate of the lack of megakaryocyte/platelet phenotype in the Recql4Δ/Δ animals is the very low/negligible expression of RECQL4 during normal megakaryopoiesis.
Our preferred model is that the major determinant of the cellular phenotype is the relative cell division rate of the respective populations (61). The increased division of committed progenitors causes an increased rate of dilution of the RECQL4 protein present after recombination of the locus, to the critical point after which aberrant DNA replication occurs. The dilution model also hints at why we observed no requirement for RECQL4 in terminal erythroid differentiation using Epor-Cre. Erythroblasts undergo a limited number of cell divisions prior to enucleation (4 to 5 cell divisions from the proerythroblast stage), eliminating the need for further DNA replication (24, 62). Thus, viable erythroid cells were found in the Epor-Cre Recql4Δ/Δ that had genomically deleted Recql4 but did not divide sufficiently after this point to dilute the remaining protein to a critical point. Consistent with this is that only the last stages of erythroid differentiation demonstrated any reduction. This model accounts for the apparent sparing of the primitive HSC fractions, as these populations are known to be relatively quiescent (61, 63). This explanation is consistent with recent evidence from the Xenopus midblastula transition, where it was shown that dilution of limiting RECQL4 protein levels restricts the initiation of DNA replication (16). Thus, the in vivo phenotype is driven by the rates of dilution of RECQL4 protein after gene recombination, and the cells that divide most rapidly, such as the MPPs and committed progenitors, are most severely impacted by the absence of RECQL4. These results differentiate RECQL4 from the other mammalian RecQ helicases and demonstrate a unique role for RECQL4 in the maintenance of hematopoiesis.
The phenotype of the Recql4Δ/Δ hematopoietic system, with the exception of the sparing of the megakaryocyte lineage, shares many of the key features of aplastic anemia in humans (64, 65). The genetic causes of aplastic anemia are largely unknown, and the diagnosis is largely by exclusion. While it remains to be determined whether mutations in RECQL4 may contribute to the genesis of aplastic anemia, the striking overlap of symptoms warrants further investigation of RECQL4 in these diseases. It may be warranted to assess the status of RECQL4 in patients with unexplained BM failure and in syndromes with clinical overlap with RTS, such as xeroderma pigmentosum, ataxia telangiectasia, FA, and dyskeratosis congenita. The RecQ helicase BLM interacts with the FA pathway (49, 66). However, the loss of RECQL4 resulted in a phenotype distinct from that associated with loss of Blm or reported in FA. Concurrent loss of p53 does not modify the Recql4Δ/Δ phenotype, whereas reducing the levels of p53 can normalize HSC function in FA cells (48). Additionally, at a cellular level, Recql4Δ/Δ cells display an S-phase accumulation, whereas FA cells accumulate intrastrand crosslinks and arrest in the G2/M phase of the cell cycle (66). One striking feature of the loss of RECQL4 function is the profound disruption to immune development. B cell differentiation is impaired from the pre-proB cell stage onward in the BM, and T cell development appears to be compromised by the loss of BM-derived thymic reseeding cell populations. There are reports of immune compromise in patients with RTS (67, 68). The hematopoietic abnormality in 1 of these patients was corrected by an allogeneic cord blood stem cell transplant (68), confirming that, as in the mouse, this is a hematopoietic-intrinsic requirement for RECQL4. The range of severity of immune phenotypes and lineages affected in RTS patients may be reflective of retained function of the mutant RECQL4 protein.
The majority of mutations in RTS patients are located in the helicase domain of the protein and, most likely, given the early lethality of Recql4 null alleles, result in the expression of a C-terminal truncated hypomorphic RECQL4 protein with intact Sld2-like function but absent/reduced helicase activity. Consistent with a hypomorphic allele, Drosophila mutants of Recql4 (RecQ4) have been isolated that are viable, display a range of phenotypes, and express only approximately 6% of the WT protein levels (69). The mutant protein is sufficient for DNA replication, but not for participation in DNA repair, potentially accounting for the aneuploidy reported in mice containing the C-terminal truncated alleles (21, 70, 71). The analysis of the conditional allele has demonstrated that the essential in vivo function of Recql4 is in DNA replication rather than genome stability. This analysis allows a better understanding of the consequences of RECQL4 mutation and accounts for the prevalence of RTS mutations found within the helicase domain and their absence from the Sld2-like N-terminal region of RECQL4.
Methods
Mice.
Recql4fl/fl mice (C57BL/6-Recql4tm2272Arte) were generated by TaconicArtemis GmbH. Full details of the allele are provided in the Supplemental Methods.
Rosa26-CreERT2 mice (22) on a C57BL/6 background were purchased from The Jackson Laboratory and were used as heterozygotes for all experiments (B6.129-Gt[ROSA]26Sortm1(cre/ERT2)Tyj/J; stock number: 008463). Epor-Cre, p53fl/fl, R26eYFPki/ki and hScl-CreERT (provided by J. Göthert, University Hospital of Essen, Essen, Germany) have been previously described (37, 38, 43, 44, 72, 73). All lines were on a backcrossed C57BL/6 background.
Tamoxifen-containing food was prepared by Specialty Feeds at 400 mg/kg tamoxifen citrate (Sigma-Aldrich) in a base of standard mouse chow.
Flow cytometry analysis.
PB was analyzed on a hematological analyzer (Sysmex KX-21N; Roche Diagnostics). Bones were flushed, spleens crushed, and single-cell suspensions were prepared. Antibodies against murine Ter119, CD71, B220, IgM, Mac-1, Gr1, F4/80, CD43, CD19, CD4, CD8, CD44, Sca-1, c-Kit, CD34, FLT3, FcγR (CD16/32), CD41, CD51, and CD150, either biotinylated or conjugated with FITC, phycoerythrin, phycoerythrin-Cy5, peridinin chlorophyll protein–Cy5.5, phycoerythrin-Cy7, allophycocyanin, or allophycocyanin Alexa Fluor 750 were all obtained from eBioscience. CD105 and CD150 were from BioLegend. Biotinylated antibodies were detected with streptavidin conjugated with Alexa Fluor 405 or Brilliant Violet-605 (Invitrogen or BioLegend, respectively) (43). AnnexinV-APC (eBioscience) and 7-AAD (Molecular Probes) were used to assess viability as previously described (37). Cells were sorted on a BD FACSAria Cell Sorter (BD Biosciences) or analyzed on a BD LSRIIFortessa (BD Biosciences). Results were analyzed with FlowJo software version 9.0 (TreeStar).
Hematopoietic CFU assays.
50,000 whole BM cells, 300 LKS+ cells, 500 CMPs, or 1000 GMPs were plated per ml of methylcellulose medium containing 50 ng/ml murine Stem Cell Factor (mSCF) (PeproTech), 10 ng/ml murine IL-3 (mIL-3) (PeproTech), 50 ng/ml human IL-6 (hIL-6) (Amgen), and 3 U/ml rhEpo (Jansen-Cilag). The methylcellulose medium used for colony assays contained 1% methylcellulose (Methocel MC no. 64630; Fluka), IMDM (Invitrogen), 20% FBS for mouse myeloid CFC (Stem Cell Technologies), penicillin/streptomycin (P/S) (Gibco; Invitrogen), GlutaMAX (Gibco; Invitrogen) and cytokines as described. Colonies were counted on day 7 and day 12 as described (32, 43).
Flow cytometric analysis of intracellular γH2AX (phospho-histone H2A.XSer139) and active caspase-3.
1–2 × 106 cells were stained for cell-surface markers for 30 minutes at 4°C, washed once in PBS, then fixed and permeabilized for 20 minutes at room temperature (RT) using the BD Cytofix/Cytoperm (cat. No. 554714). Cells were pelleted, washed with BD Perm/Wash, and stained with biotin rabbit anti-active caspase-3 antibody clone C92-605 (BD) in BD Perm/Wash buffer for 1 hour at RT. Cells were next washed and resuspended in BD Perm/Wash buffer containing Brilliant Violet 605 Streptavidin and Alexa Fluor 647 rabbit anti-γH2AX antibody clone 20E3 (Cell Signaling) for 1 hour at RT, washed with BD Perm/Wash buffer, and resuspended in FACS buffer before analyzing by FACS. Results were analyzed with FlowJo Version 9.0 (TreeStar).
Flow cytometric analysis of intracellular γH2AX (phospho-histone H2A.XSer139) and cell cycle.
T and B cell precursors were collected from the cocultures by filtration over 40 micron filters, washed with PBS, then fixed with 2% PFA for 20 minutes at RT. Cells were permeabilized with ice-cold methanol for 30 minutes on ice, washed with PBS/BSA, and resuspended in Alexa Fluor 647 rabbit anti-γH2AX antibody clone 20E3 (Cell Signaling) for 1 hour at RT. Cell were washed, resuspended in PBS containing 20 g/ml propidium iodide (PI) and 100 g/ml RNase A, and incubated for 1 hour before analyzing by flow cytometry. Results were analyzed with FlowJo software Version 9.0 (TreeStar). γH2AX foci were visualized on a per-cell basis using a laser scanning cytometer (Compucyte).
In vitro B and T cell assay.
Cells were initially treated with 500 nM 4-hydroxytamoxifen (4OH-T) (Merck) for 5 days to induce Cre activity and maintained in 100 nM of 4OH-T to enforce gene deletion. B cell cultures were performed in α-MEM (Sigma-Aldrich) with 20% FBS (PAA Gold), P/S, GlutaMAX, 5 × 10–5 M 2-mercaptoethanol (Sigma-Aldrich), 5 ng/ml Flt3L (PeproTech), and 1 ng/ml mIL-7 (R&D Systems) and 100 nM 4-OH tamoxifen. B cell development was analyzed by flow cytometry on days 7, 14, and 21 (n = 4 separate donors/genotype). For T cell cultures, 20,000 OP9-DL1 cells (42) were plated in a 24-well plate 48 hours before infected LKS+ cells were added. T cell cultures were performed in α-MEM (Sigma-Aldrich) with 20% FBS (PAA Gold), P/S, GlutaMAX, 5 × 10–5 M 2-mercaptoethanol (Sigma-Aldrich), 5 ng/ml Flt3L (PeproTech), 0.25 ng/ml mIL-7 (R&D Systems), and 100 nM 4-OH tamoxifen (74, 75). T cell development was analyzed by flow cytometry on days 7, 14, and 21 (n = 4 separate donors/genotype).
Transplantation assays.
Transplant configurations are outlined in the relevant figures. Transplants depicted in Figure 6A were performed once with fetal liver–derived cells and a second time with adult BM–derived donor cells. For transplant of cells from tamoxifen-fed donors, 5 × 105 unfractionated BM cells from R26-CreERT2ki/+Recql4+/+, R26-CreERT2ki/+Recql4fl/+, or R26-CreERT2ki/+Recql4fl/fl were competitively transplanted with 2 × 105 WT BM cells (CD45.1/CD45.2). For transplant of non–tamoxifen-treated samples, either E14.5 fetal liver or adult BM of the indicated genotypes was isolated and transplanted into recipients. Recipients were allowed to recover for 4 weeks then assessed for chimerism. Recipients were placed on tamoxifen-containing food for 30 days, and then PB was analyzed at the indicated times. For all experiments, CD45.2+ve BM cells were isolated at end point to use for genomic PCR to assess Recql4 deletion.
All transplants were performed into lethally irradiated (2 × 5 Gy dose 3 hours apart; total of 10 Gy) recipients (CD45.1) through intravenous injections. All recipients received antibiotics for 3 weeks after transplant (Baytril). A total of 5 to 6 recipients per treatment were transplanted; the experiment was performed in duplicate. PB was taken at the indicated times to monitor multilineage donor reconstitution and PB counts.
Retroviral complementation.
Human EGFP-RECQL4 and EGFP-RECQL4K508A (provided by T. Enomoto, Musashino University, Tokyo, Japan; ref. 17) were cloned into MSCV. All plasmids were sequence confirmed. Retrovirus was produced by transient transfection of 293T cells using calcium phosphate precipitation. LKS+ cells were isolated and cultured in StemSpan SFEM (Stem Cell Technologies) with 20 ng/ml rhIL-6 (Amgen), 10 ng/ml rmIL-3 (Peprotech), 50 ng/ml rmSCF (Peprotech), and 5 ng/ml rhFlt3L (Peprotech) for 48 hours. Cells were then centrifuged with 50% (v/v) retroviral supernatant at 1100 g for 90 minutes in 8 μg/ml polybrene. Following infection, cells were cultured for a further 48 hours before being GFP sorted or used for in vitro culture.
Analysis of Recql4 expression in murine megakaryocytes.
The expression of Recql4 in murine megakaryocytes was assessed via analysis of publicly available microarray data sets (GEO, NCBI). In 2 independent sets, Recql4 was below the detection threshold in the megakaryocyte populations (GEO GSE49664 and GSE6593).
Statistics.
The significance of results was analyzed using the unpaired 2-tailed Student’s t test on the basis of highly inbred littermate mice having been used in all experiments using Prism software. P < 0.05 was considered significant. All data are presented as mean ± SEM.
Study approval.
All animal experiments were approved by the Animal Ethics Committee at St. Vincent’s Hospital.
Supplementary Material
Acknowledgments
The authors would like to thank V. Sankaran, R. Dickins, and S. Lane for comments and discussion; S. Dewamitta and B. Liddicoat for technical assistance; and the SVH BioResources Centre for care of experimental animals. This work was supported by grants from the Leukaemia Foundation (to C.R. Walkley); a National Health and Medical Research Council (NHMRC) project grant (to C.R. Walkley); an NHMRC Career Development Award (to C.R. Walkley); an NHMRC Senior Research Fellowship (to J. Heierhorst and L.E. Purton); a Victorian Cancer Agency Clinical Research Fellowship (to M. Wall); and in part by the Victorian State Government Operational Infrastructure Support Program (to St. Vincent’s Institute). C.R. Walkley is the Phillip Desbrow Senior Research Fellow of the Leukaemia Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(8):3551–3565. doi:10.1172/JCI75334.
References
- 1.Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A. Rothmund-Thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999;61(3):268–276. doi: 10.1006/geno.1999.5959. [DOI] [PubMed] [Google Scholar]
- 2.Kitao S, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22(1):82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- 3.Siitonen HA, et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003;12(21):2837–2844. doi: 10.1093/hmg/ddg306. [DOI] [PubMed] [Google Scholar]
- 4.Wang LL, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95(9):669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- 5.Wang LL, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001;102(1):11–17. doi: 10.1002/1096-8628(20010722)102:1<11::AID-AJMG1413>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 6.Siitonen HA, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17(2):151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. Am J Med Genet A. 2010;152A(6):1575–1579. doi: 10.1002/ajmg.a.33427. [DOI] [PubMed] [Google Scholar]
- 8.Carlson AM, Lindor NM, Litzow MR. Therapy-related myelodysplasia in a patient with Rothmund-Thomson syndrome. Eur J Haematol. 2011;86(6):536–540. doi: 10.1111/j.1600-0609.2011.01609.x. [DOI] [PubMed] [Google Scholar]
- 9.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9(9):644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 10.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374(pt 3):577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009;28(5):568–577. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sangrithi MN, et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121(6):887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009;28(19):3005–3014. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marino F, Vindigni A, Onesti S. Bioinformatic analysis of RecQ4 helicases reveals the presence of a RQC domain and a Zn knuckle. Biophys Chem. 2013;177–178:34–39. doi: 10.1016/j.bpc.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445(7125):281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- 16.Collart C, Allen GE, Bradshaw CR, Smith JC, Zegerman P. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science. 2013;341(6148):893–896. doi: 10.1126/science.1241530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe T, Yoshimura A, Hosono Y, Tada S, Seki M, Enomoto T. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells. Role of the N-terminal region of RECQL4 in cells. Biochim Biophys Acta. 2011;1813(3):473–479. doi: 10.1016/j.bbamcr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci. 2005;118(pt 18):4261–4269. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- 19.Hoki Y, et al. Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum Mol Genet. 2003;12(18):2293–2299. doi: 10.1093/hmg/ddg254. [DOI] [PubMed] [Google Scholar]
- 20.Ichikawa K, Noda T, Furuichi Y. [Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases]. Nihon Yakurigaku Zasshi. 2002;119(4):219–226. doi: 10.1254/fpj.119.219. [DOI] [PubMed] [Google Scholar]
- 21.Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14(6):813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- 22.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 23.Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259–263. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev. 2008;22(4):463–475. doi: 10.1101/gad.1627208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, et al. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood. 2013;121(8):e43–e49. doi: 10.1182/blood-2012-09-456079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seita J, et al. Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS One. 2012;7(7):e40321. doi: 10.1371/journal.pone.0040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pronk CJ, et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 2007;1(4):428–442. doi: 10.1016/j.stem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, et al. Identification of Lin-Sca1+kit+CD34+Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated recipients. Blood. 2005;105(7):2717–2723. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- 29.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 30.Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273(5272):242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 31.Purton LE, Scadden DT. Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell. 2007;1(3):263–270. doi: 10.1016/j.stem.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–1095. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walkley CR, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. 2007;129(6):1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell. 2013;13(5):520–533. doi: 10.1016/j.stem.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Hock H, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18(19):2336–2341. doi: 10.1101/gad.1239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1(3):324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gothert JR, et al. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood. 2005;105(7):2724–2732. doi: 10.1182/blood-2004-08-3037. [DOI] [PubMed] [Google Scholar]
- 39.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265(5175):1098–1101. doi: 10.1126/science.8066449. [DOI] [PubMed] [Google Scholar]
- 41.Cho SK, Webber TD, Carlyle JR, Nakano T, Lewis SM, Zuniga-Pflucker JC. Functional characterization of B lymphocytes generated in vitro from embryonic stem cells. Proc Natl Acad Sci U S A. 1999;96(17):9797–9802. doi: 10.1073/pnas.96.17.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17(6):749–756. doi: 10.1016/S1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- 43.Singbrant S, et al. Erythropoietin couples erythropoiesis, B-lymphopoiesis, and bone homeostasis within the bone marrow microenvironment. Blood. 2011;117(21):5631–5642. doi: 10.1182/blood-2010-11-320564. [DOI] [PubMed] [Google Scholar]
- 44.Heinrich AC, Pelanda R, Klingmuller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004;104(3):659–666. doi: 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 45.Lombard DB, et al. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol. 2000;20(9):3286–3291. doi: 10.1128/MCB.20.9.3286-3291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babbe H, et al. Genomic instability resulting from Blm deficiency compromises development, maintenance, and function of the B cell lineage. J Immunol. 2009;182(1):347–360. doi: 10.4049/jimmunol.182.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Babbe H, Chester N, Leder P, Reizis B. The Bloom’s syndrome helicase is critical for development and function of the αβ T-cell lineage. Mol Cell Biol. 2007;27(5):1947–1959. doi: 10.1128/MCB.01402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceccaldi R, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s syndrome and Fanconi anemia. Mol Cell. 2009;36(6):943–953. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst). 2010;9(7):796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohzaki M, et al. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis. 2012;33(6):1203–1210. doi: 10.1093/carcin/bgs149. [DOI] [PubMed] [Google Scholar]
- 52.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang S, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36(8):877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 54.Du X, et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24(19):8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chester N, Kuo F, Kozak C, O’Hara CD, Leder P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998;12(21):3382–3393. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu Y, et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21(23):3073–3084. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanjuan-Pla A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502(7470):232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto R, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154(5):1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 59.Raslova H, et al. Interrelation between polyploidization and megakaryocyte differentiation: a gene profiling approach. Blood. 2007;109(8):3225–3234. doi: 10.1182/blood-2006-07-037838. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z, Hu M, Shivdasani RA. Expression analysis of primary mouse megakaryocyte differentiation and its application in identifying stage-specific molecular markers and a novel transcriptional target of NF-E2. Blood. 2007;109(4):1451–1459. doi: 10.1182/blood-2006-08-038901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sankaran VG, et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 2012;26(18):2075–2087. doi: 10.1101/gad.197020.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. 1997;25(5):445–453. [PubMed] [Google Scholar]
- 64.Rovo A, Tichelli A, Dufour C. Diagnosis of acquired aplastic anemia. Bone Marrow Transplant. 2013;48(2):162–167. doi: 10.1038/bmt.2012.230. [DOI] [PubMed] [Google Scholar]
- 65.Guinan EC. Diagnosis and management of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:76–81. doi: 10.1182/asheducation-2011.1.76. [DOI] [PubMed] [Google Scholar]
- 66.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122(11):3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Somer L, et al. Granulomatous skin lesions complicating Varicella infection in a patient with Rothmund-Thomson syndrome and immune deficiency: case report. Orphanet J Rare Dis. 2010;5:37. doi: 10.1186/1750-1172-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Broom MA, et al. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. Clin Genet. 2006;69(4):337–343. doi: 10.1111/j.1399-0004.2006.00592.x. [DOI] [PubMed] [Google Scholar]
- 69.Wu J, Capp C, Feng L, Hsieh TS. Drosophila homologue of the Rothmund-Thomson syndrome gene: essential function in DNA replication during development. Dev Biol. 2008;323(1):130–142. doi: 10.1016/j.ydbio.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Werner SR, Prahalad AK, Yang J, Hock JM. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun. 2006;345(1):403–409. doi: 10.1016/j.bbrc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- 71.Yang J, et al. Recql4 haploinsufficiency in mice leads to defects in osteoblast progenitors: Implications for low bone mass phenotype. Biochem Biophys Res Commun. 2006;344(1):346–352. doi: 10.1016/j.bbrc.2006.03.104. [DOI] [PubMed] [Google Scholar]
- 72.Walkley CR, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008;22(12):1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994–1004. [PMC free article] [PubMed] [Google Scholar]
- 74.Smeets MF, Mackenzie-Kludas C, Mohtashami M, Zhang HH, Zuniga-Pflucker JC, Izon DJ. Removal of myeloid cytokines from the cellular environment enhances T-cell development in vitro. Int Immunol. 2013;25(10):589–599. doi: 10.1093/intimm/dxt025. [DOI] [PubMed] [Google Scholar]
- 75.Smeets MF, Chan AC, Dagger S, Bradley CK, Wei A, Izon DJ. Fli-1 overexpression in hematopoietic progenitors deregulates T cell development and induces pre-T cell lymphobl wild-type astic leukaemia/lymphoma. PLoS One. 2013;8(5):e62346. doi: 10.1371/journal.pone.0062346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.